

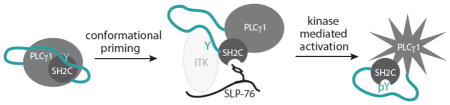
Abstract
Activation of the phospholipase, PLCγ1, is critical for proper T cell signaling following antigen receptor engagement. In T cells, the Tec family kinase, ITK, phosphorylates PLCγ1 at tyrosine 783 (Y783) leading to activation of phospholipase function and subsequent production of the second messengers IP3 and DAG. In this work we demonstrate that PLCγ1 can be primed for ITK mediated phosphorylation on Y783 by a specific region of the adaptor protein, SLP-76. The SLP-76 phosphotyrosine containing sequence, pY173IDR, does not conform to the canonical recognition motif for an SH2 domain yet binds with significant affinity to the C-terminal SH2 domain of PLCγ1 (SH2C). The SLP-76 pY173 motif competes with the autoinhibited conformation surrounding the SH2C domain of PLCγ1 leading to exposure of the ITK recognition element on the PLCγ1 SH2 domain and release of the target tyrosine, Y783. These data contribute to the evolving model for the molecular events occurring early in the T cell activation process.
Keywords: substrate priming, T cell signaling, SH2 domain, phosphotyrosine, autoinhibitory
Graphical abstract
Introduction
Activation of phospholipase Cγ1 (PLCγ1) by phosphorylation of Y783 is mediated by Interleukin-2 induced tyrosine kinase (ITK) and is a key event following T-cell receptor (TCR) stimulation (1, 2). Once active, PLCγ1 hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) to produce inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG); second messengers that control calcium flux from the endoplasmic reticulum and activation of Protein kinase C, respectively (3). A number of additional signaling molecules participate in activation of PLCγ1, in particular the scaffolding functions of SLP-76 (SH2 domain-containing leucocyte protein of 76 kDa) (4), LAT (linker for activation of T cells) (5) and Gads (Grb2-related adaptor downstream of Shc) (6) play important roles in co-localization of ITK and PLCγ1 (7). Specific contacts among the proteins in this T cell signaling complex have been identified and characterized providing glimpses into the molecular mechanisms by which TCR signals are propagated.
ITK and PLCγ1 are both multi-domain containing proteins that have Src homology 2 (SH2) and Src homology 3 (SH3) domains in addition to their catalytic domains (Fig. 1a). Binding sites for the SH2 and SH3 adaptor domains of ITK and PLCγ1 have been identified on both SLP-76 and LAT. Specifically, the PLCγ1 SH3 domain and N-terminal SH2 domain (SH2N) bind to the proline-rich region of SLP-76 and the LAT phosphotyrosine 132 (pY132), respectively (8, 9). The ITK SH2 domain binds one of three well-characterized phosphotyrosine sites (10) in the N-terminal half of SLP-76 (Fig. 1); pY145 is the target of ITK SH2 (11, 12) while SLP-76 pY112 and pY128 bind to SH2 domains within the signaling molecules, Vav and Nck (13–16). In addition to these canonical Src homology domain mediated interactions, a direct substrate docking interaction between ITK and PLCγ1 occurs via a phosphotyrosine independent mechanism involving the C-terminal SH2 domain (SH2C) of PLCγ1 and the ITK kinase domain (17). The PLCγ1 SH2C domain docks onto the C-lobe of the ITK kinase domain creating an enzyme/substrate recognition complex required for efficient phosphorylation of PLCγ1 Y783 (18, 19). Clearly, a large number of distinct protein-protein interactions (Fig. 1b) are present within the complex web of signaling interactions required for proper signal transduction in the T cell (20–22) and it is likely that additional interactions, including those not yet described, also contribute to the pathway. Indeed, a recent finding points to another regulatory phosphorylation site in the N-terminal region of SLP-76 at Y173 (23); this newly recognized phosphotyrosine is adjacent to the pY112, pY128 and pY145 sites.
Figure 1.

Domain structures and schematic of interactions for the signaling proteins that assemble downstream of the T cell receptor. (a) ITK, PLCγ1 and SLP-76 domain maps, PLCγ1 Y783 is indicated within the 33-residue linker between the SH2C and SH3 domains of PLCγ1. The fourth phosphorylation site on SLP-76, Y173, is circled and the Proline-Rich Region of SLP-76 is indicated as PRR (SLP-76 Y112, Y128 and Y145 have been previously characterized and serve as binding sites for Vav, Nck and Itk when phosphorylated). (b) Schematic of the T cell signaling complex involving the proteins in the current study: ITK, PLCγ1 and SLP-76 as well as LAT, Vav, Nck and Gads (not included in this study). The dashed double-headed arrow indicates the SLP-76 pY173/PLCγ1 SH2C interaction described in the current study. Solid, double-headed arrows indicate characterized interactions between specific domains of ITK, PLCγ1, SLP-76 and LAT that are discussed in the text. Asterisks (*) denote canonical SH3 or SH2 mediated interactions involving proline-rich (PXXP) or phosphotyrosine (pY) motifs, respectively. The lighter gray arrow indicates the phosphotyrosine-independent docking interaction between PLCγ1 SH2C and ITK kinase domains that is required for phosphorylation of PLCγ1 by ITK.
Sela et al. (23) demonstrate that phosphorylation at Y173 within SLP-76 is required for TCR induced phosphorylation of PLCγ1. A detailed mechanistic explanation for this observation is currently lacking but the authors in the original work speculate that SLP-76 pY173 may bind to the SH2C domain of PLCγ1 to facilitate docking of PLCγ1 onto the ITK domain (23). The kinase docking site and phosphotyrosine binding pocket form distinct surfaces on the PLCγ1 SH2C domain and it is therefore possible that binding of SLP-76 pY173 to PLCγ1 SH2C may enhance ITK/PLCγ1 docking. We have previously shown, however, that the interaction of PLCγ1 SH2C with the ITK kinase domain is unaffected by phosphopeptide binding to PLCγ1 SH2C (18) and so an allosteric role for SLP-76 pY173 facilitating docking can be ruled out. Co-localization of ITK and PLCγ1 at adjacent sites on SLP-76 may enhance ITK mediated phosphorylation of PLCγ1 at Y783 but PLCγ1 makes multiple contacts within the signaling complex (PLCγ1 SH3/SLP-76; PLCγ1 SH2N/LAT) and Sela et al. show that mutation of Y173 does not reduce association of ITK or PLCγ1 with SLP-76 in T cells (23). Moreover, it has been observed that a loss of function mutation in the SH2C domain within full length PLCγ1 does not significantly affect recruitment of PLCγ1 to the SLP-76 signaling complex yet PLCγ1 Y783 phosphorylation, and hence PLCγ1 activity, is decreased significantly (24).
Here we pursue a detailed model to explain the role of SLP-76 pY173 in T cell signal transduction. We test the hypothesis that SLP-76 pY173 alters the inhibitory conformation of PLCγ1 (25) thereby priming PLCγ1 for activation by ITK. Our findings show that, in spite of a non-classical SH2 binding motif, the SLP-76 derived phosphopeptide surrounding pY173 binds to PLCγ1 SH2C, disrupting an intramolecular PLCγ1 inhibitory complex, leading to release of PLCγ1 Y783 for more efficient phosphorylation by ITK. The data provided here add to our evolving view of the molecular events controlling antigen receptor stimulated activation of PLCγ1.
Results
The unphosphorylated Y783 containing linker in PLCγ1 associates intramolecularly with the adjacent SH2C domain in solution
The structure of the SH2N-SH2C-linker fragment of PLCγ1 has been recently solved (26) and reveals a specific intramolecular contact between the PLCγ1 linker region that contains Y783 and the SH2C domain (Fig. 2a). A large stretch of the PLCγ1 linker region is not defined by the electron density, but Y783 and the following six amino acids (Y783VEANPM) are defined and form contacts with the pY+3 binding pocket of SH2C (Fig. 2a). We first wished to assess the extent to which the Y783 linker region associates with PLCγ1 SH2C in solution. Using NMR spectroscopy, we acquired 1H-15N HSQC data for the PLCγ1 SH2C domain alone and the longer PLCγ1 SH2C-linker protein (Fig 2b). Extensive differences in the chemical shifts of the SH2C NMR resonances are observed upon comparing spectra of PLCγ1 SH2C and SH2C-linker, consistent with association between the linker region and the SH2C domain. Linewidths (and therefore rotational correlation times) are very similar for both proteins indicating the linker region contacts SH2C in an intramolecular manner. Thus, solution NMR data appears consistent with the SH2C/linker association observed in the crystal structure.
Figure 2.

Solution data is consistent with the autoinhibited PLCγ1 conformation observed in the crystal structure. (a) Crystal structure of the PLCγ1 SH2N-SH2C-linker fragment (PDB ID: 4FBN). The SH2N and SH2C domains are gray and the linker region is cyan. The resolved linker residues surrounding and including Y783 are labeled. The N-terminal region of the PLCγ1 linker is also resolved and adopts a helical segment. The intervening eleven residues in the linker are not resolved in the crystal structure and are indicated by a dashed line. (b) Superposition of 1H-15N HSQC spectra for PLCγ1 SH2C (blue) and PLCγ1 SH2C-linker (cyan). (c) Structure of the PLCγ1 SH2C-linker region extracted from the structure shown in (a). Linker is cyan, black indicates SH2C residues for which no chemical shift difference is observed and light gray shows SH2C residues for which chemical shift changes are observed when spectra of SH2C and SH2C-linker are compared (see (b)). (d) Superposition of three 1H-15N HSQC spectra: PLCγ1 SH2C-linker (cyan), mutant PLCγ1 SH2C-linker NPM_AAA (red), and PLCγ1 SH2C (blue). The resonances for two representative residues (H714 and G727; highlighted in green on the PLCγ1 SH2N-SH2C-linker structure in (a)), are shown to illustrate the similarity between spectra of the mutant PLCγ1 SH2C-linker NPM_AAA and the isolated SH2C domain. Apparent doubling of the Gly727 resonance in the spectrum of SH2C-linker NPM_AAA (also see Supp. Fig. S1) may result from multiple conformational states perhaps arising from cis/trans prolyl isomerization occurring in the mutant protein. (e) Size exclusion chromatography (top) showing the elution volume for PLCγ1 SH2C (solid line), SH2C-linker (dotted line) and mutant SH2C-linker NPM_AAA (dashed line). SDS-PAGE (bottom) showing the purity of the wild type and mutant SH2C-linker proteins.
Extensive chemical shift differences between the backbone amide resonances of SH2C and SH2C-linker are observed (Fig. 2b). It is likely that, in addition to intramolecular contacts between linker and SH2C, covalent extension of the C-terminus of SH2C to form SH2C-linker contributes to the observed spectral differences. Nevertheless, we can unequivocally identify the SH2C resonances that do not change when compared to SH2C-linker and the remaining SH2C resonances are therefore either directly or indirectly affected by the presence of the Y783 containing linker. Mapping the spectral changes onto the SH2C domain within the PLCγ1 SH2N-SH2C-linker crystal structure shows that the most contiguous regions giving rise to spectral differences upon linker association correspond to regions of the SH2C domain that directly contact the linker in the crystal structure (Fig. 2c).
To further examine the solution conformation of PLCγ1 SH2C-linker, we probed the contribution of specific residues C-terminal to Y783 since these side chains make contacts to SH2C in the crystal structure (Fig. 2a). We constructed an SH2C-linker variant where N787, P788 and M789 are mutated to alanine (PLCγ1 SH2C-linker NPM_AAA) and acquired 1H-15N HSQC data. The SH2C-linker resonances shift significantly in the mutant protein to positions that are nearly coincident with the resonances of PLCγ1 SH2C alone (Fig. 2d and Supplementary Figure S1) suggesting that the C-terminal residues of SH2C-linker (N787P788M789) stabilize linker association in solution. Finally, using size exclusion chromatography, we find that the retention time of the mutant PLCγ1 SH2C-linker (N787P788M789_AAA) is less than wild type SH2C-linker, suggesting the NPM to AAA mutation releases the linker from the SH2C interaction, increasing the apparent size of the SH2C-linker protein (Fig. 2e). Together, these data provide evidence that the SH2C-linker region in PLCγ1 adopts an intramolecular complex in solution that resembles that observed in the available crystal structure.
Intramolecular SH2C-linker association limits docking of the PLCγ1 substrate to the ITK kinase domain
Efficient PLCγ1 phosphorylation by ITK in T cells depends on a docking interaction between PLCγ1 SH2C and the ITK kinase domain; PLCγ1 SH2C binds to the C-lobe of the ITK kinase domain in a phosphotyrosine independent manner via side chains in the CD loop and C-terminus of SH2C (Fig. 3a). The PLCγ1 linker region associates with SH2C in a region that is nearly coincident with the kinase-docking site we have mapped previously (18). The proximity of PLCγ1 linker bound to SH2C and the recognition motif required for ITK docking prompted us to test the effect of the PLCγ1 linker on ITK kinase domain/PLCγ1 SH2C domain association. In a pull down experiment using immobilized GST fusions of PLCγ1 SH2C and SH2C-linker, we find that the ITK kinase/PLCγ1 SH2C domain interaction is stronger in the absence of the PLCγ1 linker sequence (Fig. 3b). Taken together with the data supporting the intramolecular complex seen in the PLCγ1 crystal structure, we interpret these data to suggest that the intramolecular association between SH2C and linker region competes with binding of PLCγ1 SH2C to the ITK kinase domain.
Figure 3.

Kinase docking site and intramolecular linker association site on PLCγ1 SH2C are partially coincident. (a) Surface rendition of PLCγ1 SH2N-SH2C-linker showing the kinase docking site on SH2C (orange, comprised of the CD loop and C-terminal region of the SH2 domain) required for ITK mediated phosphorylation of PLCγ1 Y783, location of the intramolecular linker association with SH2C (cyan), and the SH2N domain (receding). (b) Duplicate pull-down experiments showing diminished association of the FLAG tagged ITK kinase domain with PLCγ1 SH2C-linker compared to PLCγ1 SH2C. Anti-FLAG antibody shows level of associated ITK kinase domain and Ponceau S shows total levels of GST-SH2C and GST-SH2C-linker. (c) Ribbon structure of PLCγ1 SH2C domain bound to a canonical phosphotyrosine containing peptide (PDB ID: 2PLD). Phosphopeptide ligand is red and the pY and pY+3 residues are labeled. (d) Overlay of the phospholigand bound SH2C structure shown in (c) and the SH2C-linker portion of the PLCγ1 SH2N-SH2C-linker structure. The phosphopeptide, pYIIP, is red and the PLCγ1 linker region containing Y783 is cyan. The pY+3 pocket is circled and the pY pocket is boxed and enlarged in the inset showing the different conformations of the pY and Y783 side chains.
Despite unusual flanking sequence, the SLP-76 pY173 peptide binds to the PLCγ1 SH2C domain
Like the PLCγ1/ITK substrate/kinase docking interaction, the intramolecular interaction between the Y783 linker and SH2C domain is independent of tyrosine phosphorylation. In spite of the absence of phosphorylation on Y783, the interaction of the linker with SH2C bears a resemblance to the structure of a canonical phosphotyrosine ligand bound to the PLCγ1 SH2C domain (Figs. 2a, 3c). The bound linker and pY containing peptide are nearly superimposable and in both cases the ‘specificity pocket’ or pY+3 pocket of SH2C mediates direct contacts with residues C-terminal to the tyrosine (pY or Y783) (Fig. 3d). A difference between the two complexes occurs at the pY-binding pocket. In the SH2/phosphopeptide structure this pocket is filled by the pY side chain of the peptide ligand but the SH2C pY pocket remains empty in the structure of PLCγ1 SH2N-SH2C-linker without Y783 phosphorylation (Fig. 3d, inset).
Given the overlap in binding sites on the PLCγ1 SH2C domain for the unphosphorylated linker and a phosphopeptide ligand, we hypothesize that the pY173 containing phosphopeptide derived from SLP-76 competes with intramolecular linker binding to destabilize the autoinhibited form of PLCγ1. The SLP-76 sequence, pY173-I-D-R, contains an arginine instead of the canonical/preferred hydrophobic residue in the pY+3 position (27) and so we first tested binding of the SLP-76 sequence to PLCγ1 SH2C. Titration of unlabeled SLP-76 peptide, N-S-M-pY173-I-D-R-P-P-T-G-K, into 15N labeled PLCγ1 SH2C was carried out with acquisition of 1H-15N HSQC spectra at each point in the titration (Fig. 4a). Chemical shift changes are measured for each amide NH resonance in the SH2C domain (Fig. 4b) and the largest changes are visualized on the structure of PLCγ1 SH2C (Fig. 4c). The resonance changes induced by addition of the SLP-76 peptide localize to the pY and pY+3 pockets as would be expected for a canonical interaction between phosphotyrosine peptide and SH2 domain (Fig. 4c). The unusual presence of arginine in the pY+3 position prompted us to model this residue into the structure of PLCγ1 SH2C bound to a canonical phosphopeptide. Superposition of the Cα-Cβ bond of arginine onto that of proline in the structure shown in Figure 3c suggests that the hydrophobic residues lining the pY+3 pocket may create favorable interactions with the aliphatic portion of the arginine side chain much as they do with the proline in the pY+3 position (Fig. 4d). In addition, the model suggests the possibility of favorable cation-pi interactions (28) between the guanidinium group of the arginine side chain and two adjacent tyrosines, Y740 and Y741, deep in the pY+3 pocket of SH2C (Fig. 4d). Experimental structure determination will be needed to confirm this model but it is interesting to speculate that certain SH2 domains may be capable of broader ligand recognition within the pY+3 pocket. The only other report of an arginine occupying the pY+3 position in an SH2 domain phospholigand comes from a large proteomics study testing the binding propensity of predicted ligands to various SH2 domains (Supp Fig S4 in (29)). The sequence pYDTR binds to the Crk SH2 domain, which contains FY at the position corresponding to Y740 and Y741 in PLCγ1 SH2C. It is also notable that only a subset of SH2 domains contain the YY or FY motif, suggesting the potential for a specific recognition mechanism for pY target sequences with arginine in pY+3 position (30). Finally, to quantify the affinity of the unusual SLP-76 pY173 peptide binding to PLCγ1 SH2C, we measured fluorescence as a function of increasing pY173 peptide concentration and determined a dissociation constant of 1.6 μM (Fig. 4e).
Figure 4.

The SLP-76 pY173 containing peptide binds the PLCγ1 SH2C domain. (a) Superposition of HSQC spectra from the NMR titration of PLCγ1 SH2C with increasing concentration of the SLP-76 pY173 peptide. Spectra are overlaid from light to dark blue corresponding to increasing SLP-76 pY173 peptide concentration. (b) Chemical shift deviations (Δδ) for each of the PLCγ1 SH2C domain residues upon binding of pY173 phosphopeptide. The average deviation (0.1 ppm) is indicated by the horizontal dashed line. (c) Residues for which Δδ > 0.1 ppm are indicated in red on the structure of PLCγ1 SH2C domain. (d) left panel shows the pY+3 pocket in the structure of PLCγ1 SH2C bound to a peptide containing proline at the pY+3 position (PDB ID: 2PLD, peptide ligand is cyan). Right, model of SH2C/peptide complex containing arginine in place of proline in the pY+3 position. Y740 and Y741 are labeled. (e) Fluorescence titration curve of the SLP-76 pY173 phosphopeptide binding to PLCγ1 SH2C.
Binding of SLP-76 pY173 to PLCγ1 SH2C releases the Y783 containing linker and unmasks the kinase docking site on SH2C
Having determined that the Slp76 derived pY173 phosphopeptide binds to the PLCγ1 SH2C domain, we next examined the effect of this phosphopeptide on the intramolecular PLCγ1 SH2C/linker association. Using dynamic light scattering, we compared the size distribution for PLCγ1 SH2C-linker and SH2C-linker plus the SLP-76 pY173 peptide. The mutant SH2C-linker (N787P788M789_AAA) is used as a control for near complete linker displacement based on the data in Figure 2. Of the three PLCγ1 fragments, SH2C-linker, peptide bound SH2C-linker and the mutant SH2C-linker, the PLCγ1 SH2C-linker protein is the most compact structure with an average hydrodynamic radius of 2.0 nm (Fig. 5a). The size distributions for both the mutant SH2C-linker (N787P788M789_AAA) and SH2C-linker plus SLP-76 pY173 peptide shift to larger particle size; an increase of 12–25% over the SH2C-linker protein (average hydrodynamic radii of 2.25 and 2.5 nm). These data suggest that the pY173 peptide displaces the linker in a manner similar to mutation of the linker itself leading to a more disengaged stretch of amino acids surrounding PLCγ1 Y783. In a separate experiment, addition of increasing SLP-76 pY173 peptide to the 15N-labeled sample of PLCγ1 SH2C-linker (Fig. 5b) reveals NMR chemical shift changes toward the resonances of the pY173 peptide bound SH2C spectrum (Fig. 5c). Thus, the SLP_76 pY173 peptide ligand binds to PLCγ1 SH2C-linker in a manner that is nearly identical to the isolated SH2C domain albeit with slightly lower affinity, Kd = 2.9 μM compared to a Kd = 1.6 μM for the pY173/SH2C interaction (Fig. 5d, 4e). All of these data are consistent with a model where SLP-76 phosphopeptide binding to PLCγ1 SH2C directly competes with and releases the PLCγ1 linker region containing Y783 resulting in exposure of the kinase docking site on PLCγ1 SH2C and increased accessibility of PLCγ1 Y783 for the ITK kinase.
Figure 5.

(a) Dynamic light scattering data for PLCγ1 SH2C-linker (filled diamonds, solid line), mutant SH2C-linker NPM_AAA (filled squares, dotted line) and SH2C-linker + SLP-76 pY173 peptide (filled triangles, dashed line). (b) Superposition of HSQC spectra from the NMR titration of PLCγ1 SH2C-linker with increasing concentration of the SLP-76 pY173 peptide. Spectra are overlaid from light to dark blue corresponding to increasing SLP-76 pY173 peptide concentration (c) Peaks corresponding to the residues of SH2C in the context of SH2C-linker with increasing concentration of SLP-76 pY173 peptide (light to dark) superimposed with peak of same residue (red) when SH2C is fully bound to SLP-76 pY173 peptide. (d) Fluorescence titration curve of the SLP-76 pY173 phosphopeptide binding to PLCγ1 SH2C-linker.
SLP-76 pY173 enhances the ITK phosphorylation of PLCγ1
To assess the effect of SLP-76 pY173 on the ITK mediated phosphorylation of PLCγ1 Y783, we carried out an in vitro kinase assay subjecting full length PLCγ1 to phosphorylation by full length ITK. An antibody specific to PLCγ1 pY783 is used to detect phosphorylation at the PLCγ1 783 position following incubation of the PLCγ1 substrate with active ITK (Fig. 6). Consistent with PLCγ1 adopting an autoinhibited form, we find that in vitro phosphorylation efficiency of Y783 in full length PLCγ1 is quite poor; pY783 levels are not different than background levels detected for the PLCγ1 substrate alone (Fig. 6a, lanes 2 & 3). The same in vitro kinase assay is next carried out with increasing concentration of the SLP-76 derived phosphopeptide, N-S-M-pY173-I-D-R-P-P-T-G-K (Fig. 6a, lanes 4–7). Quantification of the pY783 levels shows that increasing SLP-76 pY173 peptide increases the extent of PLCγ1 pY783 phosphorylation by ITK (Fig. 6b). Smaller fragments of PLCγ1, P-SH2N-SH2C-linker-SH3-H and SH2C-linker, show the same trend (Fig. 6c–f). Increased concentration of the SLP-76 pY173 peptide causes an increase in ITK mediated PLCγ1 phosphorylation on Y783. For the smallest Y783 containing PLCγ1 fragment, SH2C-linker, we tested the importance of the pY binding pocket by mutation of R694 to alanine (Fig. 6e,f). Disruption of the PLCγ1 SH2C binding pocket by mutation results in no increase in Y783 phosphorylation by ITK upon addition of the SLP-76 pY173 peptide (Fig. 6e,f). These data directly implicate the PLCγ1 SH2C domain in the observed enhancement and show that addition of the SLP-76 pY173 peptide to the kinase assay is not simply activating ITK (by binding to the ITK SH2 domain). This latter point is consistent with our previous observations that pY containing peptides do not increase ITK kinase activity toward a generic peptide substrate (unpublished data). Collectively, our findings point to a SLP-76 dependent priming model for PLCγ1 activation by ITK. The SLP-76 pY173 sequence binds to the PLCγ1 SH2C domain, disfavoring the autoinhibited conformation, which renders PLCγ1 a better substrate for ITK.
Figure 6.

SLP-76 pY173 enhances ITK mediated phosphorylation of PLCγ1 at Y783. (a) Lanes 1 and 2 contain enzyme alone and substrate alone, respectively. Phosphorylation of full length PLCγ1 by full length ITK in the absence of SLP-76 pY173 peptide (lane 3) and in the presence of increasing concentration of the SLP-76 pY173 peptide (lanes 4–7). In (a), (c) and (e), phosphorylation of Y783, ITK enzyme levels and substrate levels are detected using anti pY783, anti ITK, and either anti Strep antibody (a) or Coomassie stain (c,e). (b) Histogram representation of pY783 level from three independent experiments; error bars indicate the standard deviation at each concentration of pY173 peptide. Intensity of pY783 level in the kinase reaction that lacks SLP-76 pY173 is normalized to 1. (c) Kinase assay using the smaller PLCγ1 substrate fragment that encompasses the ‘specific array’ or SA and includes P-SH2N-SH2C-linker-SH3-H, with increasing concentration of SLP-76 pY173 peptide as in (a). (d) Histogram representation of pY783 level from three independent experiments using PLCγ1 P-SH2N-SH2C-linker-SH3-H as substrate. (e) Comparison of pY783 level in PLCγ1 SH2C-linker (wt) and the SH2-linker R694A mutant with increasing concentration of the SLP-76 pY173 peptide. (f) Histogram representation of pY783 level from three independent experiments using SH2C-linker wild type and SH2C-linker (R694A) mutant.
Discussion
ITK mediated phosphorylation of Y783 in PLCγ1 occurs in the context of a multi-component signaling cluster that includes the scaffold protein SLP-76 in addition to a number of others proteins (LAT, Vav, Nck, GADS) (Fig. 7). Our data provide support for an autoinhibited conformation of PLCγ1 in solution that is consistent with the reported crystal structure of the SH2N-SH2C-linker region of PLCγ1 (26). We find that the autoinhibited form of PLCγ1 shields the kinase-docking site on the PLCγ1 SH2C domain (18) and limits access to the substrate tyrosine, Y783 (Fig. 7a). Our data also suggest that the most recently characterized phosphorylation site on SLP-76, pY173, can prime PLCγ1 for phosphorylation by ITK.
Figure 7.

Model for priming of PLCγ1 by SLP-76 for phosphorylation and activation by ITK. (a) PLCγ1 is depicted in its autoinhibited conformation. The only domain explicitly depicted is that of SH2C-linker, which is part of the ‘specific array’ (SA) of PLCγ1 that includes the split PH domain, N-terminal SH2 domain (SH2N), SH2C, linker and SH3 (P-SH2N-SH2C-linker-SH3-H). In the autoinhibited form the linker between SH2C and SH3 domains is intramolecularly engaged with SH2C, shielding Y783 and the kinase docking site on SH2C. In this depiction, T cell activation leads to phosphorylation of SLP-76 tyrosines such as Y145 creating a binding site for the ITK SH2 domain. Subsequent phosphorylation of SLP-76 Y173 occurs (possibly by ITK) creating a putative binding site for PLCγ1 SH2C. Additional protein-protein associations involving Nck, Vav, LAT and GADS occur and are included here for context. (b) Binding of PLCγ1 SH2C to SLP-76 pY173 would open the autoinhibited form of PLCγ1, revealing the kinase docking site on PLCγ1 SH2C and priming Y783 for phosphorylation by ITK. (c) Phosphorylated pY783 could compete in an intramolecular fashion with SLP-76 pY173 for the PLCγ1 SH2C binding pocket displacing activated PLCγ1 from SLP-76 and creating an open pY173 binding site on Slp-76 for priming another molecule of PLCγ1. It should be noted that disengaging the SH2C domain from the PLCγ1 catalytic domain could be accomplished by either pY173 (from Slp-76) or pY783 (within PLCγ1) as suggested by Bunney et al. (26).
The SLP-76 pY173 containing phosphopeptide can bind to the PLCγ1 SH2C domain (in spite of the non-canonical arginine in the pY+3 position), releasing the PLCγ1 linker region from SH2C to reveal both the ITK kinase-docking site on PLCγ1 SH2C and the substrate tyrosine, Y783 (Fig. 7a,b). These conformational changes away from the autoinhibited form of PLCγ1 would predispose the PLCγ1 molecule for efficient phosphorylation by ITK. Once PLCγ1 is phosphorylated, an intramolecular interaction between pY783 and SH2C (31, 32) can compete with the PLCγ1 SH2C/SLP-76 pY173 interaction and displace PLCγ1 from the SLP-76/ITK signaling complex allowing another round of ITK mediated catalysis to proceed (Fig. 7c). This final step is consistent with recent observations that activated PLCγ1 rapidly moves away from the ITK/LAT/SLP-76 complex to the TCR cluster (33) and consistent with the relatively low affinity of the pY173 sequence for the PLCγ1 SH2C domain (Fig. 4e). The extent to which the other PLCγ1 adaptor domains (PH, SH2N, SH3, C2) play a role in mediating release of active PLCγ1 from the ITK/SLP-76/LAT complex is not clear and must be considered in any evolving model of T cell activation. Finally, it is notable that complete loss of the SLP-76 pY173 site has no measureable effect on assembly of the signaling complex in T cells yet has a profound effect on the extent to which PLCγ1 is activated. Our biochemical results underscore the importance of how a substrate is presented to its cognate kinase; signaling molecules must not only assemble into a productive complex but must also be able to access the precise conformational state required for productive signal transduction.
The SLP-76 phosphoprotein combines its well-known scaffold function with a regulatory role in the form of conformational priming of PLCγ1. Previously published findings suggested that SLP-76 is required to activate ITK by maintaining an active conformation of the kinase (34). While interactions between SLP-76 pY145 and the ITK SH2 domain are required for proper T cell signaling (11), we have never been able to observe any direct effect of SLP-76 derived phosphopeptides on the in vitro kinase activity of ITK (unpublished data, A.H.A. and Xiaoguang Qu). Since the previously mentioned experiments pointing to a role for SLP-76 in activating ITK (34) made use the PLCγ1 SH2N-SH2C-linker-SH3 substrate to probe ITK catalytic activity, it is possible that the activating effect that was observed is due to the role of SLP-76 in priming PLCγ1 for phosphorylation rather than activation of ITK catalytic activity per se.
There are numerous additional domain interactions that mediate formation of the signaling complexes involving PLCγ1 and ITK and so the evolving picture for PLCγ1 SH2C and SLP-76 pY173 must be considered as part of a larger set of regulatory interactions. A newly described protein-protein interaction regulating B-cell signaling implicates calcium dependent binding of the PLCγ2 C2 domain and pY119 in Slp-65 (the B-cell scaffolding protein related to the T cell expressed SLP-76) (35). Given the sequence similarities surrounding Slp-65 pY119 (EpY119IDNR) and SLP-76 pY173, (MpY173IDRP), the finding in B cells prompted us to consider whether the conformational priming of PLCγ1 we have characterized here could involve the PLCγ1 C2 domain. First we note that the SLP-76 driven amplification of PLCγ1 phosphorylation occurs even for the smallest fragments of PLCγ1 containing just SH2C and linker suggesting that C2 is not mediating the observed increase in pY783 levels (Fig. 6). It is also of note that the Slp-65 derived phosphopeptide does not bind full length PLCγ1 (35) suggesting that the Slp-65 phosphotyrosine motif, pYIDN, does not bind the PLCγ1 C2 domain and may not even bind well to PLC SH2 domain(s) leaving this Slp-65 site available to regulate B cell signaling in a manner quite different from the related site in SLP-76. Our data further suggest that the motif surrounding pY173 in SLP-76 (pYIDR) is tuned to bind specific SH2 domains; in particular the unusual presence of arginine in the pY+3 position may steer the SLP-76 phosphotyrosine motif toward binding SH2 domains (such as PLCγ1 SH2C) that can accommodate the long and positively charged arginine in this position. Thus, despite what appear to be sequence similarities in the proteins that regulate T- and B-cell signaling cascades, we are finding that very different mechanistic rules may apply to these distinct immune signaling systems.
Substrate priming has not, to our knowledge, been previously ascribed to the SLP-76 molecule. Since its identification in 1995 (36), SLP-76 has been well characterized as a scaffold protein and its role in recruiting and co-localizing multiple signaling proteins in T cells is undisputed. Our findings now suggest that SLP-76 may also play a role in directly regulating the apparent enzymatic activity emanating from the T cell receptor proximal Tec kinase, ITK. Substrate priming has been well described in other systems. One example is glycogen synthase; hierarchical phosphorylation events prime glycogen synthase for phosphorylation (and activation) by glycogen synthase kinase (37–39). Another example involves the RING ubiquitin ligases; a conformational shift away from an autoinhibited state coupled with tyrosine phosphorylation comprise integral components of the regulatory mechanisms controlling ubiquitin transfer (40–43). Dissecting the biochemical steps within the trio of T cell signaling proteins, SLP-76, ITK and PLCγ1, has uncovered a possible role for SLP-76 pY173 in promoting PLCγ1 activation in T cells by conformational priming. The next stage in advancing our understanding of how productive signaling complexes are formed in T cells will require significant effort in reconstituting larger, multi-component complexes that can recapitulate the signaling steps present in the cell.
Materials and Methods
Constructs
Baculoviral expression constructs of full-length ITK (mouse) and rat full-length PLCγ1 (rat) have been described previously (44). The fragments of PLCγ1 SH2C, SH2C-linker and P-SH2N-SH2C-linker-SH3-H were amplified by polymerase chain reaction and cloned to pGEX-4T-1 expression vector (GE Healthcare). The SH2C R694A mutation was introduced by site directed mutagenesis (Stratagene). All sequences were verified by sequencing at the Iowa State University DNA synthesis and sequencing facility.
Protein/peptide Production and Purification
Full-length ITK and PLCγ1 were expressed in Sf9 cells and were purified as described previously (44). Similarly, PLCγ1 SH2C and SH2C-linker were expressed in E. coli BL21 cells and purified as described previously (17). For P-SH2N-SH2C-linker-H, bacterial cell pellets were re-suspended in lysis buffer consisting of 50mM Tries, pH 8 and 150 mM NaCl and lysed by treating with lysozyme (0.5 mg/ml). Cleared lysate was applied to a Glutathione Sepharose column (GE Healthcare) equilibrated with lysis buffer. The GST-tagged P-SH2N-SH2C-linker-H protein was eluted with lysis buffer containing 10 mM glutathione and the GST tag was cleaved by treatment with Thrombin (Sigma) for 5 minutes at room temperature. The cleavage reaction was stopped with 1mM PMSF, and the cleaved protein was applied to a HiLoad Superdex 200 prep grade column to separate GST from the desired PLCγ1 P-SH2N-SH2C-linker-H protein. The SLP-76 pY173 peptide with sequence Ac-NSMpY173IDRPPTGK-NH2 was synthesized and purified by GenScript.
NMR Spectroscopy
NMR spectra were collected on a Bruker AVII 700 spectrometer with a 5mm HCN z-gradient cryoprobe operating at a 1H frequency 700.13 MHz at 298K. NMR titration experiments were carried out by acquiring 1H-15N HSQC spectra of 250 μM SH2C or SH2C-linker within increasing SLP-76 pY173 peptide concentration: 0, 39.9, 79.9, 159.1, 238, 355.7, 722.18, 1273, 1982 μM. The chemical shift changes in PLCγ1 SH2C induced by addition the SLP-76 pY173 peptide were quantified by using the following equation: (45). Residues with chemical shift changes above the mean plus one standard deviation were considered significant. Buffer composition used to solubilize the SLP-76 pY173 peptide is identical to the protein buffer (50mM KH2PO4, pH 6.4, 150 mM NaCl, 2mM DTT and 0.02% NaN3).
Fluorescence Spectroscopy
Tryptophan fluorescence for 3 μM PLCγ1 SH2C or SH2C-linker with increasing concentration of the SLP-76 pY173 peptide was measured using a Cary eclipse spectrophotometer at 25°C. The wavelength used for tryptophan excitation was 295nm while emission was recorded between 300–400 nm. All measurements were carried out in triplicate. The maximum fluorescence intensity for each titration point was plotted against ligand concentration and the dissociation constant (Kd) was calculated by fitting the data in MATLAB using the equation: (46), where I is fluorescence intensity, I0 is fluorescence intensity of protein without any ligand, ΔI is change in fluorescence intensity on ligand binding, Kd is dissociation constant, P is protein concentration and L is ligand concentration.
Dynamic light scattering
Hydrodynamic radii of 50 μM PLCγ1 SH2C-linker, 1:1 ratio of SH2C-linker + SLP-76 pY173 peptide, and SH2C-linker NPM_AAA mutant in buffer containing 50mM KH2PO4 pH 6.4, 150 mM NaCl, 2mM DTT and 0.02% NaN3 were measured using a DynaPro® NanoStar™ light scattering instrument (Wyatt Technology) at 25°C.
Kinase assays and western blotting
In vitro kinase assays were performed using full-length ITK and various substrate constructs of PLCγ1 (0.1 μM Full length PLCγ1, 0.1 μM P–SH2N-SH2C-linker-SH3-H, 1 μM SH2C-linker, 1 μM SH2C-linker R694A) with the increasing concentration of synthetic SLP-76 pY173 peptide. Kinase assay buffer consisted of 50mM HEPES pH 7,10mM MgCl2, 1 mM DTT, 1mg/ml BSA, 1mM Pefabloc, and 200 μM ATP. PLCγ1 substrates and SLP-76 pY173 peptide were preincubated for 20 minutes prior to addition of the ITK enzyme. Kinase reactions were carried out for one minute for full length PLCγ1 and P–SH2N-SH2C-linker-SH3-H substrates and five minutes for SH2C-linker and SH2C-linker R694A. Upon completion of the kinase reaction, samples were boiled, separated by SDS-PAGE and western blotted with the anti pY783 antibody (EMD Millipore) for the detection of pY783 levels in each PLCγ1 substrate. Anti ITK (2F12) (Thermo Scientific) and Strep Tag II Monoclonal antibody (EMD Millipore) were used to detect total level of ITK and full length PLCγ1 respectively. Smaller fragments of PLCγ1 were detected by Coomassie staining.
Supplementary Material
Research Highlights.
Examining the role of SLP-76 pY173 in ITK mediated activation/phosphorylation of PLCγ1.
PLCγ1 adopts an autoinhibited conformation in solution consistent with the reported crystal structure of tandem SH2 domains.
The SLP-76 pY173 peptide contains an unusual arginine in the pY+3 position yet binds to the C-terminal SH2 domain of PLCγ1 disfavoring the autoinhibited form.
Binding of SLP-76 pY173 enhances ITK mediated phosphorylation of PLCγ1
The results add a new function – substrate priming – to the important scaffolding functions of the T cell signaling protein SLP-76.
Acknowledgments
This work is supported by grants from the National Institutes of Health (National Institute of Allergy and Infectious Diseases, AI43957 & AI075150) to A.H.A. and by the Gary Roewe Research Award to S. Devkota. We also acknowledge technical assistance from Thamothanaran Subbiah in DNA construction and protein purification for the PLCγ1 P–SH2N-SH2C-linker-SH3-H fragment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Houtman JC, Houghtling RA, Barda-Saad M, Toda Y, Samelson LE. Early phosphorylation kinetics of proteins involved in proximal TCR-mediated signaling pathways. J Immunol. 2005;175:2449–2458. doi: 10.4049/jimmunol.175.4.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Villar JJ, Kanner SB. Regulated association between the tyrosine kinase Emt/Itk/Tsk and phospholipase-C gamma 1 in human T lymphocytes. J Immunol. 1999;163:6435–6441. [PubMed] [Google Scholar]
- 3.Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev. 2009;228:93–114. doi: 10.1111/j.1600-065X.2008.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu JN, Koretzky GA. The SLP-76 family of adapter proteins. Semin Immunol. 2004;16:379–393. doi: 10.1016/j.smim.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Yablonski D, Weiss A. Mechanisms of signaling by the hematopoietic-specific adaptor proteins, SLP-76 and LAT and their B cell counterpart, BLNK/SLP-65. Adv Immunol. 2001;79:93–128. doi: 10.1016/s0065-2776(01)79003-7. [DOI] [PubMed] [Google Scholar]
- 6.Yoder J, Pham C, Iizuka YM, Kanagawa O, Liu SK, McGlade J, Cheng AM. Requirement for the SLP-76 adaptor GADS in T cell development. Science. 2001;291:1987–1991. doi: 10.1126/science.1057176. [DOI] [PubMed] [Google Scholar]
- 7.Koretzky GA, Abtahian F, Silverman MA. SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat Rev Immunol. 2006;6:67–78. doi: 10.1038/nri1750. [DOI] [PubMed] [Google Scholar]
- 8.Deng L, Velikovsky CA, Swaminathan CP, Cho S, Mariuzza RA. Structural basis for recognition of the T cell adaptor protein SLP-76 by the SH3 domain of phospholipase Cgamma1. J Mol Biol. 2005;352:1–10. doi: 10.1016/j.jmb.2005.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paz PE, Wang S, Clarke H, Lu X, Stokoe D, Abo A. Mapping the Zap-70 phosphorylation sites on LAT (linker for activation of T cells) required for recruitment and activation of signalling proteins in T cells. Biochem J. 2001;356:461–471. doi: 10.1042/0264-6021:3560461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan MS, Sadler J, Austin JE, Finkelstein LD, Singer AL, Schwartzberg PL, Koretzky GA. Functional hierarchy of the N-terminal tyrosines of SLP-76. J Immunol. 2006;176:2430–2438. doi: 10.4049/jimmunol.176.4.2430. [DOI] [PubMed] [Google Scholar]
- 11.Bunnell SC, Diehn M, Yaffe MB, Findell PR, Cantley LC, Berg LJ. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J Biol Chem. 2000;275:2219–2230. doi: 10.1074/jbc.275.3.2219. [DOI] [PubMed] [Google Scholar]
- 12.Su YW, Zhang Y, Schweikert J, Koretzky GA, Reth M, Wienands J. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur J Immunol. 1999;29:3702–3711. doi: 10.1002/(SICI)1521-4141(199911)29:11<3702::AID-IMMU3702>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 13.Wu J, Motto DG, Koretzky GA, Weiss A. Vav and SLP-76 interact and functionally cooperate in IL-2 gene activation. Immunity. 1996;4:593–602. doi: 10.1016/s1074-7613(00)80485-9. [DOI] [PubMed] [Google Scholar]
- 14.Wunderlich L, Farago A, Downward J, Buday L. Association of Nck with tyrosine-phosphorylated SLP-76 in activated T lymphocytes. Eur J Immunol. 1999;29:1068–1075. doi: 10.1002/(SICI)1521-4141(199904)29:04<1068::AID-IMMU1068>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 15.Raab M, da Silva AJ, Findell PR, Rudd CE. Regulation of Vav-SLP-76 binding by ZAP-70 and its relevance to TCR zeta/CD3 induction of interleukin-2. Immunity. 1997;6:155–164. doi: 10.1016/s1074-7613(00)80422-7. [DOI] [PubMed] [Google Scholar]
- 16.Fang N, Motto DG, Ross SE, Koretzky GA. Tyrosines 113, 128, and 145 of SLP-76 are required for optimal augmentation of NFAT promoter activity. J Immunol. 1996;157:3769–3773. [PubMed] [Google Scholar]
- 17.Joseph RE, Min L, Xu R, Musselman ED, Andreotti AH. A remote substrate docking mechanism for the tec family tyrosine kinases. Biochemistry. 2007;46:5595–5603. doi: 10.1021/bi700127c. [DOI] [PubMed] [Google Scholar]
- 18.Min L, Joseph RE, Fulton DB, Andreotti AH. Itk tyrosine kinase substrate docking is mediated by a nonclassical SH2 domain surface of PLCgamma1. Proc Natl Acad Sci U S A. 2009;106:21143–21148. doi: 10.1073/pnas.0911309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Q, Joseph RE, Fulton DB, Andreotti AH. Substrate recognition of PLCgamma1 via a specific docking surface on Itk. J Mol Biol. 2013;425:683–696. doi: 10.1016/j.jmb.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol. 2014;15:798–807. doi: 10.1038/ni.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherman E, Barr V, Samelson LE. Super-resolution characterization of TCR-dependent signaling clusters. Immunol Rev. 2013;251:21–35. doi: 10.1111/imr.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samelson LE. Immunoreceptor signaling. Cold Spring Harb Perspect Bioll. 2011;3:a011510. doi: 10.1101/cshperspect.a011510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sela M, Bogin Y, Beach D, Oellerich T, Lehne J, Smith-Garvin JE, Okumura M, Starosvetsky E, Kosoff R, Libman E, Koretzky G, Kambayashi T, Urlaub H, Wienands J, Chernoff J, Yablonski D. Sequential phosphorylation of SLP-76 at tyrosine 173 is required for activation of T and mast cells. EMBO J. 2011;30:3160–3172. doi: 10.1038/emboj.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braiman A, Barda-Saad M, Sommers CL, Samelson LE. Recruitment and activation of PLCgamma1 in T cells: a new insight into old domains. Embo J. 2006;25:774–784. doi: 10.1038/sj.emboj.7600978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBell K, Graham L, Reischl I, Serrano C, Bonvini E, Rellahan B. Intramolecular regulation of phospholipase C-gamma1 by its C-terminal Src homology 2 domain. Mol Cell Biol. 2007;27:854–863. doi: 10.1128/MCB.01400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunney TD, Esposito D, Mas-Droux C, Lamber E, Baxendale RW, Martins M, Cole A, Svergun D, Driscoll PC, Katan M. Structural and functional integration of the PLCgamma interaction domains critical for regulatory mechanisms and signaling deregulation. Structure. 2012;20:2062–2075. doi: 10.1016/j.str.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneko T, Joshi R, Feller SM, Li SS. Phosphotyrosine recognition domains: the typical, the atypical and the versatile. Cell Commun Signal. 2012;10:32. doi: 10.1186/1478-811X-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crowley PB, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 29.Liu BA, Jablonowski K, Shah EE, Engelmann BW, Jones RB, Nash PD. SH2 domains recognize contextual peptide sequence information to determine selectivity. Mol Cell Proteomics. 2010;9:2391–2404. doi: 10.1074/mcp.M110.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell SJ, Jackson RM. Diversity in the SH2 domain family phosphotyrosyl peptide binding site. Protein Eng. 2003;16:217–227. doi: 10.1093/proeng/gzg025. [DOI] [PubMed] [Google Scholar]
- 31.Hajicek N, Charpentier TH, Rush JR, Harden TK, Sondek J. Autoinhibition and phosphorylation-induced activation of phospholipase C-gamma isozymes. Biochemistry. 2013;52:4810–4819. doi: 10.1021/bi400433b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gresset A, Hicks SN, Harden TK, Sondek J. Mechanism of phosphorylation-induced activation of phospholipase C-gamma isozymes. J Biol Chem. 2010;285:35836–35847. doi: 10.1074/jbc.M110.166512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruz-Orcutt N, Vacaflores A, Connolly SF, Bunnell SC, Houtman JC. Activated PLC-gamma1 is catalytically induced at LAT but activated PLC-gamma1 is localized at both LAT- and TCR-containing complexes. Cell Signal. 2014;26:797–805. doi: 10.1016/j.cellsig.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogin Y, Ainey C, Beach D, Yablonski D. SLP-76 mediates and maintains activation of the Tec family kinase ITK via the T cell antigen receptor-induced association between SLP-76 and ITK. Proc Natl Acad Sci U S A. 2007;104:6638–6643. doi: 10.1073/pnas.0609771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelke M, Oellerich T, Dittmann K, Hsiao HH, Urlaub H, Serve H, Griesinger C, Wienands J. Cutting edge: feed-forward activation of phospholipase Cgamma2 via C2 domain-mediated binding to SLP65. J Immunol. 2013;191:5354–5358. doi: 10.4049/jimmunol.1301326. [DOI] [PubMed] [Google Scholar]
- 36.Jackman JK, Motto DG, Sun Q, Tanemoto M, Turck CW, Peltz GA, Koretzky GA, Findell PR. Molecular cloning of SLP-76, a 76-kDa tyrosine phosphoprotein associated with Grb2 in T cells. J Biol Chem. 1995;270:7029–7032. doi: 10.1074/jbc.270.13.7029. [DOI] [PubMed] [Google Scholar]
- 37.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- 38.Williams DD, Marin O, Pinna LA, Proud CG. Phosphorylated seryl and threonyl, but not tyrosyl, residues are efficient specificity determinants for GSK-3beta and Shaggy. FEBS Lett. 1999;448:86–90. doi: 10.1016/s0014-5793(99)00342-7. [DOI] [PubMed] [Google Scholar]
- 39.Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 40.Dou H, Buetow L, Hock A, Sibbet GJ, Vousden KH, Huang DT. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat Struct Mol Biol. 2012;19:184–192. doi: 10.1038/nsmb.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. Essentiality of a non-RING element in priming donor ubiquitin for catalysis by a monomeric E3. Nat Struct Mol Biol. 2013;20:982–986. doi: 10.1038/nsmb.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozkan E, Yu H, Deisenhofer J. Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci U S A. 2005;102:18890–18895. doi: 10.1073/pnas.0509418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489:115–120. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joseph RE, Fulton DB, Andreotti AH. Mechanism and functional significance of itk autophosphorylation. J Mol Biol. 2007;373:1281–1292. doi: 10.1016/j.jmb.2007.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang YG, Song AX, Gao YG, Shi YH, Lin XJ, Cao XT, Lin DH, Hu HY. Solution structure of the ubiquitin-associated domain of human BMSC-UbP and its complex with ubiquitin. Protein Sci. 2006;15:1248–1259. doi: 10.1110/ps.051995006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bodenreider C, Beer D, Keller TH, Sonntag S, Wen D, Yap L, Yau YH, Shochat SG, Huang D, Zhou T, Caflisch A, Su XC, Ozawa K, Otting G, Vasudevan SG, Lescar J, Lim SP. A fluorescence quenching assay to discriminate between specific and nonspecific inhibitors of dengue virus protease. Anal Biochem. 2009;395:195–204. doi: 10.1016/j.ab.2009.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.