

Abstract
The 60 nucleotide GTPase center (GAC) of 23S rRNA has a phylogenetically conserved secondary structure with two hairpin loops and a 3-way junction. It folds into an intricate tertiary structure upon addition of Mg2+ ions, which is stabilized by the L11 protein in cocrystal structures. Here we monitor the kinetics of its tertiary folding and Mg2+-dependent intermediate states by observation of selected nucleobases that contribute specific interactions to the GAC tertiary structure in the cocrystals. The fluorescent nucleobase 2-aminopurine (2AP) replaced three individual adenines, two of which make long-range stacking interactions, and one that also forms hydrogen bonds. Each site reveals a unique response to Mg2+ addition and temperature, reflecting its environmental change from secondary to tertiary structure. Stopped-flow fluorescence experiments revealed that kinetics of tertiary structure formation upon addition of MgCl2 are also site-specific, with local conformational changes occurring from 5 ms – 4 s, and with global folding from 1- 5 s. Site-specific substitution with 15N-nucleobases allowed observation of stable hydrogen bond formation by NMR experiments. Equilibrium titration experiments indicate that a stable folding intermediate is present at stoichiometric concentrations of Mg2+, and suggest that there are two initial sites of Mg2+ ion association.
Keywords: 2-aminopurine, stopped-flow fluorescence, RNA folding kinetics, Mg2+-dependent RNA folding, NMR
Graphical Abstract
INTRODUCTION
RNA molecules fold into their active tertiary structures, but the rules for folding are fuzzy[1,2][3–5]. The interactions among nucleotides that lead to the correct tertiary structure typically cannot be predicted by examination of the secondary structure. Unlike proteins, RNA can and does fold into alternative inactive tertiary structures[6–8], which sometimes can be rescued but sometime lead to degradation of the misfolded molecule. To study RNA tertiary structure, model systems are essential, and especially those that utilize a range of interactions that can be probed for their energetic and kinetic contributions to tertiary structure formation[9,10][11,12] . Here, we introduce a 60 nucleotide fragment from prokaryotic 23S rRNA as a new model system for the mechanics of tertiary structure formation.
In the large ribosomal subunit, the 60 nucleotide GTPase center (GAC) binds to the L11 protein[13],[14][15]. L11+GAC is part of a complex that is responsible for recruiting cofactors that provide the energy for the ribosome enzyme as it synthesizes protein[16]. The structure of the GAC is intricate as shown in cocrystals with L11[17],[18]; there is no structure of the GAC alone.
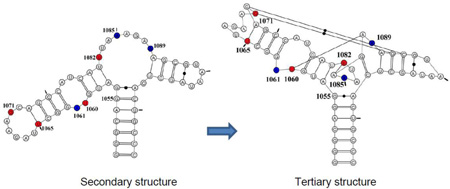
The secondary structure of the GAC is a dumbbell, comprised of two hairpin loops, connected to the remainder of 23S rRNA via a 3-way junction (Figure 1). Within the 60 nucleotides are 18 sites that are phylogenetically invariant, many of which are involved in unusual nucleotide interactions in the tertiary structure. The prokaryotic GAC (nt 1050–1108 using E coli numbering) requires monovalent ions (NH4+ or K+ are preferred[19],[20]) and/or divalent ions (Mg2+)[21] to adopt its tertiary structure. A variant of the E coli GAC containing a single nucleobase substitution (U1061A) is not able to adopt its tertiary structure without Mg2+ ions [20], and this GAC has been extensively studied for its thermodynamics of ion binding and subsequent RNA folding[17,22–24]. In particular, studies of Mg2+ ions and their functions in GAC stability and formation of tertiary structure led to a thermodynamic description of a unique chelated Mg2+ binding site[24].
Figure 1.


E coli U1061A GAC. A: secondary structure model. B: tertiary structure model. Nucleotides U1065-A1073 form a T-loop; nucleotides U1082-A1086 form a triloop within the junction region; nucleotides G1093-A1098 form a U-turn. Green connecting lines indicate long-range hydrogen bonds. Blue: 2AP-substituted 1061, 1085, 1089. Red: 15N-nucleotides U1060, U1065, G1071, U1082. Tic marks every 10 nucleotides.
Two cocrystals of GAC bound to the C-terminal domain of L11 proteins were solved: E. coli U1061A GAC bound to B stearothermophilus L11 (1hc8 [17]) and T. maritima U1061 GAC and L11 (1mms[18]). The GAC structures in the two cocrystals are nearly identical, despite their sequence variations. Each structure was solved with Mg2+ ions; in addition, the E coli GAC structure contained K+ and T maritima contained Cd2+. The presence of a chelated K+ ion in the E coli GAC structure at the base of the 1065–1073 stemloop suggested that this could be the site of the chelated Mg2+ ion[24]. We describe the structure of this seven nucleotide loop as a T-loop[25], consisting of a 5′ UNR four nucleotide U-turn [26] and a 3′ bulge; the bulged nucleotides A1070, G1071, and C1072, are splayed out and G1071 and C1072 make base triples with the stem of the 1092–1098 hairpin. The 3-way junction acts as a hinge and within that junction is a triloop formed by U1082-A1086. Also within that junction, at the end of the stem of the 1092–1098 hairpin, is a tight turn made of nucleotides 1087–1090 (Figure 1B). The hinge region of the GAC bends to juxtapose the two hairpin stems which are then connected via base triples (Figure 1B).
Here, in the context of U1061A GAC, we examine the formation of several tertiary interactions that use the nucleobases for stacking or hydrogen bonding. The structure of the GAC found in the cocrystals is our model for the final folded form of the RNA. Conn et al., [17] and Leipply and Draper [27] compared the structure of this GAC with Mg2+ ions with/without L11 protein using hydroxyl radical probing, and concluded that L11 did neither induce new structure nor alter existing structure, although protein binding did protect sites on the backbone. We therefore use GAC U1061A to study the temporal and equilibrium formation of 1) the long-range stacking interactions of nucleobase A1061 with nucleobase A1070; 2) stacking of nucleobase A1089 with G1071 and A1087; and 3) stacking of A1085 with its adjacent nucleobases in the triloop (Figure 1B). We substitute A1061, A1085, or A1089, with the fluorescent nucleobase 2-aminopurine (2AP) to measure the kinetics of Mg2+-dependent tertiary structure formation using stopped-flow fluorescence experiments. We observe multiexponential kinetic traces, which we interpret as reporting on local conformational changes unique to each probe, often on timescales from 10–90 ms, and global folding with time constants of 1–5 seconds. Investigations of the GAC structures at equilibrium during Mg2+-titrations used 15N-selectively-labeled RNAs in NMR experiments. At equimolar [GAC]/[Mg2+], we observe formation of hydrogen bonding by U1082 and U1065, which we posit are reporting on formation of the triloop in the junction (U1082:A1087) and the T-loop (U1065:A1073). Subsequently, we see the G1071 imino proton, which should reflect formation of base triples, and finally we see the imino proton of U1060. Folding pathways at equilibrium could reflect kinetic pathways, but more data are needed to compare them.
Results
2AP substitutions
In our experiments, we use the U1061A E coli GAC variant in order to unambiguously separate secondary and tertiary structure formations. In the U1061A GAC secondary structure, A1061 is in a symmetric internal loop where it could form a noncanonical base pair with A1077. However, in the tertiary structure seen in the cocrystals, U1061 (1mms) and A1061 (1hc8) stack with A1070 in a long range stacking interaction (Figure 2), but make no hydrogen bonding interactions. We replaced A1061 with 2AP (A1061AP) in the GAC.
Figure 2.

Adenine sites replaced with 2AP. Structures taken from 1hc8 cocrystal [17]. Structural representation by VMD[53].
A1089 is in the 3-way junction of the GAC secondary structure, but in the U1061A E coli GAC cocrystal tertiary structure (1hc8), A1089 is nearly coplanar with A1090, while A1090 forms a base pair with U1101 (Figure 2). A1089 is sandwiched between nucleobases G1087 (nonconserved) and G1071 (invariant). We replaced A1089AP, with the potential consequence that the planar interaction with A1090 would be distorted by the close proximity of the two amino groups, or alternatively, that U1101 could hydrogen bond to the 2-NH2 group of 2AP as well as the 6-NH2 of A1090 (Figure 2).
A1085 is invariant, and in the secondary structure is in the 3-way junction. In the cocrystals, it forms a minor groove base triple with conserved G1055:C1104 through its N1 position (Figure 2). It also makes hydrogen bonding interactions with several proximal riboses. In its position within the U1082-A1086 triloop, it stacks with A1084 and A1086. We substituted A1085AP.
Thermal denaturation experiments of GAC RNAs measured by UV absorbance were shown to differentially report on secondary structure (280 nm) and tertiary structure (260 nm) denaturation (melting) [20]. Comparable thermal denaturation experiments with the 2AP GAC RNAs are shown in Figure 3 as plots of dAbs/dT, and compared to the unmodified GAC RNA. In 100 mM KCl, in the absence of Mg2+, all RNAs have a broad transition at 260 nm, from ~43° to 60° C, and a weaker transition observed at 280 nm centered near 60° C. With addition of 3 mM MgCl2, two transitions are clearly visible at 260 nm, one near 56°-60° C (tertiary structures) and the other near 75° C (secondary structures)[21]. A1061AP and A1085AP RNAs both have a 260 nm shoulder near 45° C, while the corresponding A1089AP transition is broad. The tertiary structure transition in the unmodified GAC RNA is more intense than in the 2AP-RNAs, and though we conclude that all RNAs have adopted the tertiary structure in 3 mM MgCl2, the A1085AP transition has lower intensity, suggesting that its tertiary structure has been destabilized.
Figure 3.

Temperature-dependence of absorbance and fluorescence. UV thermal denaturation plotted dAbs/dT, at 260 nm (black) and 280 nm (red) in 100 mM KCl, 10 mM sodium cacodylate, pH 6.5, ±3 mM MgCl2, 2 μM RNA. A/E: unmodified E coli U1061A without/with 3 mM MgCl2. B/F: A1061AP without/with 3 mM MgCl2. C/G: A1085AP without/with 3 mM MgCl2. D/H: A1089AP without/with 3 mM MgCl2.
2AP Steady-state Fluorescence
We compared the fluorescence intensity of the three 2AP-GAC molecules as a function of temperature in 100 mM KCl ± 3 mM MgCl2, and found that each site has a unique temperature-dependence (Figure 4). Stacking interactions with other nucleobases (on the 3′ and/or 5′ side) will result in loss of 2AP fluorescence intensity (quenching)[28] leading to a preliminary physical interpretation of intensity changes.
Figure 4.

Temperature-dependence of GAC steady state 2AP fluorescence intensity. A1061AP (left), A1085AP (mid), A1089AP (right). Blue: no Mg2+. Red: 3 mM Mg2+. 2 μM RNA, 100 mM KCl, 10 mM sodium cacodylate pH 6.5.
The initial fluorescence intensity of A1085AP is relatively high, indicating that it is not in a stable stacking interaction. In the secondary structure, A1085 is in the hinge region, which hydroxyl radical probing has shown to be accessible in the absence of Mg2+ [17]. We suspect that the hinge is disordered in the secondary structure. The very similar temperature dependence of A1085AP fluorescence intensity with and without Mg2+ indicates that its environments are similar, although absorbance data show that there is clearly a Mg2+-dependent conformational change of the RNA.
The initial fluorescence intensity of A1061AP is quite low in the absence of Mg2+, consistent with base stacking. Its fluorescence intensity increases slightly upon addition of Mg2+, suggesting that its stacking interactions have altered, although overall it remains in a stacked environment. Perhaps addition of Mg2+ alters the structure of the internal loop around A1061AP, or perhaps it changes its stacking partners, since the cocrystals show it stacked with A1070 in the tertiary structure.
A1089AP in KCl maintains a high fluorescence intensity until the GAC starts to melt, then its fluorescence is quenched by intramolecular collisions and by solvent. With addition of 3 mM MgCl2, its fluorescence intensity decreases by half, consistent with moving into a more stacked environment. It remains in this environment until the tertiary structure starts to melt. However, its fluorescence intensity is not as low as that of A1061AP, suggesting that its stacking geometry or neighboring nucleobases are different.
Time-resolved fluorescence
Time-correlated single photon counting (TCSPC) experiments reveal the environments of 2AP; for example, does it exist in one geometry (such as a static stacked structure) or does it sample conformational space (is it dynamic?). The time-dependence of its fluorescence decay is fit to multiexponential lifetimes, where we interpret their amplitudes as fraction of time spent in different environments, and their lifetimes as the physical state of the nucleobase. 2AP nucleotide in solution has a single fluorescence lifetime τ ~10–11 ns, but in an RNA it will typically decay with several lifetimes unique to its physical environments. We assign long lifetimes (7–9 ns) to an unstacked state, and short lifetimes (< 0.5 ns) to a stacked environment. When 2AP is sampling local conformations or is perhaps in a non-canonical stacked orientation, its lifetime(s) is between those extremes.
Time-resolved anisotropy or fluorescence depolarization reports on molecular tumbling (rotational correlation time) and/or local motions of the fluorophore. In solution, 2AP nucleotide tumbles very rapidly, so its fluorescence anisotropy (depolarization) decay φ ~ 9 ps, but in an RNA, its depolarization will be longer. We typically fit the longest anisotropy component to the global rotational time of the molecule and the other to local motions. Based on the dimensions of the cocrystal, we estimated the global tumbling time of the compact folded GAC as <τR> = 5.6 ns at 30° C, but its secondary structure could tumble much slower if the structure is rigid, or alternatively, each arm could have independent motion.
We measured time-resolved fluorescence parameters for A1061AP, A1085AP and A1089AP RNAs at 30° C. Recall that steady-state fluorescence intensity of A1061AP and A1089 is constant from 10° to ~40° C, while that of A1085AP is decreasing. UV absorption of all GAC RNAs increases in 100 mM KCl at 30° C, indicating that some part of the GAC is beginning to melt, but with addition of 3 mM MgCl2, absorbance is near baseline. TCSPC and anisotropy data are given in Table 1.
Table 1.
Time-resolved fluorescence decay and anisotropy parameters
| A1061AP TCSPC |
amplitude 1 (%) |
τ1 ns | amplitude 2 (%) |
τ2ns | amplitude 3 (%) |
τ3 ns |
|---|---|---|---|---|---|---|
| 30 °C, no Mg2+ |
28% | 6.4 | 21% | 2.3 | 51% | 0.19 |
| 30 °C, 8 mM Mg2+ |
6.4% | 7.2 | 23% | 1.8 | 71% | 0.34 |
| TRA | Ro | β1 | φ1 ns | β2 | φ2 ns | χ2 |
| 30 °C, no Mg2+ |
0.27 | 48% | 4.6 | 52% | 0.42 | 1.2 |
| 30 °C, 8 mM Mg2+ |
0.30 | 79% | 5.8 | 21% | 1.6 | 1.3 |
|
A1089AP TCSPC |
amplitude 1 (%) |
τ1 ns |
amplitude 2 (%) |
τ2ns |
amplitude 3 (%) |
τ3 ns |
| 30 °C, no Mg2+ |
24% | 7.6 | 26% | 2.3 | 50% | 0.3 |
| 30 °C, 8 mM Mg2+ |
16% | 7.7 | 30% | 2.1 | 55% | 0.25 |
| TRA | Ro | β1 | φ1 ns | β2 | φ2 ns | χ2 |
| 30 °C, no Mg2+ |
0.28 | 56% | 4.3 | 44% | 0.66 | 1.1 |
| 30 °C, 8 mM Mg2+ |
0.26 | 76% | 5.6 | 24% | 0.78 | 1.2 |
|
A1085AP TCSPC |
amplitude 1 (%) |
τ1 ns |
amplitude 2 (%) |
τ2ns |
amplitude 3 (%) |
τ3 ns |
| 30 °C, no Mg2+ |
20% | 7.8 | 37% | 2.3 | 42% | 0.34 |
| 30 °C, 8 mM Mg2+ |
23% | 7.5 | 37% | 3.0 | 40% | 0.59 |
| TRA | Ro | β1 | φ1 ns | β2 | φ2 ns | χ2 |
| 30 °C, no Mg2+ |
0.27 | 48% | 6.8 | 52% | 0.81 | 1.4 |
| 30 °C, 8 mM Mg2+ |
0.26 | 79% | 6.3 | 21% | 0.53 | 1.2 |
TCSPC Time-correlated single photon counting. TRA Time-resolved anisotropy. 10 mM sodium cacodylate, pH 6.5, 100 mM KCl. [RNA] = 2 μM. 2-aminopurine nucleotide alone has a single fluorescence lifetime of 10–11 ns, and a single time-resolved anisotropy decay of 9 ps. Support-plane data analysis provided errors for the decay lifetimes, which are not shown here. Typically, < 10% uncertainties are symmetric around the given lifetime.
The internal symmetric loop
A1061AP is located in an internal loop in the stem of the 1065–1073 hairpin. Steady-state fluorescence intensity of A1061AP is quite low, ± Mg2+, suggesting that it spends most of its time stacked. Time-resolved fluorescence parameters support this interpretation (Table 1), since the dominant fluorescence decay has a lifetime τ3 = 190 ps (no Mg2+) or 340 ps (+ Mg2+). The anisotropy data provide a more nuanced picture: without Mg2+, there are equal contributions from global (φ1 = 4.6 ns) and local (φ2 = 0.42 ns) motions, while with Mg2+, ~80% of the depolarization occurs during global tumbling of the RNA (φ1 = 5.8 ns). Curiously, 20% of the depolarization decay is due to a local rotational correlation time of A1061AP φ2 = 1.6 ns, which is quite long. Considering steady-state fluorescence intensity and time-resolved fluorescence decay, we speculate that this nucleobase is jumping between alternative environments in the tertiary structure.
The Hinge Region
In the secondary structure, A1089 might be stacked with its neighbors, but this region of the GAC acts as a hinge, so it could be flexible. In the GAC tertiary structure, A1089 is stacked on both sides with other nucleobases, so we anticipated that its conformational mobility would be restricted. The surprise is that there are three virtually identical fluorescence lifetimes with and without Mg2+ (Table 1). We do observe a change in the proportion of each component: in Mg2+, the fraction of time spent in the unstacked environment is reduced. When GAC has adopted its tertiary fold in 8 mM Mg2+, we interpret the longest depolarization decay (φ1 = 5.6 ns) as global tumbling of the RNA, in agreement with the theoretical calculations. Global tumbling contributes the dominant component to the anisotropy, suggesting that A1089 is predominantly fixed in position (76%) in the folded GAC. The longest depolarization time of A1089AP in GAC without Mg2+ should also report on global tumbling. As with A1061AP, this decay time is significantly shorter (φ1 = 4.3 ns) than in the tertiary folded GAC; these data are consistent with a GAC with floppy arms that move independently of each other.
A1085 is part of a triloop in the tertiary structure, where it forms a minor groove base triple with G1055 and C1104. That hydrogen bonding pattern would be disrupted by substitution with 2AP. Its fluorescence lifetimes ± 8 mM Mg2+ are very similar in their relative amplitudes and decay times (Table 1). Only the anisotropy data suggest that the environment of the nucleobase changes with added Mg2+, since the fraction of depolarization due to global tumbling becomes the dominant mechanism. The depolarization decay with/without Mg2+ is longer than observed at other sites, perhaps due to its central position in the dumbbell that reports on anisotropic motion of the structure.
To summarize the steady-state and time-resolved fluorescence data, we find that A1061AP is stacked in the secondary structure without Mg2+, although it does have independent motion. If A1061AP can form a noncanonical pair with the opposite A1077 in the secondary structure, then it could be held in the internal loop. When Mg2+ is present, its stacked environment is dominant. A1089AP stacking is dependent on Mg2+, which we attribute to a conformational change in the larger hinge region. A1089AP nucleobase is not locked into one position, but retains local motion. A1085AP fluorescence properties indicate that the junction is disordered without Mg2+, but it appears to have destabilized the tertiary structure of this region of the GAC.
Hydrogen bond formation in the tertiary structure
The GAC tertiary structure in cocrystals is stabilized by nucleobase stacking and hydrogen bonds, many of the latter coming from noncanonical interactions between nucleobases. We have selected four interactions at different positions in the GAC where imino proton hydrogen bonds make critical contributions to the tertiary structure. 1) G1071 in the 1065–1073 loop uses its N1H imino proton to make a major groove base triple with G1091:C1100 in the stem of the 1092–1096 hairpin. This base triple is one of two that anchor the stem to the loop (Figure 5; 1mms, 1hc8). 2) U1060 in the symmetric internal bulge can form a U1060:U1078 pair in the structure of the hairpin alone (data not shown), and presumably can do the same in the GAC secondary structure. In the tertiary structure, U1060 forms a long-range U1060:A1088 Hoogsteen base pair, through its imino proton to AN7 (Figure 5). 3) U1065 appears to form an unstable Watson-Crick base pair with A1073 in the isolated 1065–1073 hairpin loop without Mg2+ [29], since in NMR experiments, its probable imino proton resonance was observed only at 10° C. In the cocrystals, U1065 could make a single hydrogen bond to A1073, but its imino proton is juxtaposed to A1069 C2′-OH (Figure 5). 4) U1082 in the GAC secondary structure should have no interactions with other bases if the hinge is disordered. In the tertiary structure, it forms a reverse WC base pair with A1086 N7 through its imino proton (Figure 5). In the absence of stable hydrogen bonds, all these imino protons will be in exchange with 1H2O in NMR experiments, and therefore not observable. When they form their hydrogen bonds in the tertiary structure, they should become observable in NMR experiments in 90% H2O, and thus report on Mg2+ dependent structure formation.
Figure 5.

Structural environments of U1060, U1065, G1071, and U1082 showing the hydrogen bonding orientations of the nucleobase imino protons from the cocrystal 1hc8[17]. Structural representation by VMD[53].
For NMR Mg2+-titration experiments, (15N-G1071, 15N-U1065, 15N-U1060, 15N-U1082)-GAC with a folded secondary structure was prepared in 100 mM KCl, 10 mM sodium cacodylate, pH 6.5. At 10° and 20° C, we observed two imino proton resonances, one from a U at 11.2 ppm 1H, and the other from the G1071 at 13.2 ppm 1H (Figure 6A). These resonances were temperature sensitive, and not visible over 20° C; the U resonance was especially weak, and only observable after many scans. We assign this imino resonance to that from U1060:U1078, based on our NMR data for the isolated hairpin (data not shown). Our observation of the G1071 imino proton resonance was unexpected, and we hypothesize that it arises from an alternative structure of this hairpin.
Figure 6.

1H/15N-SOFAST 1–1 HMQC spectra of (15N-G1071, 15N-U1065, 15N-U1060, 15N-U1082)-GAC as a function of added MgCl2. A. 500 μM GAC in 100 mM KCl, 10 mM sodium cacodylate, pH 6.5, 20° C. B. 500 μM GAC with 500 μM MgCl2, 20° C. C. 500 μM GAC with 1 mM MgCl2, 30° C. D. 500 μM GAC with 8 mM MgCl2 at 30° C. See text for assignment rationale.
MgCl2 was titrated into 500 μM GAC to final concentrations of 200 μM, 500 μM, 1 mM, 3 mM, and 8 mM, over the course of a week (15N-SO-FAST HMQC spectra were acquired after each addition, and since peak intensity was very low, the data were signal-averaged using 3840 scans.) Spectra were acquired at 10° C, 20° C, and 30° C. The spectrum at 20° C with 200 μM MgCl2 was nearly identical to that of the starting state, but in 500 μM MgCl2, two new U imino proton peaks appeared, one at 14.5 ppm and another more intense peak at 9.5 ppm (Figure 6B).
Addition of 1 mM MgCl2 resulted in the appearance of several new imino proton resonances at 20° C. First, the G1071 imino proton appeared in a second position, the original near 13.3 ppm (which became resolved as two peaks) and a second peak near 11.2 ppm; the peak intensities were nearly equal. A more intense U imino proton resonance appeared at 13.8 ppm, and several weak peaks were observed (at 14.1, 12.2, and a barely detectable one at 11.2 that is more intense at 10° C). At 30° C, only five peaks are visible: the two G1071 imino proton resonances, and U imino protons at 9.5, 14.5, and 13.8 ppm (Figure 6C).
GAC 15N/1H HMQC spectra in the presence of 3 mM and 8 mM MgCl2 at 30° C show that the G1071 imino proton resonance at 13.2–13.4 ppm is very weak, while the one at 11.2 is intense but is a doublet, indicating a heterogeneous environment. U imino protons at 9.5, 14.0, and 14.7 are intense (Figure 6D). We assign the four intense resonances (G1071, and three U imino protons) to the correctly folded tertiary structure. Also in these spectra, we see two U imino proton resonances at 13.6 and 12.2 ppm, and together with the weak G1071 resonances, we assign them to an alternative structure of the GAC hairpin.
We identify the three intense U imino proton resonances based on their chemical shifts and their pattern of appearance. The resonances at 14.5 and 9.5 ppm appear together at a stoichiometric concentration of Mg2+, suggesting that they represent structures that preferentially bind Mg2+ ions. We assign the peak at 14.5 ppm to U1082 which forms a reverse WC pair when the triloop forms in the junction, and the peak at 9.5 ppm to U1065 when the T-loop forms (in the cocrystals, U1065 imino proton is proximal to a ribose hydroxyl oxygen when it forms the noncanonical loop-closing base pair).
We assign the U imino proton resonance at 14.0–14.1 ppm (1–8 mM MgCl2) to U1060 in its position in a long-range Hoogsteen base pair. This is the last resonance to appear in the spectrum, and while it is very weak (near baseline) in 1 mM MgCl2 at 20° C and missing at 30° C, it is an intense resonance in 3 and 8 mM MgCl2 at 30° C. This interaction depends on excess Mg2+ ions (2- to 6-fold), but whether the conformational change required to bring U1060 and A1088 into proximity requires one weakly bound Mg2+ ion or more distributed ions cannot be determined from this experiment.
At the end of the titration, the RNA was removed and run on a denaturing gel to assess the extent of degradation that had occurred due to inevitable Mg2+-cleavage. There were several smaller bands, consistent with backbone scission, but their relative intensity was low compared to the main band of the GAC. The RNA was recovered from the gel, repurified, and NMR data acquired, which showed the same pattern of peak resonances. Therefore, we are confident that the NMR data report conformational changes of the intact GAC.
To summarize the NMR data, we have assigned the U imino protons inferentially, based on their chemical shifts and our knowledge of the tertiary structure. We see that there is a hierarchy of GAC folding in these equilibrium experiments. It begins with a conformational change of the 1065–1073 loop and independent folding of the U1082–1086 triloop at sub-stoichiometric concentrations of Mg2+. In stoichiometric concentrations of Mg2+, the two hairpin arms are juxtaposed and held together by the G1071/C1072 base triples, which we infer by the presence of the G1071 imino proton, but only when an excess of Mg2+ is present does U1060 find A1088 to anchor the hinge structure.
Kinetics of GAC Folding
We used stopped-flow experiments to observe the kinetics of Mg2+-dependent global folding (absorbance) or local/global folding (fluorescence). To monitor global folding, we took advantage of the Mg2+-dependent UV absorbance decrease at 260 nm. The UV kinetic traces of unmodified GAC RNA are shown in Figure 7 as a function of temperature, with fits given in Table 2. (Quantities of 2AP RNAs were too limited to use in the absorption stopped-flow mode). The time-resolution (dead time) of these experiments is ~50 ms, due to instrument parameters. There are consistent trends in the data: folding is more rapid upon addition of more MgCl2, and folding is more rapid at higher temperature. The one caveat here is that in the absence of Mg2+ at 40° C, the GAC structure is beginning to melt, so it would be folding from a more disordered state. We assign the dominant transition (2–3 sec at 10° C and 200–400 ms at 40° C) to GAC global folding.
Figure 7.

Stopped-flow absorbance (260 nm) traces of unmodified U1061A GAC. From top: 10°, 20°, 30°, 40° C. Black is addition of 3 mM MgCl2, purple + 8 mM MgCl2, green + 20 mM MgCl2. [GAC]= 100 nM. 100 mM KCl, 10 mM sodium cacodylate, pH 6.5. Gray lines within the data points are the calculated fits (Origin). The dead time of the instrument in this mode is ~ 50 ms.
Table 2.
Stopped-flow absorbance data for unlabeled GAC RNA
| [Mg2+] mM |
Temp (C) |
T1 (s) |
β1 (fraction) |
|---|---|---|---|
| 3 | 10 | 3.01 | 0.93 |
| 8 | 10 | 2.13 | 0.92 |
| 20 | 10 | 1.87 | 0.89 |
| 3 | 20 | 1.12 | 0.86 |
| 8 | 20 | 0.73 | 0.88 |
| 20 | 20 | 0.66 | 0.85 |
| 3 | 30 | 0.5 | 0.69 |
| 8 | 30 | 0.31 | 0.73 |
| 20 | 30 | 0.26 | 0.74 |
| 3 | 40 | 0.38 | 0.98 |
| 8 | 40 | 0.21 | 0.92 |
| 20 | 40 | 0.19 | 0.86 |
Calculated time constants (T) and amplitude (β). Amplitude is normalized to 1.0.
In all stopped-flow fluorescence experiments, there is a Mg2+-dependent event that occurs in the instrument dead time (~1 ms) that is manifested as a percentage change in the fluorescence from its initial intensity (Figure 8). Mock addition experiments with 100 mM KCl, 10 mM sodium cacodylate show no change of fluorescence intensity from the initial value: the traces are flat. (The dead time for fluorescence detection is shorter than the dead time for absorbance detection). Each site has a unique rapid (< 1 ms) response: A1061AP GAC consistently shows a fluorescence increase, while A1089AP and A1085AP intensity change is temperature-dependent. We assign this very rapid event to a ‘global relaxation’ of the RNA as it becomes electrostatically shielded by the Mg2+ ions.
Figure 8.

Stopped flow fluorescence traces of A1061AP, A1085AP, and A1089AP (left to right). From top: 10°, 20°, 30°, 40° C. Black is addition of 3 mM MgCl2, purple + 8 mM MgCl2, green + 20 mM MgCl2. Gray lines are calculated fits to the data (Origin). There is ~1 ms deadtime of the instrument, during which all RNAs show a change in their fluorescence intensity. Fluorescence intensity is reported as percentage of starting intensity (without Mg2+). [GAC]= 100 nM. 100 mM KCl, 10 mM sodium cacodylate, pH 6.5. λex = 305 nm.
Subsequent fluorescence changes are site-specific, and one of the most dramatic occurs at A1061AP (Figure 8). Recall that steady-state fluorescence intensity of A1061AP is low ± Mg2+, consistent with quenching by stacking. Observation of the time-course of its fluorescence changes as the GAC adopts its tertiary structure reveals a complex pathway of conformational/environmental changes. At all temperatures, there is an almost immediate transient increase in A1061AP fluorescence intensity that is most pronounced with addition of 20 mM MgCl2: at 10° C, the time constant T1 = 90 ms (exchange rate R1 = 11 s−1); at 20° C, T1 = 27 ms; at 30° C, T1 = 11 ms; at 40° C, T1 = 7 ms. We interpret this transient fluorescence increase as a conformational transition of A1061AP that results in a loss of base stacking. However, subsequently, A1061AP fluorescence intensity decreases, with a final value that is temperature-dependent, indicating that it has been stacked again. The time constant of this second component is similar to that of the absorbance trace that we assign to global folding, but not identical, indicating that there are local contributions from A1061AP to this transition. The end state of the transition, reported by the final value of A1061AP fluorescence, is both Mg2+ and temperature dependent and in some traces, there is a third time constant.
The Hinge
A1089AP reports on conformational changes involving the hinge, recalling that its steady-state fluorescence intensity is reduced by 50% in 3 mM MgCl2, indicative of a more stacked environment. In the ~ 1 ms deadtime of the instrument, A1089AP fluorescence changes from its starting value, most significantly at 30° C. The major component of the traces is a fluorescence decrease, with time constants from 15 s at 10° C with addition of 3 mM MgCl2 to 470 ms at 40° C with addition of 20 mM MgCl2 (Figure 8 and Table 3). At all temperatures and Mg2+ additions, this component is significantly longer than the global folding reported by absorption kinetics and longer than the major component of A1061AP folding kinetics. A1089 is stacked with A1071 and G1087 in the cocrystal structures, which will occur only when the 1065–1073 loop has adopted its T-loop structure, and when the hinge has collapsed to allow the two hairpins to become juxtaposed. We conclude that A1089AP folding kinetics include a large-scale conformational change as well as local rearrangements.
Table 3.
Stopped-flow fluorescence curve fits.
| 2AP position |
MgCl2 | T (°C) |
T1 (s) |
β1 (%) |
T2 (s) |
β2 (%) |
T3 (s) |
β3 (%) |
|---|---|---|---|---|---|---|---|---|
| 1061 | 3 | 10 | 0.04 | −18 | 8.9 | −8 | ||
| 8 | 10 | 0.07 | −35 | 2.2 | 5 | 14.5 | −3 | |
| 20 | 10 | 0.09 | −50 | 2.6 | 15 | 21 | −2 | |
| 3 | 20 | 0.018 | −13 | 1.2 | 5 | 99 | 6 | |
| 8 | 20 | 0.024 | −25 | 1 | 15 | 245 | 10 | |
| 20 | 20 | 0.027 | −37 | 0.93 | 25 | 372 | 15 | |
| 3 | 30 | 0.013 | −8 | 0.9 | 9 | 3.3 | 11 | |
| 8 | 30 | 0.009 | −18 | 0.57 | 19 | 4.9 | 10 | |
| 20 | 30 | 0.011 | −30 | 0.43 | 30 | 5.4 | 12 | |
| 3 | 40 | 0.55 | 20 | |||||
| 8 | 40 | 0.003 | −11 | 0.37 | 32 | |||
| 20 | 40 | 0.007 | −28 | 0.03 | 12 | 0.32 | 42 | |
| 1085 | 3 | 10 | 1.2 | 1 | 0.14 | −0.7 | 25 | −2 |
| 8 | 10 | 0.002 | 4 | 0.5 | −0.5 | 18 | −6 | |
| 20 | 10 | 0.007 | 2 | 0.16 | −0.4 | 12 | −8 | |
| 3 | 20 | 0.2 | 0.6 | 46 | 2 | |||
| 8 | 20 | 0.02 | 1 | 12 | −3 | 45 | 2 | |
| 20 | 20 | 0.004 | 1.5 | 6.4 | −5 | 53 | 2 | |
| 3 | 30 | 1.5 | 1 | 13 | 5 | |||
| 8 | 30 | 3.7 | 3 | 13 | 2 | |||
| 20 | 30 | 3.4 | 2 | 15 | 2 | |||
| 3 | 40 | 0.1 | 1 | 6 | 4 | |||
| 8 | 40 | 0.14 | 1 | 4 | 6 | |||
| 20 | 40 | 0.21 | 2 | 2.8 | 6 | |||
| 1089 | 3 | 10 | 0.007 | 7 | 0.026 | −4 | 15 | 34 |
| 8 | 10 | 0.004 | 5 | 6.5 | 34 | 33 | 5 | |
| 20 | 10 | 0.008 | 6 | 4.8 | 35 | 33 | 6 | |
| 3 | 20 | 0.001 | 4 | 0.038 | −2 | 4.6 | 29 | |
| 8 | 20 | 0.002 | 4 | 2.2 | 31 | 32 | 5 | |
| 20 | 20 | 0.01 | 5 | 1.6 | 31 | 28 | 6 | |
| 3 | 30 | 2 | 23 | 10 | 0.5 | |||
| 8 | 30 | 0.003 | 3 | 1 | 22 | 7 | 4 | |
| 20 | 30 | 0.005 | 5 | 0.7 | 23 | 7 | 6 | |
| 3 | 40 | 0.001 | 4 | 0.05 | −5 | 2.1 | 23 | |
| 8 | 40 | 0.001 | 1 | 0.67 | 21 | 4.7 | 1 | |
| 20 | 40 | 0.001 | 4 | 0.47 | 21 | 3.9 | 1 | |
Calculated time constants (T) and amplitudes (β). Amplitudes < 0 indicate an increase in fluorescence intensity. Amplitude reports the percent contribution to the total intensity, from 100% initial (before Mg2+ addition), to the final non-zero intensity that results from folding.
Although ideally A1085AP could also report on hinge structure formation, this substitution appears to destabilize the tertiary structure of the hinge. Its fluorescence intensity also changes within the < 1 ms instrument dead time upon addition of MgCl2, decreasing from 10–20% at 10° and 20° C (Figure 8 and Table 3) but then its traces at 10°, 20°, and 30° C show a long lag time before another transition occurs. At 10° C, there is a ~1 s lag time, followed by a fluorescence increase with T3 = 12 s with 20 mM MgCl2 addition, while at 20° C, the traces are almost flat following the very rapid (< 1 ms) loss. At 30°, there is a 1 s lag, followed by fluorescence decrease (T2 = 13–15 s) while at 40° C, there is a progressive loss of fluorescence with time constants T1 = 140 ms and T2 = 4 s after 8 mM MgCl2 addition.
To summarize the stopped-flow fluorescence data, we assign the major A1061AP transition to the global fold of the GAC, while the major transition of A1089AP occurs after global folding. A1085AP predominantly reports on slower processes that could reflect a destabilized hinge. These data clearly show that there is a hierarchy of folding events that includes local conformational rearrangements before and after the global fold has occurred.
Discussion
We have investigated Mg2+-dependent U1061A GAC tertiary folding in both equilibrium titrations and by stopped-flow kinetics. We find that formation of stable tertiary hydrogen bonding interactions occurs in an ordered process, starting with formation of the 1065–1073 T-loop and the 1082–1086 triloop, then base triples from the T-loop, and finally the long-range U1060-A1088 hydrogen bond. These interactions can be differentially stabilized in a Mg2+ equilibrium titration, which suggests that there are several specific sites of Mg2+ ion association with different affinities, or perhaps secondary sites that need to be created by a conformational change. Whatever the mechanism of Mg2+ association, there are stable intermediates in the folding pathway. Kinetics of GAC folding also suggests a hierarchy of assembly steps, since there is a significant difference in the rates of fluorescence change at positions A1061AP and A1089AP. We cannot know if the pathway for tertiary folding is identical in equilibrium titrations and kinetic traces.
The first conformational transition
Stopped-flow fluorescence experiments revealed very rapid (< 1 ms) fluorescence intensity changes in all three 2AP-GACs. We assign this event to global electrostatic relaxation by nonspecific association of Mg2+ ions. The phenomenon of rapid Mg2+-dependent (electrostatic) global collapse of large RNAs has been reported for several systems[30][31,32][33,34], even for RNAs that cannot form tertiary structure (e.g., the 303 nucleotide bI5core RNA [35]). For now, we will continue to refer to the very rapid fluorescence changes of GAC as electrostatic relaxation, where addition of Mg2+ ions reduces the local charge density of the RNA and facilitates its exploration of local and global structures.
The interplay between phosphate charge neutralization by multivalent ions (typically Mg2+) of an unfolded RNA and subsequent (or consequent) tertiary folding has been examined in several large RNAs. The Tetrahymena Group I intron has been extensively investigated, and described as undergoing a first electrostatic relaxation followed by electrostatic compaction to intermediate (mis)folded states, to finally find its correct structure [31][33]. Other large RNAs, such as RNase P RNA[36,37], are reported to also undergo a first electrostatic relaxation followed by electrostatic compaction. The smaller Azoarcus Group I intron (195 nt) [38][39] has been shown to fold into an intermediate in < 10 ms then into its tertiary structure in < 30 ms[40]. Experiments with a 2AP-Azoarcus RNA revealed several additional transitions, however[40]. Many of these studies used SAXS, which gives a scattering envelope of the global structure; it can distinguish collapsed, intermediate, and native structures in time-resolved experiments[41].
Certainly the addition of Mg2+ does lead to a compaction of the GAC, as shown by calculation of GAC U1061A scattering envelopes in SAXS experiments ± Mg2+ [22]. In 40 mM K+, addition of 0.1 mM Mg2+ caused a compaction of the GAC: at 15° C, the radius of gyration (Rg) for GAC in its secondary structure in K+ alone was ~25 Å, while in Mg2+, Rg of the tertiary structure was 15 Å. These data led to the interpretation of first formation of an Intermediate structure, characterized by a decrease in Rg upon addition of low concentrations of Mg2+, but then proceeding to a Native tertiary structure with an additional decrease in Rg[22]. A time-resolved SAXS experiment might be able to determine if global compaction corresponds to the rapid (< 1 ms) fluorescence change that we observe.
Local folding
Internal Loop
Nucleotides in the 1060/1061 internal loop contribute several critical interactions to the tertiary structure, but in the GAC secondary structure, they are likely to participate in two noncanonical base pairs formed by U1060:U1078 and A1061:A1077, based on NMR data of isolated hairpin loops[29]. This existing structure must be disrupted to allow A1061 and U1060 nucleobases to make tertiary contacts.
A1061AP has a complicated kinetics trace. It first undergoes a large amplitude fluorescence increase with a time constant of T1 = 18 – 27 ms at 20° C with addition of 3 – 20 mM MgCl2. The fluorescence increase indicates a loss of stacking, but this phase is transient, since it is always followed by a large amplitude fluorescence decrease with a time constant T2 at least 10-fold longer (at 20° C, T2 = 1.2 – 0.9 s with addition of 3 – 20 mM MgCl2). Addition of 20 mM MgCl2 elicits the most dramatic response, suggesting that the conformational change reported by this transition uses weakly associated Mg2+ ions or that it requires multiple associated ions. The initial increase in fluorescence could correspond to a disruption of an A1061AP:A1077 base pair that allows A1061AP to move out of the internal loop, and then finds a new position in the GAC structure. We note that the final fluorescence of A1061AP is temperature-dependent as well as Mg2+ dependent, suggesting that it may occupy different sites within the GAC structure.
In NMR experiments, the U1060 imino proton is the last to appear in the titration experiments, and becomes stable at [Mg2+]/[RNA] = 6. U1060 forms an unstable U:U pair that could stack with the adjacent A1061, as suggested by NMR experiments with the isolated hairpin (data not shown). This U:U pair must be disrupted and as U1060 forms a long-range hydrogen bond to A1088 as seen in the cocrystals. A1061 must undergo an exchange of position in the GAC to stack with A1070, but it seems that A1061 and U1060 interactions with their partners occur independently of each other.
The 1065–1073 hairpin loop
The secondary structure of this isolated hairpin loop was probed by NMR experiments in KCl without Mg2+ ions. Wang et al.[29] observed a weak imino proton resonance that could correspond to a WC base pair from U1065:A1073, but also noted unusual ribose puckers of several of the seven loop nucleotides. No internal structure of the loop was reported. Our data suggest that in the GAC secondary structure, this loop is also unstructured, although our NMR data indicate that it could adopt an alternative structure. When Mg2+ is added, NMR spectra are consistent with a conformational change of the loop, and we assign the U1065 imino proton to the resonance at 9.5 ppm, consistent with a hydrogen bond to a ribose 2′ hydroxyl oxygen. The now classic example of an unusual imino proton chemical shift is that of G in the UU2CG4 tetraloop. The G4 imino proton makes a hydrogen bond with the U2 C2O carbonyl oxygen, and has a chemical shift of 9.9 ppm[42], analogous to our assignment of U1065. The early appearance of this proton in the Mg2+ NMR titration experiments supports the suggestion of Leipply & Draper[24] that there is a chelated Mg2+ near A1073.
When the GAC has only its secondary structure in 100 mM KCl, we observe an imino proton resonance from G1071 at 13.6 ppm which is unexpected, since fluorescence data show that the loop is disordered. We assign this resonance, and two other weak peaks that appear, to an alternative structure of the hairpin. [Curiously, these peaks disappear when L11 protein is added, and only the four strong peaks remain, a result that we are investigating.]
G1071 in the hairpin loop makes one of the two base triples with the opposite hairpin stem, but the 1065–1073 loop structure has to change from its unstructured state to a structured conformation to display the base. In the cocrystals, this loop is a T-loop [25], where the 5’ UNR sequences form a U-turn and the 3′ A1070, G1071, C1072 nucleotides are extruded. In other experiments, we find that formation of this loop structure requires Mg2+ (data not shown), and the NMR data presented here show that formation of the 1065–1073 loop-closing base pair is an early event in the folding pathway.
The hinge structures
The 3-way junction becomes the hinge that juxtaposes the two hairpins, but in the secondary structure, we describe it as unstructured. The triloop made from nucleotides U1082 to A1086 within the junction is apparently formed early in the folding path. In equilibrium NMR experiments, appearance of the U1082:A1086 imino proton hydrogen bond is co-incident with appearance of the U1065:A1073 hydrogen bond, while kinetic traces show a conformational transition of A1089AP at 30–50 ms. We suggest that both conformational changes occur within the same time window, but that they are not correlated. Rather, we suspect that there are two Mg2+ binding sites nearby to each element that allow formation of the unusual 1065–1073 hydrogen bond and the U1082-A1086 triloop.
Long-range interactions
We have monitored two nucleotides that participate in long-range hydrogen bonding interactions: G1071 and U1060. Our NMR data show that the G1071 imino proton hydrogen bond is observed only after the 1065–1073 loop structure has been altered by Mg2+ (as shown by the U1065 imino proton resonance). Also in the NMR experiments, the final resonance to be observed from U1060 appears at [Mg2+]/[GAC] = 6. This long-range hydrogen bond will anchor U1060 to A1088, and likely restrict the relative movements of the two hairpin stems.
A1089 occupies a complicated position in the tertiary structure (Figure 2), as it is stacked between nucleobases G1087 and G1071. We have seen that G1071 makes a stable base triple hydrogen bond only after its stemloop structure has been formed, so it will be positioned to make a stacking interaction at a later step in the folding pathway. Formation of the triloop that is adjacent to A1089 occurs early, which could constrain A1089 to stack with G1087. However, the major kinetic transition of A1089AP is slower than the global folding transition reported by absorbance, consistent with late formation of the structure around this region.
When enough Mg2+ is available, the GAC proceeds to fold into its final tertiary structure. In kinetics experiments, the need for more Mg2+ ions is apparent from the decrease in the time constants when 20 mM MgCl2 is added. This concentration is in 105-fold excess of the GAC in stopped-flow experiments, so the effect of the addition is to rapidly populate all sites within the GAC. While this is a saturating concentration of Mg2+, the GAC still shows some slower transitions, particularly as noted in the continued fluorescence intensity changes beyond 100 s for the A1089AP GAC. Whether this trace is due to a misfolded form or a slow transition to the correct structure will require additional probing.
Unknowns
We have no probes in the G1093-A1098 hairpin loop which forms a canonical four nucleotide 5′-UNR U-turn, with a sheared G:A loop closing base pair in the folded RNA. In the secondary structure, it could potentially also adopt this structure, since its nucleotides make no tertiary contacts. However, in two solution NMR studies of this isolated hairpin loop (both in the absence of Mg2+) the reported structures were not identical [43],[44] and only one was consistent with a U-turn. These data suggest that Mg2+ might be required to stabilize the U-turn seen in the cocrystal structure, although other RNA 5′UNR U-turns are not Mg2+-dependent[45].
We are missing unambiguous probes within the triloop. A1085AP would have been an excellent reporter on the kinetics of loop formation and then on base stacking, but we find that it induces instability into this loop which we attribute to loss of specific hydrogen bonds. This triloop sequence comprises 5% of triloops found in rRNAs [46], but its thermodynamics of formation are not known.
GAC Folding
This essential RNA element is replete with intricate and noncanonical interactions. Its strict Mg2+-dependence of folding, previous biochemical experiments that alter its sequence and measure thermodynamics of ion binding, and its known crystal structures make the GAC a perfect subject for study of its folding pathway. We have already discovered that its kinetics of folding are complex and multistate, and have captured at least one folding intermediate in Mg2+ titrations at equilibrium. By using selective labeling, we can use complementary techniques to observe formation of nucleobase-specific interactions both temporally and at equilibrium. With further study of the GAC RNA, new insights into the interplay of local and global folding will be found, and the contribution of its L11 protein to its folding and stability can be explored in detail.
Materials and Methods
RNA samples
RNAs were either transcribed using T7 RNA polymerase via run-off transcription from a plasmid[47] or chemically synthesized by Agilent labs [48] to insert 2-aminopurine (2AP) or 15N-nucleotides. All RNAs in solution were dialyzed against 0.5 M EDTA, then two or three times against MilliQ water with the final exchange overnight. Samples were lyophilized and rehydrated as needed in buffer. We use the Draper lab folding protocol for the GAC[24]: heat at 65°C for 30 minutes in 10 mM sodium cacodylate pH 6.5 and 100 mM KCl its buffer (without Mg2+), then sit at room temperature for about 15 minutes.
UV thermal denaturation
UV melting was performed on a Gilford 260 fitted with a Gilford thermoprogrammer 2527. RNA concentrations were 2 μM. Sample temperature was ramped at 0.5°C/min from 6–90°C while absorbance was recorded at 260 nm or 280 nm.
Steady-state fluorescence
experiments were performed on a PTI (Photon Technology international) fitted with a Peltier controlled 4-cuvette turret. RNA concentration for these experiments was 2 μM. Emission and excitation scans were measured using λem = 368 nm and λex = 308 nm. Lamp power and PMT voltage were kept constant at 70 Watts and 1000 Volts, respectively. Slit widths were constant. Temperature was increased with a ramp of 1°C/min; points were averaged for 5 seconds with an integration time of 1 second.
Free 2-aminopurine-triphosphate fluorescence was monitored versus temperature as a standard for comparison with 2AP-GAC. 2APTP (2 μM) fluorescence intensity represents the maximum fluorescence emission for the solution conditions and fluorometer configuration. At high temperatures collisional quenching with solvent will decrease the fluorescence[49].
TCSPC and TRA
Time-correlated single photon counting (TCSPC) and time-resolved anisotropy (TRA) measurements were performed with our custom instrument[50]. A Ti:Sapphire laser was pulse-picked and tripled to excite the 2AP at 300 nm. Samples were heated in a water-jacketed cuvette holder; temperature was monitored using a thermistor. Lifetime and IRF decay curves were collected until overflow (65536 counts) with the polarizer at 55°. IRF was collected with a 1/100 dilution of LUDOX (Aldrich) solution; its FWHM was measured every day and varied from 215–222 ps. Anisotropy decays were collected in separate runs where vertical polarization was collected first to overflow then horizontal polarization was collected with the same acquisition time. Fluofit Pro 4.4 (Picoquant) was used to fit both lifetime and anisotropy curves. Fluorescence lifetime decays were typically best fit with a three component (i) exponential model with reconvolution.
Where It is the intensity at time t, a is the pre-exponential factor, and τi is the lifetime of the ith component. The initial steady-state value of the anisotropy (Ro) is without any motion of the fluorophore. Time-resolved anisotropy decays from Ro were fit using only a two component exponential model to give depolarization decay times φ1,2 and their amplitudes β1,2. The G-factor was calculated using tail matching.
Buffer scattering was insignificant. Goodness of fit was assessed by support plane analysis.
The theoretical rotational correlation time[49] of GAC was calculated for interpretation of 2AP anisotropy components. GAC RNA can be classified as a prolate ellipsoid with an axial ratio of 1.8 when it adopts its tertiary fold, based on dimensions of the cocrystal structures. For this shape, three different rotational correlation times are needed: ϴ1=5.7, ϴ2=5.2, ϴ3=5.9 ns at 30°C. These predicted values assume a viscosity of 0.797 cP, molecular weight of 21 kDa, specific volume of 0.6 ml/g, and a hydration of 0.2 ml/g. We calculated a simple average value <τR> = 5.6 ns for experimental evaluations.
Stopped-flow fluorescence
An Applied Photophysics SX-20 Stopped-Flow spectrometer was used for all stopped-flow measurements. This instrument has two syringes for mixing; one holds the RNA in buffer and KCl, the other contains MgCl2 in buffer and KCl. For absorbance, λex = 260 nm; for fluorescence, λex = 305 nm, with excitation slit widths of 2 mm resulting in a bandwidth of 9.3 nm. Temperature was regulated with a water bath and sample temperature was monitored through Applied Photophysics software. The number of points collected per time interval was adjusted to achieve the best resolution; from t=0 to t= 1 second, we sampled 5000 points, while from 1 to 100 seconds, we collected 10000 points. The dead time of the instrument in fluorescence mode is ~ 1 msec, but in absorbance mode, is ~ 50 ms.
GAC was 100 nM final concentration, while MgCl2 addition was in increments of 3, 8, and 20 mM final concentration. Buffer was 10 mM sodium cacodylate pH 6.5, 100 mM KCl. The RNA secondary structure was formed as described before it was loaded into the syringe. Buffer with no RNA or MgCl2 was also prepared for control.
In each Mg2+ condition, raw data from 8 to 12 measurements were first averaged. The buffer/buffer background control trace was subtracted from the [RNA without Mg2+] control and each of the [RNA + Mg2+] experiments. The corrected [RNA without Mg2+] trace was then smoothed using a 20-point running average. This trace was essentially flat and is a measure of 100% fluorescence. To compare the fluorescence change to the steady-state fluorescence, the lower ‘baseline’ is adjusted to the final fluorescence value after addition of Mg2+ (this is the Y offset or Y0 in the fitting equation). Fitting the decay curves to the exponential model in Origin (Equation 1) is expressed as fluorescence changes with corresponding fraction of the total fluorescence signal (βn): Y% of the signal is constant, a varying fraction is lost in < 1 ms, and the remainder is lost during the folding events.
| (1) |
NMR experiments
15N-1H SO-FAST HMQC spectra[51,52] , were recorded at 500 and 600 MHz (1H) on Varian Inova and Bruker Avance III spectrometers, respectively. Spectral widths: 12000 Hz (1H), 1600 Hz (15N). GAC RNA ([GAC] = 500 μM) was folded as described, then its spectra recorded in 100 mM KCl, 10 mM sodium cacodylate pH 6.5, from 10° to 30° C. The RNA was added into lyophilized aliquots of MgCl2 (Aldrich 99.995%) to give final concentrations of 200 μM, 500 μM, 1 mM, 3 mM, and 8 mM in a 3 mm tube. After each addition, the sample was allowed to equilibrate for an hour at 20° C, then spectra were collected at 10°, 20° and 30° C for 12 hours. 90% H2O, 10% D2O.
Highlights.
The rugged landscape of RNA folding is explored here for a conserved 60 nucleotide rRNA fragment from the perspective of individual nucleobases.
Global folding of the GTPase center RNA is Mg2+-dependent, but observing individual structural elements (hairpin, internal bulge, hinge) shows a hierarchy of Mg2+-dependent structural changes.
Kinetics of tertiary structure formation is not two-state, but rather a culmination of multiple conformational changes within the RNA on timescales ranging from < 1 ms to 4 s.
The investigation of this RNA structural transition by monitoring individual nucleobases reveals their dynamic conformational changes before, during, and after the final tertiary structure has formed.
Acknowledgements
2AP- and 15N-RNAs were synthesized by Agilent Labs, and we thank Dr Laurakay Bruhn, Dr Jeff Sampson, and Dr Doug Dellinger. We thank Professor Roberto Galletto for use of his stopped-flow spectrometer. This work was supported by a grant to KBH (NIH R01 GM098102) and a gift from Agilent.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael J Rau, Email: mjrau01@gmail.com.
Robb Welty, Email: robb@wustl.edu.
W. Tom Stump, Email: stump@biochem.wustl.edu.
References
- 1.Herschlag D, Allred BE, Gowrishankar S. From static to dynamic: the need for structural ensembles and a predictive model of RNA folding and function. Curr Opin Struct Biol. 2015;30C:125–133. doi: 10.1016/j.sbi.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Treiber DK, Rook MS, Zarrinkar PP, Williamson JR. Kinetic intermediates trapped by native interactions in RNA folding. Science. 1998;279:1943–1946. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- 3.Butcher SE, Pyle AM. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc Chem Res. 2011;44:1302–1311. doi: 10.1021/ar200098t. [DOI] [PubMed] [Google Scholar]
- 4.Chauhan S, Woodson SA. Tertiary interactions determine the accuracy of RNA folding. J Am Chem Soc. 2008;130:1296–1303. doi: 10.1021/ja076166i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitra S, Laederach A, Golden BL, Altman RB, Brenowitz M. RNA molecules with conserved catalytic cores but variable peripheries fold along unique energetically optimized pathways. RNA. 2011;17:1589–1603. doi: 10.1261/rna.2694811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinan S, Yuan X, Russell R. The Azoarcus group I intron ribozyme misfolds and is accelerated for refolding by ATP-dependent RNA chaperone proteins. J Biol Chem. 2011;286:37304–37312. doi: 10.1074/jbc.M111.287706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson SA, Koduvayur S, Woodson SA. Self-splicing of a group I intron reveals partitioning of native and misfolded RNA populations in yeast. RNA. 2006;12:2149–2159. doi: 10.1261/rna.184206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell R, Das R, Suh H, Travers KJ, Laederach A, Engelhardt MA, et al. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J Mol Biol. 2006;363:531–544. doi: 10.1016/j.jmb.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 9.Li PTX, Bustamante C, Tinoco I. Unusual mechanical stability of a minimal RNA kissing complex. Proc Natl Acad Sci U S A. 2006;103:15847–15852. doi: 10.1073/pnas.0607202103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiore JL, Holmstrom ED, Fiegland LR, Hodak JH, Nesbitt DJ. The role of counterion valence and size in GAAA tetraloop-receptor docking/undocking kinetics. J Mol Biol. 2012;423:198–216. doi: 10.1016/j.jmb.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Bassi GS, Murchie AI, Walter F, Clegg RM, Lilley DM. Ion-induced folding of the hammerhead ribozyme: a fluorescence resonance energy transfer study. EMBO J. 1997;16:7481–7489. doi: 10.1093/emboj/16.24.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rinnenthal J, Klinkert B, Narberhaus F, Schwalbe H. Direct observation of the temperature-induced melting process of the Salmonella fourU RNA thermometer at base-pair resolution. Nucleic Acids Res. 2010;38:3834–3847. doi: 10.1093/nar/gkq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beauclerk AA, Hummel H, Holmes DJ, Böck A, Cundliffe E. Studies of the GTPase domain of archaebacterial ribosomes. Eur J Biochem. 1985;151:245–255. doi: 10.1111/j.1432-1033.1985.tb09095.x. [DOI] [PubMed] [Google Scholar]
- 14.el-Baradi TT, de Regt VC, Einerhand SW, Teixido J, Planta RJ, Ballesta JP, et al. Ribosomal proteins EL11 from Escherichia coli and L15 from Saccharomyces cerevisiae bind to the same site in both yeast 26 S and mouse 28 S rRNA. J Mol Biol. 1987;195:909–917. doi: 10.1016/0022-2836(87)90494-3. [DOI] [PubMed] [Google Scholar]
- 15.Ryan PC, Draper DE. Thermodynamics of protein-RNA recognition in a highly conserved region of the large-subunit ribosomal RNA. Biochemistry. 1989;28:9949–9956. doi: 10.1021/bi00452a012. [DOI] [PubMed] [Google Scholar]
- 16.Agrawal RK, Linde J, Sengupta J, Nierhaus KH, Frank J. Localization of L11 protein on the ribosome and elucidation of its involvement in EF-G-dependent translocation. J Mol Biol. 2001;311:777–787. doi: 10.1006/jmbi.2001.4907. [DOI] [PubMed] [Google Scholar]
- 17.Conn GL, Gittis AG, Lattman EE, Misra VK, Draper DE. A compact RNA tertiary structure contains a buried backbone-K+ complex. J Mol Biol. 2002;318:963–973. doi: 10.1016/S0022-2836(02)00147-X. [DOI] [PubMed] [Google Scholar]
- 18.Wimberly BT, Guymon R, McCutcheon JP, White SW, Ramakrishnan V. A detailed view of a ribosomal active site: the structure of the L11-RNA complex. Cell. 1999;97:491–502. doi: 10.1016/s0092-8674(00)80759-x. [DOI] [PubMed] [Google Scholar]
- 19.Laing LG, Gluick TC, Draper DE. Stabilization of RNA structure by Mgions Specific and non-specific effects. J Mol Biol. 1994;237:577–587. doi: 10.1006/jmbi.1994.1256. [DOI] [PubMed] [Google Scholar]
- 20.Lu M, Draper DE. Bases defining an ammonium and magnesium ion-dependent tertiary structure within the large subunit ribosomal RNA. J Mol Biol. 1994;244:572–585. doi: 10.1006/jmbi.1994.1753. [DOI] [PubMed] [Google Scholar]
- 21.Ryan PC, Draper DE. Detection of a key tertiary interaction in the highly conserved GTPase center of large subunit ribosomal RNA. Proc Natl Acad Sci U S A. 1991;88:6308–6312. doi: 10.1073/pnas.88.14.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grilley D, Misra V, Caliskan G, Draper DE. Importance of partially unfolded conformations for Mg(2+)-induced folding of RNA tertiary structure: structural models and free energies of Mg2+ interactions. Biochemistry. 2007;46:10266–10278. doi: 10.1021/bi062284r. [DOI] [PubMed] [Google Scholar]
- 23.Shiman R, Draper DE. Stabilization of RNA tertiary structure by monovalent cations. J Mol Biol. 2000;302:79–91. doi: 10.1006/jmbi.2000.4031. [DOI] [PubMed] [Google Scholar]
- 24.Leipply D, Draper DE. Evidence for a thermodynamically distinct Mg2+ ion associated with formation of an RNA tertiary structure. J Am Chem Soc. 2011;133:13397–13405. doi: 10.1021/ja2020923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagaswamy U, Fox GE. Frequent occurrence of the T-loop RNA folding motif in ribosomal RNAs. RNA. 2002;8:1112–1119. doi: 10.1017/s135583820202006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutell RR, Cannone JJ, Konings D, Gautheret D. Predicting U-turns in ribosomal RNA with comparative sequence analysis. J Mol Biol. 2000;300:791–803. doi: 10.1006/jmbi.2000.3900. [DOI] [PubMed] [Google Scholar]
- 27.Leipply D, Draper DE. Dependence of RNA tertiary structural stability on Mg2+ concentration: interpretation of the Hill equation and coefficient. Biochemistry. 2010;49:1843–1853. doi: 10.1021/bi902036j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jean JM, Hall KB. 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. Proc Natl Acad Sci U S A. 2001;98:37–41. doi: 10.1073/pnas.011442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang YX, Huang S, Draper DE. Structure of a U.U pair within a conserved ribosomal RNA hairpin. Nucleic Acids Res. 1996;24:2666–2672. doi: 10.1093/nar/24.14.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pabit SA, Sutton JL, Chen H, Pollack L. Role of ion valence in the submillisecond collapse and folding of a small RNA domain. Biochemistry. 2013;52:1539–1546. doi: 10.1021/bi3016636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das R, Kwok LW, Millett IS, Bai Y, Mills TT, Jacob J, et al. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J Mol Biol. 2003;332:311–319. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 32.Moghaddam S, Caliskan G, Chauhan S, Hyeon C, Briber RM, Thirumalai D, et al. Metal ion dependence of cooperative collapse transitions in RNA. J Mol Biol. 2009;393:753–764. doi: 10.1016/j.jmb.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, Gruner SM, et al. Rapid compaction during RNA folding. Proc Natl Acad Sci U S A. 2002;99:4266–4271. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takamoto K, Das R, He Q, Doniach S, Brenowitz M, Herschlag D, et al. Principles of RNA compaction: insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J Mol Biol. 2004;343:1195–1206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 35.Buchmueller KL, Webb AE, Richardson DA, Weeks KM. A collapsed non-native RNA folding state. Nat Struct Biol. 2000;7:362–366. doi: 10.1038/75125. [DOI] [PubMed] [Google Scholar]
- 36.Fang X-W, Thiyagarajan P, Sosnick TR, Pan T. The rate-limiting step in the folding of a large ribozyme without kinetic traps. Proc Natl Acad Sci U S A. 2002;99:8518–8523. doi: 10.1073/pnas.142288399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang XW, Pan T, Sosnick TR. Mg2+-dependent folding of a large ribozyme without kinetic traps. Nat Struct Biol. 1999;6:1091–1095. doi: 10.1038/70016. [DOI] [PubMed] [Google Scholar]
- 38.Perez-Salas UA, Rangan P, Krueger S, Briber RM, Thirumalai D, Woodson SA. Compaction of a bacterial group I ribozyme coincides with the assembly of core helices. Biochemistry. 2004;43:1746–1753. doi: 10.1021/bi035642o. [DOI] [PubMed] [Google Scholar]
- 39.Rangan P, Woodson SA. Structural requirement for Mg2+ binding in the group I intron core. J Mol Biol. 2003;329:229–238. doi: 10.1016/s0022-2836(03)00430-3. [DOI] [PubMed] [Google Scholar]
- 40.Chauhan S, Behrouzi R, Rangan P, Woodson SA. Structural rearrangements linked to global folding pathways of the Azoarcus group I ribozyme. J Mol Biol. 2009;386:1167–1178. doi: 10.1016/j.jmb.2008.12.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roh JH, Guo L, Kilburn JD, Briber RM, Irving T, Woodson SA. Multistage collapse of a bacterial ribozyme observed by time-resolved small-angle X-ray scattering. J Am Chem Soc. 2010;132:10148–10154. doi: 10.1021/ja103867p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varani G, Cheong C, Tinoco I. Structure of an unusually stable RNA hairpin. Biochemistry. 1991;30:3280–3289. doi: 10.1021/bi00227a016. [DOI] [PubMed] [Google Scholar]
- 43.Huang S, Wang YX, Draper DE. Structure of a hexanucleotide RNA hairpin loop conserved in ribosomal RNAs. J Mol Biol. 1996;258:308–321. doi: 10.1006/jmbi.1996.0252. [DOI] [PubMed] [Google Scholar]
- 44.Fountain MA, Serra MJ, Krugh TR, Turner DH. Structural features of a six-nucleotide RNA hairpin loop found in ribosomal RNA. Biochemistry. 1996;35:6539–6548. doi: 10.1021/bi952697k. [DOI] [PubMed] [Google Scholar]
- 45.Stallings SC, Moore PB. The structure of an essential splicing element: stem loop IIa from yeast U2 snRNA. Structure. 1997;5:1173–1185. doi: 10.1016/s0969-2126(97)00268-2. [DOI] [PubMed] [Google Scholar]
- 46.Thulasi P, Pandya LK, Znosko BM. Thermodynamic characterization of RNA triloops. Biochemistry. 2010;49:9058–9062. doi: 10.1021/bi101164s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dellinger DJ, Timár Z, Myerson J, Sierzchala AB, Turner J, Ferreira F, et al. Streamlined process for the chemical synthesis of RNA using 2’-O-thionocarbamate-protected nucleoside phosphoramidites in the solid phase. J Am Chem Soc. 2011;133:11540–11556. doi: 10.1021/ja201561z. [DOI] [PubMed] [Google Scholar]
- 49.Lackowicz J. Principles of Fluorescence Spectroscopy. 3rd Editio. Springer; 2006. [Google Scholar]
- 50.Jean JM, Hall KB. Stacking-unstacking dynamics of oligodeoxynucleotide trimers. Biochemistry. 2004;43:10277–10284. doi: 10.1021/bi049701p. [DOI] [PubMed] [Google Scholar]
- 51.Schanda P, Brutscher B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc. 2005;127:8014–8015. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- 52.Schanda P, Forge V, Brutscher B. HET-SOFAST NMR for fast detection of structural compactness and heterogeneity along polypeptide chains. Magn Reson Chem. 2006;44:S177–S184. doi: 10.1002/mrc.1825. Spec No: [DOI] [PubMed] [Google Scholar]
- 53.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Mol Graph. 1996;14.1:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]