

Abstract
siRNA has the possibility to revolutionize medicine by enabling highly specific and efficient silencing of proteins involved in disease pathogenesis. Despite nearly 20 years of research dedicated to translating siRNA from a research tool into a clinically relevant therapeutic, minimal success has been had to date. Access to RNA interference machinery located in the cytoplasm is often overlooked, but must be considered when designing the next generation of siRNA delivery strategies. Peptide transduction domains (PTD) have demonstrated moderate siRNA transfection, which is primarily limited by endosomal entrapment. Strategies aimed at overcoming endosomal entrapment associated with peptide vectors are reviewed here, including osmotic methods, lipid conjugation, and fusogenic peptides. As an alternative to traditional PTD, the hemolytic peptide melittin exhibits the native capacity for endosomal disruption but causes cytotoxicity. However, appropriate packaging and protection of melittin with activation and release in the endosomal compartment has allowed melittin-based strategies to demonstrate both in vitro and in vivo safety and efficacy. These data suggest that melittin's membrane disruptive properties can enable safe and effective endosomolysis, building a case for melittin as a key component in a new generation of siRNA therapeutics.
Keywords: Melittin, siRNA delivery, endosomal escape
1 Introduction
1.1 RNA Interference by siRNA
RNA interference (RNAi) refers to an evolutionary conserved mechanism for post transcriptional control of protein expression in which short double-stranded RNA target specific messenger RNA (mRNA) for degradation, thus decreasing protein translation (Eccleston et al. 2004). Tuschl et al. were the first to demonstrate that RNAi can be artificially induced by the delivery of exogenous small interfering RNA (siRNA) (Elbashir et al. 2001). siRNA are short 21-23 base pair duplex RNA oligonucleotides with 5′-phosphorylated ends and 2-nucleotide 3′ overhangs. The “antisense” strand shares sequence complementarity to a target mRNA, while the “sense” strand serves as a bystander. When delivered into the cytoplasm of a cell, siRNA can co-opt the native RNAi machinery and induce assembly of the RNA induced silencing complex (RISC). The RISC unwinds siRNA, binds the antisense strand, and cleaves the sense strand, allowing the RISC to bind and cleave targeted mRNA based on the sequence of the antisense strand (Sakurai et al. 2011). This selective degradation of mRNA provides an avenue to decrease the expression of proteins involved in disease pathogenesis. Indeed, initial experimental siRNA therapeutics appeared just three years after the discovery of siRNA (McCaffrey et al. 2002). Recently, clinical trials have been initiated for the use of siRNA in ocular, renal, and hepatic diseases with limited success (Haussecker 2012).
1.1 Cellular Barriers to the Therapeutic Use of siRNA
On a cellular level, efficient and non-toxic siRNA delivery to the cytoplasm remains a major impediment to the use of siRNA as a therapeutic. siRNA are large (∼21kDa), highly charged macromolecules, which can not directly translocate across the hydrophobic core of the cell membrane (Overhoff et al. 2005). Moreover, the barrier provided by impermeable membrane bilayers not only applies to direct translocation from the extracellular milieu into the cytoplasm, but also applies to cytosolic access of siRNA enclosed in endocytic vesicles (Detzer et al. 2009, Gilleron et al. 2013). Entrapment of siRNA in endocytic compartments not only prevents siRNA from reaching the cytosol, but also accelerates siRNA degradation due to the harsh acidic environment encountered during endosome-lysosome trafficking (Figure 2) (Wang et al. 2010).
Figure 2.
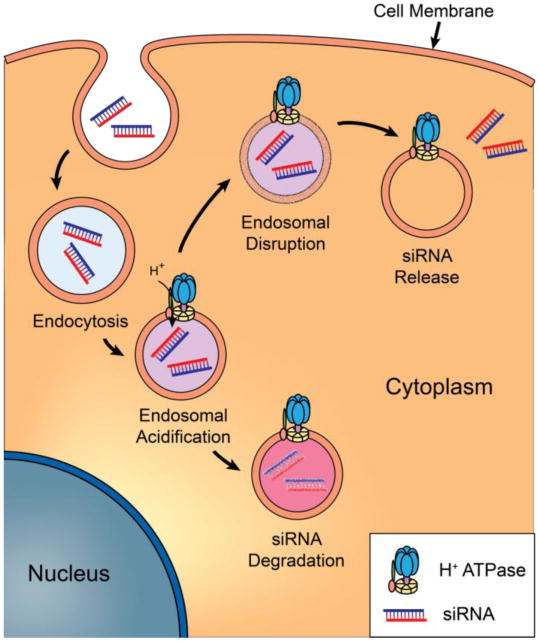
siRNA entering the cell via endocytosis is degraded in lysosomes by the acidic environment before reaching the cytoplasm due to endosomal entrapment. However, appropriate methods for achieving endosomal disruption can release siRNA into the cytoplasm.
This barrier represents a major impediment in the delivery of biological therapeutics to the cytoplasm, especially when packaged with peptide transduction domains (El-Sayed et al. 2009). Strategies to achieve endosomolysis have traditionally been based on osmotic agents, fusogenic lipids, and fusogenic peptides. The application of these methodologies to peptide-mediated siRNA delivery will be reviewed here. In addition, novel strategies enabled by the membrane-disruptive properties of melittin appear to negate the need for secondary endosomolytic methodologies and will be presented as an alternative to traditional peptide-based siRNA delivery strategies.
2 Cationic Peptides for siRNA Transfection
Given the challenges associated with traditional siRNA delivery methodology, there is clearly a need for siRNA delivery technology to enable endosomal escape with minimal cytotoxicity. With the observation that the Trans-Activator of Transcription (TAT) peptide from HIV could directly translocate across cell membranes to trans-activate the viral promoter in tissue culture, cell penetrating peptides (CPPs) have become a widely utilized tool for delivery of therapeutics (Frankel et al. 1988). CPPs have been recruited for delivery of cargoes ranging from small molecules to large proteins (Heitz et al. 2009). With their hypothesized ability to bypass the cellular membrane, CPPs were expected to enable cytoplasmic delivery of siRNA, while avoiding the endosomal compartment. Although this hope has ultimately proven unfounded, peptides remain a viable option for siRNA delivery thanks to a relative lack of cytotoxic effects.
2.1 Covalent Formulations
Initial attempts to harness peptides for siRNA transfection focused on direct chemical conjugation to known cell penetrating peptides, penetratin and transportan. These studies reported an IC50 of 25nM, and with minimal cytotoxicity (Chiu et al. 2004, Davidson et al. 2004, Muratovska et al. 2004). Unfortunately, these initial studies were later shown to be confounded by poor purification as excess unconjugated peptide augmented the transfection efficiency. Turner et al. demonstrated that after careful purification, siRNA conjugated to cell penetrating peptides had minimal transfection capacity, requiring 5μM to achieve significant knockdown (Turner et al. 2006, Turner et al. 2007, Endoh et al. 2008). They attributed the limited transfection capacity to endosomal entrapment based on the findings that endosomolytic agents such as chloroquine were able to release siRNA into the cytoplasm (Shiraishi et al. 2006). When tested for intratracheal delivery of siRNA to the lung, these purified conjugates did not exhibit any improvement over naked siRNA alone (Moschos et al. 2007). In addition, work by other groups revealed that peptide-siRNA conjugate uptake was limited in the presence of serum proteins (Turner et al. 2005). However, all studies investigating the use of peptide-oligonucleotide conjugates have shown minimal cytotoxicity with doses up to 5-10μM in vitro, indicating at least a high degree of safety on a cellular level (Lundin et al. 2008).
2.2 Noncovalent Formulations
Due to the limited efficacy of peptide-siRNA conjugates and the finding that excess peptide imbues improved transfection efficiency, peptide-based siRNA vectors have more commonly been utilized in non-covalent formulations (Table 1). Initial studies examining siRNA delivery via electrostatic packaging by TAT, penetratin, and transportan all gave minimal siRNA mediated knockdown despite high levels of siRNA internalization (Lundberg et al. 2007, Endoh et al. 2008, Meade et al. 2008). Further treatment with chloroquine increased siRNA-mediated knockdown, revealing that some of these peptides were unable to achieve sufficient endosomal escape (Mäe et al. 2009). Interestingly, penetratin and poly-arginine peptides continued to exhibit minimal knockdown in the presence of chloroquine indicating that endocytosed siRNA was not released from the peptide vector. These initial studies point out the requirement for peptide vectors to both release siRNA and promote endosomal escape in order to achieve maximal siRNA transfection efficiency.
Table 1. Cell penetrating peptides for siRNA transfection.
| Peptide | Target Gene | IC50 | Ref |
|---|---|---|---|
| TAT47-57 (YGRKKRRQRRR) | eGFP | none | (Meade et al. 2007, Endoh et al. 2008) |
| Penetratin (RQIKIWFQNRRMKWKK-amide) | Luciferase | none | (Lundberg et al. 2007, Åmand et al. 2008, Mäe et al. 2009) |
| Transportan (GWTLNSAGYLLGKINLKALAALAKKIL-amide) | Luciferase | none | (Lundberg et al. 2007) |
| TP10 (AGYLLGKINLKALAALAKKIL-amide) | Luciferase | none | (Lundberg et al. 2007, Mäe et al. 2009) |
| Rn (8<n<15) | |||
| R9-RVG | GFP | 100nM | (Kumar et al. 2007) |
| R9 | VEGF | none | (Kim et al. 2006, Mäe et al. 2009) |
| R15 | VEGF | 100nM | (Kim et al. 2010) |
| Dermaseptin S4 | -- | none | (Trabulo et al. 2010) |
| MPG (Ac-GALFLGFLGAAGSTMGAWSQPKKKRKV-amide) | Luciferase | >100nM | (Simeoni et al. 2003) |
| MPGΔNLS (Ac-GALFLGFLGAAGSTMGAWSQPKSKRKV-amide) | Luciferase, GAPDH, Cyclin B1 | 30-50nM | (Simeoni et al. 2003, Crombez et al. 2009) |
| MPG8 (Ac-βAFLGWLGAWGTMGWSPKKKRK-amide) | Cyclin B1 | 1nM | (Crombez et al. 2009) |
| MPGα (Ac-GALFLAFLAAALSLMGLWSQPKKKRKV-amide) | Luciferase | 1nM | (Veldhoen et al. 2006) |
| CADY (GLWRALWRLLRSLWRLLWRA-amide) | GAPDH | <1nM | (Crombez et al. 2008, Crombez et al. 2011, Rydström et al. 2011, Deshayes et al. 2012) |
| dsRBD | none | (Kim et al. 2009) | |
| TAT-dsRBD | 400nM | (Eguchi et al. 2009) |
Despite these initial difficulties, noncovalent siRNA delivery via cell penetrating peptides has achieved some success, with much higher efficiencies (IC50<1nM) than covalent means (IC50>5μM) (Meade et al. 2008, Okuda et al. 2009, Hassane et al. 2010). Moreover, peptide vectors have demonstrated efficacy in a variety of cell types in tissue culture as well as successful abatement of cancer progression in mouse models (Endoh et al. 2009). Importantly, peptide vectors have demonstrated remarkable safety, with minimal toxicity even at high μM concentrations. Unfortunately, the limiting factor for peptide-based vectors appears to be endosomal entrapment as well as decreased transfection in the presence of serum proteins (Mäe et al. 2009, Crombez et al. 2011). Selected peptides for non-covalent siRNA delivery are reviewed below.
2.2.1 Noncovalent Formulations: MPG
Initial success with peptide mediated siRNA delivery was achieved with MPG, a hybrid peptide consisting of the fusion sequence from HIV glycoprotein 41 and the nuclear localization sequence of the SV40 virus. This peptide, originally developed for plasmid DNA transfection, achieved knockdown of target mRNA with an IC50 of 20-50nM (Simeoni et al. 2003, Deshayes et al. 2004, Deshayes et al. 2008). Initial work indicated that MPG mediated siRNA transfection via direct translocation through the cell membrane. However, later studies concluded that MPG/siRNA complexes enter cells via macropinocytosis (Veldhoen et al. 2008). Further sequence refinement led to a truncated form, MPG-8, as well as MPGα which has increased membrane inserting properties (Veldhoen et al. 2006, Crombez et al. 2009). These peptides are characterized by sub-nanomolar IC50 when targeting luciferase. However, detailed work by Veldhoen et al. demonstrated that MPGα is not maximally efficient, required almost two orders of magnitude more siRNA to achieve the same knockdown as Lipofectamine 2000 (Veldhoen et al. 2006). This finding may be attributable to endosomal entrapment, as treatment with chloroquine improved transfection by nearly 20%. Despite these difficulties, MPG has been licensed to Sigma-Aldrich as the N-TER siRNA transfection system designed for in vitro research applications (Sigma Aldrich, 2015).
2.2.2 Noncovalent Formulations: CADY
CADY is the first peptide designed to specifically promote both siRNA binding and membrane permeability. With an alpha-helical secondary structure, CADY has a siRNA binding face with cationic residues, and a membrane binding face with tryptophan residues (Deshayes et al. 2012). To date, CADY is the most efficient peptide based siRNA vector with an IC50 <0.5nM (Crombez et al. 2008). Interestingly, CADY appears to function by direct membrane translocation as neither ATP depletion nor incubation at 4°C inhibits siRNA transfection (Rydström et al. 2011). Nevertheless, CADY may experience limited in vivo utility due to decreased transfection in the presence of serum proteins (Crombez et al. 2011).
2.2.3 Noncovalent Formulations: dsRBD
Dowdy et al. further improved upon peptide-mediated transfection by utilizing conjugates of TAT and double stranded RNA binding domains (dsRBD) (Eguchi et al. 2009, Kim et al. 2009). dsRBD sequences were modified from protein kinase R, a cytoplasmic protein which plays a crucial role in the detection of viral infection. These TAT-dsRBD conjugates were shown to transfect a variety of difficult to transfect cell lines such as Jurkat T-cells and endothelial cells in tissue culture with an IC50 near 400nM, and were able to yield significant siRNA knockdown in vivo. Unfortunately, later studies demonstrated that due to low binding affinities, a single dsRBD is unable to bind siRNA efficiently, suggesting that TAT-dsRBD mediated transfection is attributable to electrostatic TAT/siRNA interactions (Geoghegan et al. 2012). These peptides were initially commercialized by Traversa Therapeutics and were licensed to IDT as research tools (2009). Unfortunately, there has been limited progress after Traversa shut down in 2012 after failing to secure funding. However, progress has been made recently with further in vivo studies focused on inner ear transfection in chinchillas (Qi et al. 2014).
2.3 Modified Peptides for siRNA Transfection
Modifications to cell penetrating peptides that promote increased endosomal escape have centered on three design features: combination with buffering agents to promote osmotic rupture of endosomes, fusion to pH sensitive fusogenic viral peptides, conjugation to lipid moieties, and (Table 2) (Varkouhi et al. 2011, Erazo-Oliveras et al. 2012).
Table 2. Modified Peptide Vectors.
| Modified Peptides | Benefit | Ref |
|---|---|---|
| TAT | ||
| Stearylation | Improved siRNA uptake/Endosomal escape | (Futaki et al. 2001) |
| HA2 conjugation | pH triggered endosomal escape | (Wadia et al. 2004) |
| LK15 conjugation | Endosomal escape | (Arthanari et al. 2010) |
| Histidine10 conjugation | pH triggered endosomal escape | (Lo et al. 2008) |
| Penetratin | ||
| HA2 conjugation | pH triggered endosomal escape | (Lundberg et al. 2007) |
| Stearylation | pH triggered endosomal escape | (Mäe et al. 2009) |
| Poly-Arginine | ||
| Stearylation | Endosomal escape | (Mäe et al. 2009, Lehto et al. 2011) |
| Myristoylation | Brain targeted delivery | (Ifediba et al. 2010) |
| Cholesterol conjugation | Increased particle stability/Endosomal escape | (Kim et al. 2006) |
| Stearylation and Insertion of histidine | Endosomal escape | (Tanaka et al. 2010) |
| Poly-Lysine | ||
| Insertion of histidine | Endosomal escape | (Yu et al. 2004, Langlet-Bertin et al. 2010, Chou et al. 2011) |
| TP10 | ||
| EB1 | Endosomal Escape | (Lundberg et al. 2007) |
| Stearylation | Improved siRNA uptake/Endosomal escape | (Mäe et al. 2009) |
| Stearylation and chloroquine conjugation | pH triggered endosomal escape | (Andaloussi et al. 2011, Ezzat et al. 2011, Ezzat et al. 2012, Lindberg et al. 2013) |
| MPG8 | ||
| Cholesterol conjugation | Improved particle stability/Endosomal Escape | (Crombez et al. 2009) |
| Calcitonin-derived peptides | ||
| Myristoylation | Improved siRNA uptake/Endosomal Escape | (Hoyer et al. 2012) |
| HA2 conjugation | Improved siRNA uptake/pH triggered endosomal escape | (Hoyer et al. 2012) |
2.3.1 Osmotic Endosomolysis
Lysosomotropic agents are taken up selectively into lysosomes. One example is chloroquine, which is a weak base that accumulates in endosomes and lysosomes after being protonated (Wibo et al. 1974). Chloroquine's functions include inhibiting endosomal acidification and causing endosomolysis. Cells incubated with chloroquine exhibit extensive endosomal swelling and vacuolation suggestive of endosomal disruption (Ohkuma et al. 1981, Erbacher et al. 1996). Moreover, biologic therapeutics including plasmids exhibit greater degrees of cytoplasmic delivery when delivered in the presence of chloroquine (El-Sayed et al. 2009). The proposed mechanism involves chloroquine's ability to buffer endosomal protons leading to accumulation of counterions and osmotic rupture of endosomes.
The importance of endosomal pH buffering as a release mechanism has led researchers to take advantage of histidine residues as a potential trigger for endosomal escape (Yu et al. 2004). Histidine residues are unique for their ability to be protonated at acidic pH (pKa ∼6) while remaining uncharged at neutral pH. By incorporating high percentages of protonatable histidine residues, poly arginine and TAT have been modified to increase their ability to delivery nucleotides to the cytoplasm. For instance, the addition of 10 histidine residues to TAT improves plasmid DNA transfection by 7000 fold over TAT itself (Lo et al. 2008). Unfortunately, the use of histidine-augmented peptides has yet to provide the efficiency of lipid or polymer vectors despite their lack of cytotoxicity (Tanaka et al. 2010).
2.3.2 Fusogenic Peptides
Standard CPPs such as penetratin and TAT have been fused with portions of influenza proteins hemagluttin-2 (HA2), LK15, or N-E5L which mediate viral escape from the endosome when exposed to acidic pH (Lundberg et al. 2007, Neundorf et al. 2009, Arthanari et al. 2010, Hoyer et al. 2012). This functional pH triggered structural change increases the alpha helical content of the peptide and promotes insertion into the endosomal membrane. In vitro, HA2 fusions have shown the ability to improve siRNA transfection when packaged by CPP (Lundberg et al. 2007). Unfortunately, this modification increases the cytotoxicity of the peptide inducing significant cell death at 1μM (Lundberg et al. 2007). Furthermore, these fusion peptides are not maximally efficient as treatment with chloroquine improves siRNA transfection indicating that endosomal entrapment is a problem that is not completely addressed by these methods (Hoyer et al. 2012).
2.3.3 Lipid Conjugation
Additional attempts to increase the membrane disruptive potential of cell penetrating peptides come in the form of conjugation to lipids, most commonly stearyl moieties (Mäe et al. 2009, Nakase et al. 2012). Originally developed for plasmid DNA, stearylated peptides also improve the transfection of small oligonucleotides such as siRNA and splice correcting oligos (Futaki et al. 2001, Lehto et al. 2010). For these purposes, stearyl-TP10 has proven to be more effective than stearyl-penetratin, stearyl-TAT, or stearyl-R8 (Mäe et al. 2009, Ren et al. 2012). Studies utilizing unstearylated peptides suggest that differences in these stearylated variants may be due to the ability of the peptide to release siRNA. Moreover, in the case of stearyl-TP10, authors noted that improved transfection may be due to improved particle stability and increased siRNA uptake instead of improved endosomal release (Mäe et al. 2009). In fact, treatment with chloroquine doubled siRNA-mediated knockdown, again revealing poor endosomal release. This finding is not surprising given that stearyl moieties are not considered fusogenic lipids, with the majority of membrane disruption typically attributed to the lipid headgroup. The extent to which stearylation improves peptide disruption of membranes remains unclear. However the ability of peptide stearylation to increase siRNA transfection is clearly demonstrated across multiple studies.
2.3.4 PepFect – Combining Endosomolytic Strategies
The group of Langel et al. has further modified cell-penetrating peptides to overcome known endosomal entrapment of CPP-based oligonucleotide delivery agents. Their efforts have centered on the CPP TP10, the shortened version of transportan. Modifications made to TP10 include stearylation, replacement of lysines by ornithines, addition of trifluoromethylquinolone side chains, chemical modification with UV light responsive side chains and oligomerization (Regberg et al. 2014). These modifications have resulted in a range of “PepFect” and “NickFect” peptides, which deliver siRNA, splice correcting oligos, and even plasmid DNA through a mechanism that involves Scavenger Receptor A mediated endocytosis (Hassane et al. 2011, Ezzat et al. 2012, Arukuusk et al. 2013).
By taking advantage of stearylation and conjugation to the proton buffering agent chloroquine, Andaloussi et al. have created Pepfect6, which has an IC50 of <10nM in a variety of cell types (Andaloussi et al. 2011). They have further demonstrated that stearylation improves particle stability and also increases the peptides amphipathicity to promote insertion into model membranes. This presumably contributes to membrane destabilization. Additionally, the incorporation of three chloroquine moieties per peptide likely contributes to improved endosomal escape via the proton sponge effect.
In analogous work, PepFect14 and PepFect 15 were developed for the transfection of splice-correcting oligos (Ezzat et al. 2011, Lindberg et al. 2013). Starting with stearylated TP10, lysines were replaced with ornithines to improve siRNA binding and also provide a higher transfection efficiency. This peptide, Pepfect14, was able to transfect SCO into HeLa cells with an EC50 of 86-104nM depending no the presence of serum, however, even with these modifications, significant transfection efficacy was lost due to endosomal entrapment (Ezzat et al. 2011). Transfection in the presence of chloroquine augments SCO transfection by over 2 fold at 50nM siRNA concentration. Given this evidence supporting inadequate endosomal escape, PepFect14 was further modified to include the chloroquine analog trifluoromethylquinolone. This new peptide PepFect15 exhibits increased transfection efficacy over PepFect14 and its transfection efficiency is not augmented by the presence of additional chloroquine.
Taken together with studies of PepFect 6, which also includes stearylation of TP10 combined with chemical conjugation to 3 chloroquine moieties, obvious limitations to peptide- mediated transfection begin to emerge. Stearylation is required for improved particle stability and increased transfection efficiency, while incorporation of protonatable structures improves endosomal escape presumably via the proton sponge effect. These changes highlight the importance of endosomal escape strategies in developing peptides for siRNA delivery. Recent strategies to further increase nucleotide delivery capacity include UV light to disrupt endosomes via free radical production by incorporating hydrophobic dyes Alexa Fluor 633 or Texas Red into peptide/siRNA complexes followed by excitation with 545-580nm light in vitro (Räägel et al. 2013).
In vivo data are currently limited. However, early results reveal in vivo efficacy and limited toxicity. Langel et al. have published in vivo studies involving hydroinjection of PepFect6/siRNA particles to yield effective transfection of hepatocytes (Andaloussi et al. 2011). However, further analysis reveals that Pepfect6-based particles demonstrate an increase in size and a mild decrease in transfection in the presence of serum proteins highlighting the need for further peptide development and additional in vivo analyses (Andaloussi et al. 2011). Subsequent toxicity analyses have revealed minimal in vivo toxicity at peptide concentrations of up to 5mg/kg as witnessed by a lack of increase in IL-1B and TNFa serum levels 24 and 48 hours after a single intravenous injection (Suhorutsenko et al. 2011). In addition, work by Ren et al. has also revealed the ability to impart cell-specific uptake in cancer cells via incorporation of LyP1 ligands suggesting the promise for effective in vivo siRNA transfection (Ren et al. 2012). However, further studies to better elucidate serum stability, pharmacokinetics, biodistribution and immune response with repeated dosing are required before the ultimate utility of these agents can be established.
3 Melittin as a Basis for Endosomal Escape
It is apparent that peptide-mediated transfection is hampered by poor efficiency as a consequence of endosomal entrapment. Methods to overcome this limitation have focused on peptide modification to include membrane active peptides, lipids as well as chemical conjugation to proton buffering moieties. Other strategies focus on leveraging the membrane lytic properties of melittin to enable endosomal release of siRNA. Melittin is a 26 amino acid alpha helical peptide first purified from the European honeybee in 1958, which has a high affinity for lipid membranes and ultimately causes membrane lysis (Sessa et al. 1969). Although the mechanism of membrane disruption has not fully been clarified, melittin has shown the ability to lyse red blood cells, as well as model membranes (Pratt et al. 2005, Bogaart et al. 2008).
Currently, melittin is hypothesized to disrupt membrane bilayers via a two-step “detergent-like” mechanism (Ladokhin et al. 2001, Shai 2002, Bechinger et al. 2006, Lee et al. 2008). The first step involves electrostatic interaction of melittin with negatively charged lipid headgroups. After the concentration of the peptide on the lipid surface reaches a critical concentration, melittin peptides rearrange to form pore like structures that disrupt the membrane bilayer (Figure 3). Data for the initial electrostatic interaction are derived from studies in which melittin is shown to bind with lower affinity to membrane bilayers with a lower component of anionic headgroups (Sessa et al. 1969).
Figure 3.
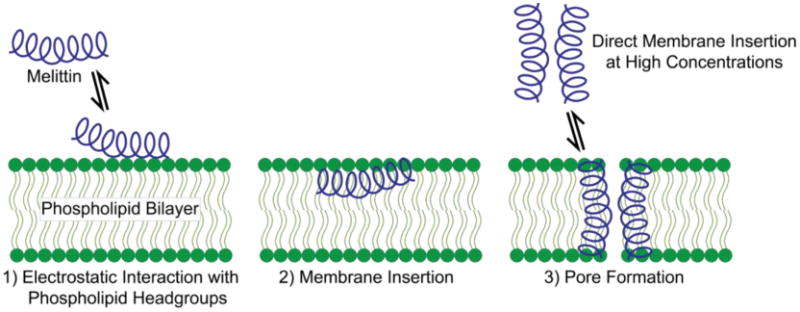
Melittin initially binds to membrane bilayers by electrostatics before inserting into the membrane and forming pores. At high melittin concentrations, there is evidence for direct membrane insertion and pore formation without the initial electrostatic interactions, which can explain continued membrane disruption even in acidic environments.
Although the efficiency with which melittin binds to and disrupts membranes depends on many variables including pH, salt concentration, and membrane composition, melittin's ability to disrupt membrane structures under myriad conditions is without question. However when considered in the context of endosomal disruption, it is critical that melittin also has the ability to disrupt membrane bilayers while exposed to an acidic environment. This could pose difficulties given melittin's initial electrostatic attraction to negatively charged lipid components. In fact, detailed biophysical studies have revealed that melittin's ability to bind to and disrupt model membranes actually decreases as pH decreases (Tan et al. 2012). Nonetheless, melittin has been shown to be capable of altering endosomal membrane integrity after cellular uptake under sub-toxic concentrations, suggesting that melittin does bind to and alter endosomal membranes despite the acidic environment (Rozema et al. 2003).
Given the known hemolytic properties of melittin, its use as a potential therapeutic may include significant in vivo toxicity. However, previous work has shown that appropriate sequestration of melittin in nanoparticles, or reversible blocking of melittin's hydrophobic residues can ameliorate any significant in vivo toxicity of naked melittin (Rozema et al. 2003, Meyer et al. 2007, Meyer et al. 2009, Soman et al. 2009). These studies suggest that if melittin can be appropriately modified or delivered to the target cell in an inactive form, its membrane disruptive properties can be harnessed for endosomal escape without either cellular or systemic toxicity given an appropriate mechanism for activation or deprotection (Figure 4).
Figure 4.
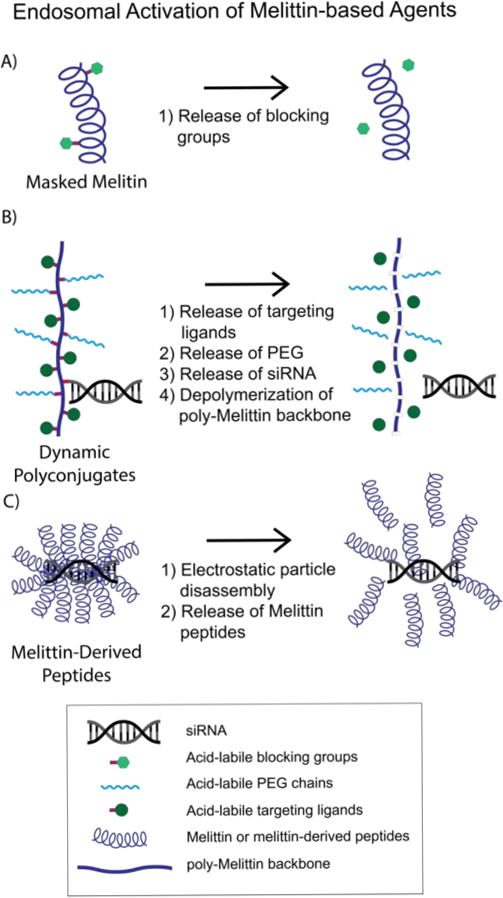
Melittin based siRNA delivery strategies must be activated in the endosome to avoid cytotoxicity. (a) Acid-activated melittin contain acid-labile blocking elements that are released in an acidic environment. (b) Dynamic polyconjugates are composed of acid-labile targeting ligands, acid-labile PEG chains, reducibly bound siRNA, and poly-melittin backbones which disassemble in the acidic, reducing environment of the endosome. (c) Melittin-derived peptides for direct siRNA binding disassemble based on electrostatic repulsion secondary to protonation in the endosome.
3.1 Melittin for the Augmentation of Oligonucleotide Delivery
Given its lytic nature, melittin itself has been recruited for the treatment of cancer, bacterial infections, and recently for augmentation of oligonucleotide delivery either indirectly as an excipient for endosomal escape of directly as an oligonucleotide transfection agent (Raghuraman et al. 2007, Oršolić 2012). Work by Wooddell et al. at Arrowhead Pharmaceuticals has provided critical evidence for the role of melittin in release of therapeutics from the endosomal compartment (Wooddell et al. 2013). In their work, melittin has been recruited for its membrane lytic properties to serve as an excipient for hepatocyte targeted siRNA therapy. In these studies, melittin or melittin like peptides are protected by acid labile groups which prevent membrane insertion until exposed to the acidic endosomal environment (Figure 4a) (Rozema et al. 2003). Activation of melittin in the hepatocyte endosome has proven to be robust and yielded a 500-fold increase in siRNA-mediated knockdown in vivo. These studies are particularly illuminating because they reveal that melittin can be delivered safely in the blood stream if protected from inserting into red blood cell membranes, and are able to avoid degradation if inaccessible to serum proteases. More importantly, these protected melittin peptides can improve endosomal escape providing definitive evidence that melittin is active within acidified endosomal compartments in an in vivo setting. This strategy is now in Phase IIa clinical trials for treatment of chronic Hepatitis B. Preliminary dose escalation data reveal HBsAg decrease of 39% and 51% at single doses of 1mg/kg and 2 mg/kg respectively without any side effects (Arrowhead 2014).
3.2 Dynamic Polyconjugates
The development of functionalized polymer vectors incorporating masked melittin derivatives for siRNA delivery was first demonstrated by Meyer et al. (2007). Their efforts yielded a functionalized polylysine backbone which could be covalently modified for acid labile PEG functionality along with masked melittin groups which could be activated in acidic compartments to impart improved endosomal escape capacity. This polylysine backbone could then be used to bind siRNA electrostatically. These constructs demonstrated a lack of cytotoxicity in vitro providing evidence for the safety of protected melittin structures. Unfortunately, these constructs cause substantial liver necrosis with abdominal bleeding in mice (Meyer et al. 2007, Meyer et al. 2008, Meyer et al. 2009). It is hypothesized that, the presence of a polycationic polymer backbone with a polyanionic oligonucleotide leads to aggregation and micron sized particles with a propensity for hepatic uptake and resultant liver toxicity.
Almost simultaneously Rozema et al. published initial studies regarding the utility of dynamic polyconjugates which follow a similar strategy (Rozema et al. 2007) (Figure 4b). One large distinction is the use of poly butyl and amino vinyl ethers to generate a masked endosomolytic polymer modeled after melittin along with PEG functionality with liver targeting via the apolipoprotein B receptor. These particles had a size of 10±2nm and single dose tail vein injections produced prolonged knockdown in hepatocytes with liver specific biodistribution. This technology was initially published with support from Mirus Bio Corporation which was later absorbed by Roche who published prolonged (>3 week) knockdown of genes in rhesus monkeys in 2009 (Babiss 2009). This technology was ultimately transferred to Arrowhead. Further development identified a masked melittin like peptide, which provided an appropriate backbone for dynamic polyconjugate assembly. This iteration of the technology has demonstrated >90% knockdown in hepatocytes at doses as low as 3mg/kg in non-human primates at a dose (Arrowhead 2012). Given the small size of DPCs, they are also being investigated for subcutaneous injections as well as antibody conjugation for additional targeting strategies. The utility of such technology is further bolstered by work initiated at Merck by Parmar et al. which focused on development of new polymer backbones via high throughput screening of a library of amphiphilic polymers (Parmar et al. 2013).
3.3 Direct siRNA Binding
Melittin's utility is not limited to acting solely as an endosomolytic agent, but can also directly serve as an oligonucleotide transfection agent. With its hydrophobic N-terminus and cationic C-terminus, melittin features a similar amphipathic sequence to that of siRNA transfecting peptides. Although melittin only contains five cationic charges in its native sequence, it has been utilized as a transfection agent to delivery plasmid DNA (Chen et al. 2006). In these studies, melittin was able to bind DNA, but only gave poor condensation due to an inadequate number of basic residues. Polymerization of melittin was found to improve DNA condensation and yielded moderate transfection. Of note, these studies relied on oxidation of free thiols with incorporation of both N and C terminal Cysteine residues allowing for depolymerization and release of free melittin peptides in the reducing environment of the endosome. This work provides another mechanism for endosomal activation of melittin to avoid the hemolytic toxicity associated with naked melittin peptides.
In separate work, melittin was modified to function as an siRNA transfection agent without requiring additional chemical modification or polymerization (Figure 4c). This work centered around the use of sequence modifications designed to decrease cytotoxicity and improve siRNA binding (Hou et al. 2013a, 2013b). The first modification involves N-terminal truncation of melittin, which is known to decrease melittin's cytotoxicity by over two orders of magnitude (Pan et al. 2010). The peptide is then augmented with additional basic residues for improved siRNA condensation. The lead candidate p5RHH was shown to bind siRNA to form peptide/siRNA particles with a diameter of <50nm with considerable temporal stability when incubated with albumin post formulation. These particles are capable of transfection in vitro in the presence of serum without any loss of efficiency and exhibit no noticeable endosomal entrapment as demonstrated by a lack of improved GFP knockdown with addition of chloroquine unlike previously published peptides. These data suggest that despite the reduced cytotoxicity of modified-melittin peptides compared to melittin itself, they retain a high capacity for endosomal disruption, likely due to the high effective endosomal concentration of peptides after macropinocytosis.
In depth analysis of the function of modified melittin demonstrate that p5RHH/siRNA particles disassemble under acidic conditions found in the endosome, likely due to protonation incorporated histidine residues, allowing liberated p5RHH to disrupt membrane bound compartments releasing siRNA to the cytoplasmic compartment (Figure 5 and 6). With the incorporation of only a two histidine residues, histidine protonation is unlikely able to generate the required osmotic forces required to disrupt endosomal integrity as histidine dependent strategies have traditionally required augmentation with 10 or more histidines (Lo et al. 2008). Instead, the authors suggest that histidine protonation drives dissociation of the peptide and siRNA allowing free peptide to disrupt the endosomal membrane (Hou et al. 2013b).
Figure 5.
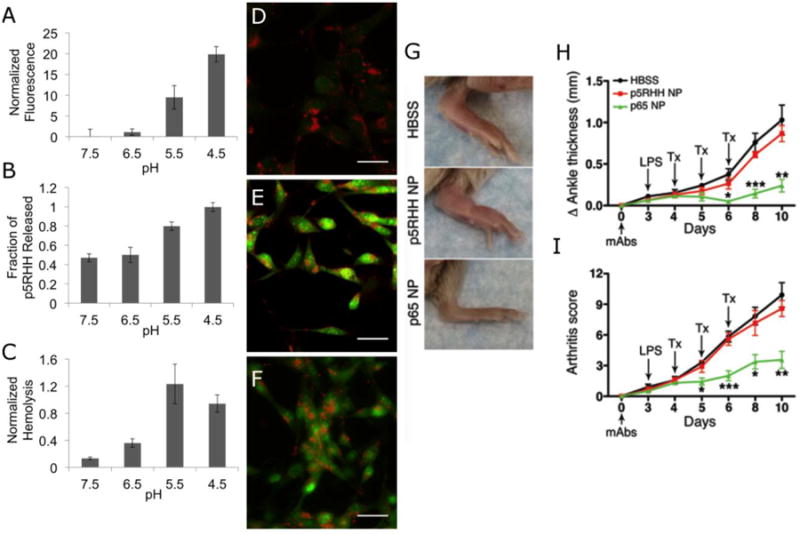
Melittin-derived peptides for siRNA delivery are pH responsive, with siRNA release (a), peptide release (b), and membrane disruption measured by hemolysis (c) all triggered by pH < 5. This pH responsiveness allows endosomal disruption in vivo as witnessed by acridine orange dye release assays. Control (d) cells only show dye accumulation in lysosomes where as chloroquine (e) and particle (f) treated cells demonstrate dye leakage as a result of endosomal disruption. p5RHH/siRNA nanoparticles are able to decrease ankle thickness (g, h) and arthritic score (i) in a murine model of rheumatoid arthritis when delivering NFkB siRNA.
Figure 6.
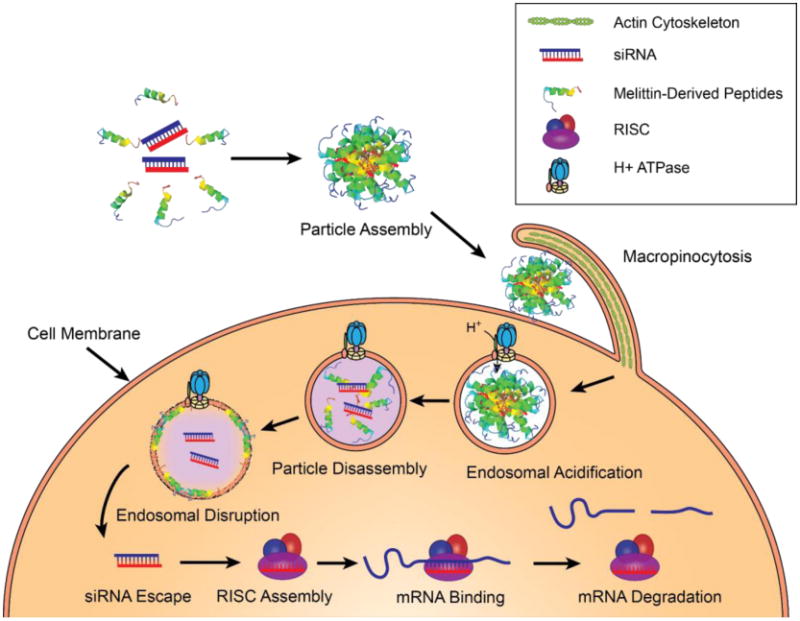
Melittin-derived peptides promote endosomal escape after particle disassembly triggered by endosomal acidification.
In additional work, p5RHH/siRNA nanoparticles were utilized to completely halt the progression of collagen antibody induced arthritis in mouse models of rheumatoid arthritis by targeting the p65 component of NFkB (Zhou et al. 2014) (Figure 5). These studies further demonstrated accumulation in arthritic paws and kidney clearance. Furthermore, the authors carefully demonstrated a lack of immunostimulatory affects of p5RHH/siRNA particles packing non-targeted siRNA. Of equal importance, this work also revealed a lack of observable decrease in the native immune function of p65 siRNA treated mice suggesting localized action in inflamed paws. A considerable amount of work remains before clinical trials can be considered including detailed pharmacokinetic and pharmacodynamics analysis in animal models. One area of interest that has yet to be clarified includes the increased renal clearance reported by these authors with a relative lack of reticuloendothelial clearance. However, the apparent in vivo efficacy make peptides derived from melittin a potential candidate for future therapeutic success in clinical settings.
5 Conclusions
Successful application of siRNA as a therapeutic requires the development of vectors that can promote cytoplasmic delivery of siRNA without inducing cytotoxicity, but providing adequate endosomal escape. Peptides are a relatively new strategy for siRNA transfection, which appear to be limited by endosomal entrapment. Traditional methods to overcome the barrier of endosomal entrapment rely on augmenting existing vectors with osmotic agents, lipid moieties, or fusogenic peptides. These strategies have provided modest improvements to peptide mediated transfection, and have provided simple guidelines to further improve peptide-mediated siRNA transfection.
With a focus on endosomal escape, melittin, a naturally occurring, membrane lytic peptide can provide an alternative to peptide transduction domains in the development of peptide-mediated transfection. Despite the hemolytic nature of naked melittin, existing data using protected or packaged melittin have demonstrated no systemic toxicity. Moreover, new strategies based on melittin have recently been shown safe and effective delivery of siRNA in in vivo settings. As a barrier to successful siRNA delivery, endosomal escape is undeniably a considerable hurdle that must be overcome to enable maximally efficient siRNA delivery. Modified peptide transduction domains and melittin-based technologies are uniquely designed to target this barrier and have enabled new nanoscale strategies to accomplish the formidable task of in vivo siRNA delivery. Ongoing development and clinical trials will ultimately determine the therapeutic relevance of these strategies.
Figure 1.
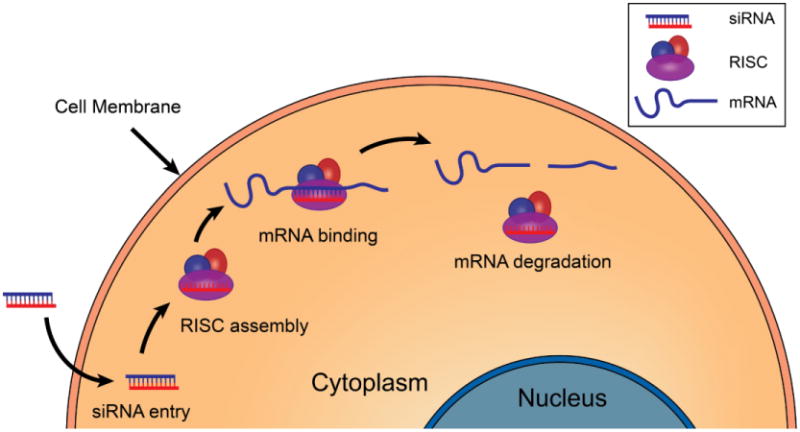
siRNA can induce mRNA degradation and gene silencing if delivered into the cytoplasm.
Acknowledgments
Research described here was supported by grants from the National Institutes of Health (U01 CA141541 to Dr. Robert Schreiber and RO1 HL073646-08 to SAW) and the Sigma Aldrich predoctoral fellowship to Dr. Kirk Hou.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Åmand HL, Fant K, Nordén B, Esbjörner EK. Stimulated endocytosis in penetratin uptake: Effect of arginine and lysine. Biochem Biophys Res Commun. 2008;371:621–625. doi: 10.1016/j.bbrc.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Andaloussi SEL, Lehto T, Mäger I, Rosenthal-Aizman K, Oprea II, Simonson OE, et al. Design of a peptide-based vector, pepfect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011;39:3972–3987. doi: 10.1093/nar/gkq1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowheadresearch.com [Internet] Arrowhead presents data showing 99% target knockdown in monkeys without toxicity using subcutaneous formulation of dynamic polyconjugate sirna delivery system. Pasadena: Arrowhead Pharmaceuticals; updated 2012 Oct 29; cited 2015 Mar 12. Available from: http://www.arrowheadresearch.com/press-releases/arrowhead-presents-data-showing-99-target-knockdown-monkeys-without-toxicity-using. [Google Scholar]
- Arrowheadresearch.com [Internet] Arrowhead presents data on ARC-520 and ARC-AAT at AASLD the liver meeting 2014. Pasadena: Arrowhead Pharmaceuticals; updated 2014 Nov 10; cited 2015 Mar 12. Available from: http://ir.arrowheadresearch.com/releasedetail.cfm?releaseid=881791. [Google Scholar]
- Arthanari Y, Pluen A, Rajendran R, Aojula H, Demonacos C. Delivery of therapeutic shRNA and siRNA by TAT fusion peptide targeting BCR–ABL fusion gene in chronic myeloid leukemia cells. J Control Release. 2010;145:272–280. doi: 10.1016/j.jconrel.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Arukuusk P, Pärnaste L, Oskolkov N, Copolovici DM, Margus H, Padari K, et al. New generation of efficient peptide-based vectors, nickfects, for the delivery of nucleic acids. Biochim Biophys Acta. 2013;1828:1365–1373. doi: 10.1016/j.bbamem.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Babiss L. Driving creativity & innovation in research [Internet] [updated 2009 Jan 20; cited 2015 Mar 12]. Available from: http://www.roche.com/bmk09slides-babiss_v2-e.pdf.
- Bechinger B, Lohner K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. BBA - Biomembranes. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Bogaart GVD, Guzmán JV, Mika JT, Poolman B. On the mechanism of pore formation by melittin. J Biol Chem. 2008;283:33854–33857. doi: 10.1074/jbc.M805171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Kim JS, Steenblock E, Liu D, Rice KG. Gene transfer with poly-melittin peptides. Bioconjug Chem. 2006;17:1057–1062. doi: 10.1021/bc060028l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a correlation between sirna localization, cellular uptake, and RNAi in living cells. Chem Biol. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Chou ST, Leng Q, Scaria P, Woodle M, Mixson AJ. Selective modification of HK peptides enhances siRNA silencing of tumor targets in vivo. Cancer Gene Ther. 2011;18:707–716. doi: 10.1038/cgt.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombez L, Aldrian-Herrada G, Konate K, Nguyen QN, McMaster GK, Brasseur R, et al. A new potent secondary amphipathic cell–penetrating peptide for sirna delivery into mammalian cells. Mol Ther. 2008;17:95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombez L, Divita G. A non-covalent peptide-based strategy for siRNA delivery. In: Langel Ü, editor. Cell-penetrating peptides. New York: Humana Press; 2011. pp. 349–360. [DOI] [PubMed] [Google Scholar]
- Crombez L, Morris MC, Dufort S, Aldrian-Herrada G, Nguyen Q, Mc Master G, et al. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009;37:4559–4569. doi: 10.1093/nar/gkp451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, et al. Highly efficient small interfering RNA delivery to primary mammalian neurons induces microRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshayes S, Gerbal-Chaloin S, Morris MC, Aldrian-Herrada G, Charnet P, Divita G, et al. On the mechanism of non-endosomal peptide-mediated cellular delivery of nucleic acids. BBA - Biomembranes. 2004;1667:141–147. doi: 10.1016/j.bbamem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Deshayes S, Konate K, Rydström A, Crombez L, Godefroy C, Milhiet PE, et al. Self-assembling peptide-based nanoparticles for siRNA delivery in primary cell lines. Small. 2012;8:2184–2188. doi: 10.1002/smll.201102413. [DOI] [PubMed] [Google Scholar]
- Deshayes S, Morris M, Heitz F, Divita G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv Drug Delivery Rev. 2008;60:537–547. doi: 10.1016/j.addr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Detzer A, Sczakiel G. Phosphorothioate-stimulated uptake of siRNA by mammalian cells: A novel route for delivery. Curr Top Med Chem. 2009;9:1109–1116. doi: 10.2174/156802609789630884. [DOI] [PubMed] [Google Scholar]
- Eccleston A, Eggleston AK. RNA interference. Nature. 2004;431:337–337. [Google Scholar]
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, et al. Efficient siRNA delivery into primary cells by a peptide transduction domain–dsRNA binding domain fusion protein. Nat Biotechnol. 2009;27:567–571. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. The AAPS Journal. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Endoh T, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv Drug Delivery Rev. 2009;61:704–709. doi: 10.1016/j.addr.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Endoh T, Sisido M, Ohtsuki T. Cellular siRNA delivery mediated by a cell-permeant RNA-binding protein and photoinduced RNA interference. Bioconjug Chem. 2008;19:1017–1024. doi: 10.1021/bc800020n. [DOI] [PubMed] [Google Scholar]
- Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals. 2012;5:1177–1209. doi: 10.3390/ph5111177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbacher P, Roche AC, Monsigny M, Midoux P. Putative role of chloroquine in gene transfer into a human hepatoma cell line by DNA/lactosylated polylysine complexes. Exp Cell Res. 1996;225:186–194. doi: 10.1006/excr.1996.0169. [DOI] [PubMed] [Google Scholar]
- Ezzat K, Andaloussi SEL, Zaghloul EM, Lehto T, Lindberg S, Moreno PMD, et al. Pepfect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011;39:5284–5298. doi: 10.1093/nar/gkr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat K, Helmfors H, Tudoran O, Juks C, Lindberg S, Padari K, et al. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012;26:1172–1180. doi: 10.1096/fj.11-191536. [DOI] [PubMed] [Google Scholar]
- Ezzat K, Zaghloul EM, EL Andaloussi S, Lehto T, El-Sayed R, Magdy T, et al. Solid formulation of cell-penetrating peptide nanocomplexes with siRNA and their stability in simulated gastric conditions. J Control Release. 2012;162:1–8. doi: 10.1016/j.jconrel.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO. Cellular uptake of the TATprotein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Futaki S, Ohashi W, Suzuki T, Niwa M, Tanaka S, Ueda K, et al. Stearylated arginine-rich peptides: A new class of transfection systems. Bioconjug Chem. 2001;12:1005–1011. doi: 10.1021/bc015508l. [DOI] [PubMed] [Google Scholar]
- Geoghegan JC, Gilmore BL, Davidson BL. Gene silencing mediated by siRNA-binding fusion proteins is attenuated by double-stranded RNA-binding domain structure. Mol Ther Nucleic Acids. 2012;1:e53. doi: 10.1038/mtna.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- Hassane FS, Abes R, El Andaloussi S, Lehto T, Sillard R, Langel U, et al. Insights into the cellular trafficking of splice redirecting oligonucleotides complexed with chemically modified cell-penetrating peptides. J Control Release. 2011;153:163–172. doi: 10.1016/j.jconrel.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Hassane FS, Saleh AF, Abes R, Gait MJ, Lebleu B. Cell penetrating peptides: Overview and applications to the delivery of oligonucleotides. Cell Mol Life Sci. 2010;67:715–726. doi: 10.1007/s00018-009-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussecker D. The business of rnai therapeutics in 2012. Mol Ther Nucleic Acids. 2012;1:e8. doi: 10.1038/mtna.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou KK, Pan H, Lanza GM, Wickline SA. Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials. 2013a;34:3110–3119. doi: 10.1016/j.biomaterials.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou KK, Pan H, Ratner L, Schlesinger PH, Wickline SA. Mechanisms of nanoparticle-mediated siRNA transfection by melittin-derived peptides. ACS nano. 2013b;7:8605–8615. doi: 10.1021/nn403311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Neundorf I. Knockdown of a G protein-coupled receptor through efficient peptide-mediated siRNA delivery. J Control Release. 2012;161:826–834. doi: 10.1016/j.jconrel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- Hoyer J, Neundorf I. Peptide vectors for the nonviral delivery of nucleic acids. Acc Chem Res. 2012;45:1048–1056. doi: 10.1021/ar2002304. [DOI] [PubMed] [Google Scholar]
- idtdna.com [Internet] IDT introduces novel peptide delivery system for dsRNA. Coralville: Integrated DNA Technologies; updated 2009 Jul 29; cited 2015 Mar 12. Available from: http://www.idtdna.com/pages/docs/press-releases/2009-transductin.pdf?sfvrsn=6. [Google Scholar]
- Ifediba MA, Medarova Z, Ng SW, Yang J, Moore A. siRNA delivery to CNS cells using a membrane translocation peptide. Bioconjug Chem. 2010;21:803–806. doi: 10.1021/bc900488e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee SH, Choe J, Park TG. Intracellular small interfering RNA delivery using genetically engineered double-stranded RNA binding protein domain. J Gene Med. 2009;11:804–812. doi: 10.1002/jgm.1365. [DOI] [PubMed] [Google Scholar]
- Kim SW, Kim NY, Choi YB, Park SH, Yang JM, Shin S. RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J Control Release. 2010;143:335–343. doi: 10.1016/j.jconrel.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Christensen LV, Jo S, Yockman JW, Jeong JH, Kim YH, et al. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther. 2006;14:343–350. doi: 10.1016/j.ymthe.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Hee Kim M, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- Ladokhin AS, White SH. ‘Detergent-like’ permeabilization of anionic lipid vesicles by melittin. BBA - Biomembranes. 2001;1514:253–260. doi: 10.1016/s0005-2736(01)00382-0. [DOI] [PubMed] [Google Scholar]
- Langlet-Bertin B, Leborgne C, Scherman D, Bechinger B, Mason AJ, Kichler A. Design and evaluation of histidine-rich amphipathic peptides for siRNA delivery. Pharm Res. 2010;27:1426–1436. doi: 10.1007/s11095-010-0138-2. [DOI] [PubMed] [Google Scholar]
- Lee MT, Hung WC, Chen FY, Huang HW. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. PNAS. 2008;105:5087–5092. doi: 10.1073/pnas.0710625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehto T, Abes R, Oskolkov N, Suhorutšenko J, Copolovici DM, Mäger I, et al. Delivery of nucleic acids with a stearylated (RXR)4 peptide using a non-covalent co-incubation strategy. J Control Release. 2010;141:42–51. doi: 10.1016/j.jconrel.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Lehto T, Simonson OE, Mäger I, Ezzat K, Sork H, Copolovici DM, et al. A peptide-based vector for efficient gene transfer in vitro and in vivo. Mol Ther. 2011;19:1457–1467. doi: 10.1038/mt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg S, Muñoz-Alarcón A, Helmfors H, Mosqueira D, Gyllborg D, Tudoran O, et al. Pepfect15, a novel endosomolytic cell-penetrating peptide for oligonucleotide delivery via scavenger receptors. Int J Pharm. 2013;441:242–247. doi: 10.1016/j.ijpharm.2012.11.037. [DOI] [PubMed] [Google Scholar]
- Lo SL, Wang S. An endosomolytic tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials. 2008;29:2408–2414. doi: 10.1016/j.biomaterials.2008.01.031. [DOI] [PubMed] [Google Scholar]
- Lundberg P, El-Andaloussi S, Sütlü T, Johansson H, Langel Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. The FASEB Journal. 2007;21:2664–2671. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- Lundin P, Johansson H, Guterstam P, Holm T, Hansen M, Langel Ü, et al. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug Chem. 2008;19:2535–2542. doi: 10.1021/bc800212j. [DOI] [PubMed] [Google Scholar]
- Mäe M, Andaloussi SE, Lehto T, Langel Ü. Chemically modified cell-penetrating peptides for the delivery of nucleic acids. Expert Opin Drug Deliv. 2009;6:1195–1205. doi: 10.1517/17425240903213688. [DOI] [PubMed] [Google Scholar]
- Mäe M, EL Andaloussi S, Lundin P, Oskolkov N, Johansson HJ, Guterstam P, et al. A stearylated cpp for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J Control Release. 2009;134:221–227. doi: 10.1016/j.jconrel.2008.11.025. [DOI] [PubMed] [Google Scholar]
- McCaffrey AP, Meuse L, Pham TTT, Conklin DS, Hannon GJ, Kay MA. Gene expression: RNA interference in adult mice. Nature. 2002;418:38–39. doi: 10.1038/418038a. [DOI] [PubMed] [Google Scholar]
- Meade BR, Dowdy SF. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv Drug Delivery Rev. 2007;59:134–140. doi: 10.1016/j.addr.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Meade BR, Dowdy SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv Drug Delivery Rev. 2008;60:530–536. doi: 10.1016/j.addr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Dohmen C, Philipp A, Kiener D, Maiwald G, Scheu C, et al. Synthesis and biological evaluation of a bioresponsive and endosomolytic siRNA–polymer conjugate. Mol Pharm. 2009;6:752–762. doi: 10.1021/mp9000124. [DOI] [PubMed] [Google Scholar]
- Meyer M, Philipp A, Oskuee R, Schmidt C, Wagner E. Breathing life into polycations: Functionalization with pH-responsive endosomolytic peptides and polyethylene glycol enables siRNA delivery. J Am Chem Soc. 2008;130:3272–3273. doi: 10.1021/ja710344v. [DOI] [PubMed] [Google Scholar]
- Meyer M, Zintchenko A, Ogris M, Wagner E. A dimethylmaleic acid–melittin-polylysine conjugate with reduced toxicity, pH-triggered endosomolytic activity and enhanced gene transfer potential. J Gene Med. 2007;9:797–805. doi: 10.1002/jgm.1075. [DOI] [PubMed] [Google Scholar]
- Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, et al. Lung delivery studies using siRNA conjugated to TAT(48–60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug Chem. 2007;18:1450–1459. doi: 10.1021/bc070077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratovska A, Eccles MR. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004;558:63–68. doi: 10.1016/S0014-5793(03)01505-9. [DOI] [PubMed] [Google Scholar]
- Nakase I, Akita H, Kogure K, Gräslund A, Langel Ü, Harashima H, et al. Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc Chem Res. 2012;45:1132–1139. doi: 10.1021/ar200256e. [DOI] [PubMed] [Google Scholar]
- Neundorf I, Rennert R, Hoyer J, Schramm F, Löbner K, Kitanovic I, et al. Fusion of a short HA2-derived peptide sequence to cell-penetrating peptides improves cytosolic uptake, but enhances cytotoxic activity. Pharmaceuticals. 2009;2:49–65. doi: 10.3390/ph2020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma S, Poole B. Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J Cell Biol. 1981;90:656–664. doi: 10.1083/jcb.90.3.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Kawaguchi Y, Okamoto H. Enhanced gene delivery and/or efficacy by functional peptide and protein. Curr Top Med Chem. 2009;9:1098–1108. doi: 10.2174/156802609789630857. [DOI] [PubMed] [Google Scholar]
- Oršolić N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012;31:173–194. doi: 10.1007/s10555-011-9339-3. [DOI] [PubMed] [Google Scholar]
- Overhoff M, Sczakiel G. Phosphorothioate-stimulated uptake of short interfering RNA by human cells. EMBO Rep. 2005;6:1176–1181. doi: 10.1038/sj.embor.7400535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Myerson JW, Ivashyna O, Soman NR, Marsh JN, Hood JL, et al. Lipid membrane editing with peptide cargo linkers in cells and synthetic nanostructures. FASEB J. 2010;24:2928–2937. doi: 10.1096/fj.09-153130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar RG, Busuek M, Walsh ES, Leander KR, Howell BJ, Sepp-Lorenzino L, et al. Endosomolytic bioreducible poly(amido amine disulfide) polymer conjugates for the in vivo systemic delivery of siRNA therapeutics. Bioconjug Chem. 2013;24:640–647. doi: 10.1021/bc300600a. [DOI] [PubMed] [Google Scholar]
- Pratt JP, Ravnic DJ, Huss HT, Jiang X, Orozco BS, Mentzer SJ. Melittin-induced membrane permeability: A nonosmotic mechanism of cell death. In Vitro Cell Dev-An. 2005;41:349–355. doi: 10.1007/s11626-005-0007-1. [DOI] [PubMed] [Google Scholar]
- Qi W, Ding D, Zhu H, Lu D, Wang Y, Ding J, et al. Efficient siRNA transfection to the inner ear through the intact round window by a novel proteidic delivery technology in the chinchilla. Gene Ther. 2014;21:10–18. doi: 10.1038/gt.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räägel H, Hein M, Kriiska A, Säälik P, Florén A, Langel Ü, et al. Cell-penetrating peptide secures an efficient endosomal escape of an intact cargo upon a brief photo-induction. Cell Mol Life Sci. 2013;70:4825–4839. doi: 10.1007/s00018-013-1416-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman H, Chattopadhyay A. Melittin: A membrane-active peptide with diverse functions. Biosci Rep. 2007;27:189–223. doi: 10.1007/s10540-006-9030-z. [DOI] [PubMed] [Google Scholar]
- Regberg J, Srimanee A, Erlandsson M, Sillard R, Dobchev DA, Karelson M, et al. Rational design of a series of novel amphipathic cell-penetrating peptides. Int J Pharm. 2014;464:111–116. doi: 10.1016/j.ijpharm.2014.01.018. [DOI] [PubMed] [Google Scholar]
- Ren Y, Cheung HW, Langel Ü, Maltzhan Gv, Agrawal A, Cowley GS, Weir BA, et al. Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Sci Transl Med. 2012;4:147ra112. doi: 10.1126/scitranslmed.3003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozema DB, Ekena K, Lewis DL, Loomis AG, Wolff JA. Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjug Chem. 2003;14:51–57. doi: 10.1021/bc0255945. [DOI] [PubMed] [Google Scholar]
- Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL, et al. Dynamic polyconjugates for targeted in vivo delivery of siRNA to hepatocytes. PNAS. 2007;104:12982–12987. doi: 10.1073/pnas.0703778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydström A, Deshayes S, Konate K, Crombez L, Padari K, Boukhaddaoui H, et al. Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles. PLoS ONE. 2011;6:e25924. doi: 10.1371/journal.pone.0025924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, Amarzguioui M, Kim DH, Alluin J, Heale B, Song MS, et al. A role for human dicer in pre-RISC loading of siRNAs. Nucleic Acids Res. 2011;39:1510–1525. doi: 10.1093/nar/gkq846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa G, Freer JH, Colacicco G, Weissmann G. Interaction of a lytic polypeptide, melittin, with lipid membrane systems. J Biol Chem. 1969;244:3575–3582. [PubMed] [Google Scholar]
- Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Nielsen PE. Enhanced delivery of cell-penetrating peptide–peptide nucleic acid conjugates by endosomal disruption. Nat Protoc. 2006;1:633–636. doi: 10.1038/nprot.2006.92. [DOI] [PubMed] [Google Scholar]
- sigmaaldrich.com [Internet] N-TER nanoparticle siRNA transfection system siRNA delivery to hard-to-transfect cell types. St. Louis: Sigma Aldrich; cited 2015 Mar 12. Available from: http://www.sigmaaldrich.com/life-science/functional-genomics-and-rnai/sirna/nter.html. [Google Scholar]
- Simeoni F, Morris MC, Heitz F, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: Implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, et al. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest. 2009;119:2830–2842. doi: 10.1172/JCI38842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhorutsenko J, Oskolkov N, Arukuusk P, Kurrikoff K, Eriste E, Copolovici DM, et al. Cell-penetrating peptides, pepfects, show no evidence of toxicity and immunogenicity in vitro and in vivo. Bioconjug Chem. 2011;22:2255–2262. doi: 10.1021/bc200293d. [DOI] [PubMed] [Google Scholar]
- Tan YX, Chen C, Wang YL, Lin S, Wang Y, Li SB, et al. Truncated peptides from melittin and its analog with high lytic activity at endosomal pH enhance branched polyethylenimine-mediated gene transfection. The Journal of Gene Medicine. 2012;14:241–250. doi: 10.1002/jgm.2609. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Kanazawa T, Ogawa T, Takashima Y, Fukuda T, Okada H. Disulfide crosslinked stearoyl carrier peptides containing arginine and histidine enhance siRNA uptake and gene silencing. Int J Pharm. 2010;398:219–224. doi: 10.1016/j.ijpharm.2010.07.038. [DOI] [PubMed] [Google Scholar]
- Trabulo S, Cardoso AL, Mano M, de Lima MCP. Cell-penetrating peptides - mechanisms of cellular uptake and generation of delivery systems. Pharmaceuticals. 2010;3:961–993. doi: 10.3390/ph3040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JJ, Arzumanov AA, Gait MJ. Synthesis, cellular uptake and HIV-1 TAT-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucleic Acids Res. 2005;33:27–42. doi: 10.1093/nar/gki142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA, Gait MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol Dis. 2007;38:1–7. doi: 10.1016/j.bcmd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Turner JJ, Williams D, Owen D, Gait MJ. Disulfide conjugation of peptides to oligonucleotides and their analogs. In: Beaucage SL, Bergstrom DE, Herdewijin P, Matsuda A, editors. Current protocols in nucleic acid chemistry. Hoboken: John Wiley & Sons, Inc; 2006. unit 4.28. [DOI] [PubMed] [Google Scholar]
- Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Veldhoen S, Laufer SD, Restle T. Recent developments in peptide-based nucleic acid delivery. Int J Mol Sci. 2008;9:1276–1320. doi: 10.3390/ijms9071276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen S, Laufer SD, Trampe A, Restle T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: Quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006;34:6561–6573. doi: 10.1093/nar/gkl941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 2010;62:90–99. doi: 10.1016/j.phrs.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Wibo M, Poole B. Protein degradation in cultured cells II. The uptake of chloroquine by rat fibroblasts and the inhibition of cellular protein degradation and cathepsin B1. J Cell Biol. 1974;63:430–440. doi: 10.1083/jcb.63.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Pirollo KF, Yu B, Rait A, Xiang L, Huang W, et al. Enhanced transfection efficiency of a systemically delivered tumor- targeting immunolipoplex by inclusion of a pH- sensitive histidylated oligolysine peptide. Nucleic Acids Res. 2004;32:e48–e48. doi: 10.1093/nar/gnh049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Hf, Yan H, Pan H, Hou KK, Akk A, Springer LE, et al. Peptide-siRNA nanocomplexes targeting NF-κB subunit p65 suppress nascent experimental arthritis. J Clin Invest. 2014;124:4363–4374. doi: 10.1172/JCI75673. [DOI] [PMC free article] [PubMed] [Google Scholar]