

Abstract
Intraperitoneal (IP) chemotherapy confers significant survival benefits in cancer patients. However, several problems, including local toxicity and ineffectiveness against bulky tumors, have prohibited it from becoming a standard-of-care. We have developed drug-loaded, tumor-penetrating microparticles (TPM) to address these problems. TPM comprises two components and uses the versatile PLGA or poly(lacticco-glycolic acid) copolymer to provide tumor-selective adherence and pharmacodynamically optimized fractionated dosing to achieve the desired tumor priming (which promotes particle penetration into tumors) plus immediate and sustained antitumor activity. Preclinical studies show that TPM is less toxic and more effective against several IP metastatic tumors with different characteristics (fast vs. slow growing, porous vs. densely packed structures, wide-spread vs. solitary tumors, early vs. late stage, with or without peritoneal carcinomatosis or ascites), compared to the intravenous paclitaxel/Cremophor micellar solution that has been used off-label in previous IP studies. TPM further requires less frequent dosing. These encouraging preclinical results have motivated the follow-up clinical development of TPM. We are working with National Institutes of Health on the IND-enabling studies.
KEY WORDS: intraperitoneal therapy, paclitaxel, peritoneal cancer, tumor priming, tumor-penetrating microparticles
INTRODUCTION
Our laboratory has a long-standing interest in developing regional cancer therapy, including intravesical therapy for bladder cancer (e.g., 1,2) and intraperitoneal (IP) cancers. The current report summarizes our findings in IP therapy; detailed information can be found in our earlier research publications and reviews (3–9).
This report comprises four parts. Part I provides general information on peritoneal cancer and the current status of IP therapy. Part II outlines the development of drug-loaded tumor-penetrating microparticles (TPM). TPM makes use of the versatility offered by polymeric carriers to achieve pharmacodynamically optimized drug delivery. Part III outlines the processes critical for transitioning academic discoveries to clinical development, i.e., IND-enabling studies. Part IV discusses the potential difficulties in the animal-to-human translation of TPM.
Part I. Peritoneal Cancer and Intraperitoneal Cancer Therapy
Cancers originating from organs in the peritoneal cavity (ovarian, pancreatic, colorectal, gastric, liver, peritoneal mesothelioma) account for about 250,000 new cases annually in the USA (10). Peritoneal metastases are common due to locoregional spread (e.g., incidence of 90, 50 and 32% in ovarian, pancreatic, and colon cancer, respectively). Peritoneal metastasis can also derive from extra-peritoneal cancers, with incidence as high as 70% in patients with advanced disease (10).
In the peritoneal cavity, movement of cells tends to follow the circulation of peritoneal fluid from the right pericolic gutter cephalad to the right hemidiaphragm. Lodging of tumor cells in diaphragmatic or abdominal lymphatic ducts causes obstruction of lymphatic drainage and decreased outflow of peritoneal fluid, leading to formation of peritoneal carcinomatosis or ascites (11–14).
In general, presence of peritoneal metastasis and carcinomatosis is an indicator of poor prognosis. Patients with carcinomatosis suffer from abdominal distention, loss of appetite, shortness of breath, abdominal pain, low blood pressure, weakness, fatigue, and intestinal obstruction due to adhesions formed between intestinal loops. The current treatment objectives for these patients are primarily palliative (e.g., pain control, repeated drainage of peritoneal fluid), as there are no meaningful therapeutic options.
Management of patients with peritoneal cancer includes using IP therapy to deliver high drug concentrations locally to tumors located in the peritoneal cavity (15). IP therapy has been under development for several decades and has shown survival benefits in selected patient groups (5,16–20). Most studies, conducted in patients with ovarian or gastrointestinal cancer, used IP therapy preoperatively to downstage the disease to facilitate surgical debulking and intraoperatively and postoperatively to treat residual small and microscopic tumors (21–36). Hyperthermia intraoperative IP chemotherapy, where the drug solution (e.g., cisplatin, mitomycin C) is heated to 41–43°C to enhance drug uptake and antitumor efficacy, has also been used. Intraoperative and early postoperative IP therapies are used with cytoreductive surgery to treat peritoneal metastases of gastric, colorectal, and appendicle cancer (37–39), whereas postoperative therapy, typically given several weeks later or after patients have recovered from surgical complications, is used in ovarian cancer.
Multiple studies have demonstrated significant pharmacokinetic advantage for IP chemotherapy in patients (summarized in 5). The ratio of drug exposure (CxT) in peritoneal fluid and systemic blood ranges from about 10 for platinated compounds, 70 for some alkylators (e.g., melphalan, etoposide, mitomycin), 400 for 5-fluoruoracil and doxorubicin, and 1000 for mitoxantrone and paclitaxel (dissolved in Cremophor/ethanol). In comparison, the tumor-to-plasma concentration ratios are much lower and the pharmacokinetic advantage is spatial dependent; e.g., IP cisplatin yielded two to three times higher concentrations in the periphery of mouse tumors with no improvement in tumor center, compared to intravenous cisplatin (40,41). Nonetheless, IP cisplatin produced histologically proven complete remission in 30% of patients who failed intravenous cisplatin (42).
The survival advantage of IP therapy has been confirmed in multiple clinical trials. For the treatment of peritoneal carcinomatosis of colorectal cancer origin, a combination of aggressive cytoreductive surgery with hyperthermia intraoperative IP chemotherapy shows substantial survival benefits compared to the standard treatment of systemic chemotherapy of 5-fluorouracil plus leucovorin with or without palliative surgery (22.3 vs. 12.6 months), although the benefit was lessened in patients with extensive residual disease (>2.5 mm diameter) (17). The hyperthermia chemotherapy is being practiced in the clinic, albeit mostly in academic centers. In ovarian cancer, adding IP chemotherapy to intravenous chemotherapy produces significantly longer progression-free and overall survival (18–20); the NCI Cooperative Group trial (GOG 172) in stage III patients with <1 cm tumors showed a 16-month longer overall survival. However, toxicities and other issues, discussed below, have led to controversy in the clinical community on using postoperative IP therapy as a standard of care for ovarian cancer (43,44).
The toxicities of IP therapy are generally related to administration procedures and/or are drug-related. The use of IP catheter is associated with higher risk of infection and fever, and occasionally physical damages to peritoneal tissues (e.g., perforation). While hematologic toxicity is a major toxicity for drugs rapidly absorbed into the systemic circulation (e.g., cisplatin, carboplatin, melphalan, etoposide), local toxicity is dose-limiting for drugs that are slowly absorbed (e.g., paclitaxel, mitoxantrone, doxorubicin) or drugs that induce chemical peritonitis (e.g., mitomycin C, 5-fluorouracil, oxaliplatin) or ileus (e.g., docetaxel) (summarized in 5). The GOG 172 trial showed that three times more patients on the IP + intravenous arm did not complete the assigned six-treatment cycle compared to the intravenous arm, in part due to catheter-related complications, other toxicities (abdominal pain or stomach cramp, dehydration, renal/metabolic, catheter-unrelated infection), and patient refusal. The IP arm showed worse quality of life shortly after treatment, in part due to receiving higher total drug dose from both intravenous plus IP therapy, but the difference diminished over time (e.g., after 1 year) (20,45).
Another major limitation of IP therapy is the lack of efficacy in larger, bulky tumors. In ovarian cancer, the post-surgical residual tumor size is the most significant prognostic indicator for IP therapy (e.g., platinum compounds, mitoxantrone, cytarabine, bleomycin, etoposide, paclitaxel), with a better prognosis and longer survival interval in patients with smaller tumors (≤0.5 cm) compared to larger tumors (≥2 cm) (16,46–50). These findings have led to the recommendation of using IP therapy in optimally surgically debulked, stage III patients with tumors of <1 cm (20,43). Several studies have shown that the tumor size restriction is likely due to the inability of a drug to penetrate and/or accumulate in the tumor mass. This notion is supported by the observations that while cisplatin and carboplatin were about equally effective in ovarian cancer patients presenting with only positive margins (<0.5 cm), the analog that shows inferior penetration and seven times lower drug levels in rodent tumors (i.e., carboplatin) also shows inferior activity in patients with larger tumors (1–3 cm) (50). Hence, improving tumor penetration will likely improve the efficacy of IP therapy.
In 2013, the European Commission approved a monoclonal antibody, catumaxomab, for treating patients with IP carcinomatosis. Catumaxomab, via its two binding arms specific for EpCAM (epithelial cell adhesion molecule) and CD3 (T lymphocytes) and its Fc region, binds simultaneously to tumor cells, T cells, and antigen-presenting cells and causes cell death (51). Catumaxomab extends the puncture-free survival (to 46 days from 11 days for the control group treated with only paracentesis) (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000972/WC500051808.pdf). Puncture-free survival is the duration over which a patient does not require paracentesis.
To date, there are no products approved by the US Food and Drug Administration (FDA) for IP therapy. The current practice is off-label use of drugs approved for intravenous administration. These are typically drug solutions and do not have the optimal properties for IP therapy. For example, we showed in rodents that >99% of an IP dose of paclitaxel dissolved in Cremophor micelles was cleared from the peritoneal cavity in less than 12 h, due to drainage through the lymphatic openings and absorption through the thin peritoneum membrane (7). Such rapid clearance of drug solution from the peritoneal cavity limits the tumor targeting advantage and creates the need for repeated administration (and use of indwelling catheters). Further, the bolus presentation of the entire dose within the peritoneal cavity introduces high local drug concentrations and local toxicity. We conclude there is a need of specialized delivery systems to optimize IP therapy and have since developed TPM tailored to the unique anatomical properties in the peritoneal cavity.
Part II. Science Behind Drug-Loaded Tumor-Penetrating Microparticles
Design of Drug-Loaded Tumor-Penetrating Microparticles: Summary
Through a series of pharmacokinetic and pharmacodynamics studies, we identified several limitations associated with the off-label use of intravenous drug solution for IP therapy (52–58). These findings were used to design TPM (Table I). TPM comprises two types of polymeric carriers, one to provide rapid release (e.g., 70% in 1 day under sink conditions) and one to provide sustained release (1% in 1 day). As discussed below, this fractionated dose presentation feature provides the pharmacodynamically optimized drug delivery to achieve several desired pharmacological properties and treatment outcomes.
Table I.
Design and Features of Tumor-Penetrating Microparticles (TPM) for IP Therapy
| Limitations of intravenous formulations | Solutions/improvements offered by TPM |
|---|---|
| 1. Not optimized for IP therapy. Intravenous formulations for systemic administration, usually solutions or suspensions of nano-sized particulates (e.g., paclitaxel in Cremophor micelles, ~13 nm), are rapidly cleared from peritoneal cavity via absorption and lymphatic drainage. This in turn results in the need of frequent treatments and indwelling catheters. 2. No tumor selectivity. Normal and tumor tissues are similarly bathed in drug solution. 3. Local toxicity due to exposure of normal tissues to high drug concentrations (e.g., abdominal pain), and use of indwelling catheter. 4. Limited penetration from peritoneal fluid into tumors. Treatment is ineffective in bulky tumors (>1 cm). 5. Not optimized to control slowly growing tumors. Due to short residence time, slowly growing tumors with low fraction of cycling cells may escape treatment. |
1. Designed to optimize pharmacokinetics and pharmacodynamics of IP therapy. The size is optimized (4–6 μm) to retard clearance from peritoneal cavity and to promote wide intracavity distribution. TPM has two components, one to release paclitaxel rapidly and the other slowly, in order to achieve optimized target site pharmacokinetics and pharmacodynamics. 2. Tumor selectivity due to selective adherence to tumor surfaces, and greater susceptibility of tumor cells to tumor priming. 3. Lower local toxicity. Fractionated drug release reduces the host tissue exposure. Sustained release eliminates the need of frequent treatments. 4. Enhanced penetration into inner parts of tumors. TPM offers tumor priming to promote penetration into large tumors. 5. Tumor growth control. The combination of rapid and slow drug presentation offers control of tumors with different growth rates. |
Pharmacokinetics of IP Therapy
Drug disposition during and after IP treatments is controlled by kinetic processes connecting several distinct anatomical and physiological compartments (Fig. 1a). Drug is removed from the peritoneal cavity by several means. Absorption (by diffusion or convection) through the peritoneum, a thin membrane (75 and 90 μm thick in rats and man, respectively), is a major path for small compounds (59,60). The rate-limiting parameter is transfer across the vessel membrane for hydrophilic drugs and blood flow for lipophilic drugs. Human peritoneum is highly permeable to molecules with molecular weight of less than 20 kD (60). Larger compounds or particulates are drained through the lymphatic ducts (61,62). For lymphatic transport, the two most important determinants are lipophilicity and molecular/particle size; compounds with molecular weight >500 and high lipophilicity (log Poctanol:water >4) and small particulates (e.g., liposomes, micelles) are absorbed (63). Within the lymphatic system, smaller particulates (<50 nm diameter) can pass through lymph nodes while larger particulates (>500 nm) are mostly trapped in lymph nodes (61). The main lymphatic drainage from the peritoneal cavity is through the stomatas on the subdiaphragmic surface, which connects to the lymphatic vessels located in deep diaphragmic tissues (64,65). In peritoneal tissues including tumors, a drug can enter via diffusion and convection and, after entering the interstitium, gain entry to blood vessels. The drug absorbed into the systemic circulation is then cleared from the body or recirculates to the tumor. In short, the drug delivery and residence in peritoneal tumors are affected by pharmacokinetics in three compartments (intratumor, intraperitoneal, systemic) where the CxT in each compartment is determined by different kinetic processes. In addition, the well-known size-dependent clinical efficacy (i.e., no activity in bulky tumors) indicates the need to elucidate the role of intratumoral transport on the spatial-dependent pharmacodynamics.
Fig. 1.
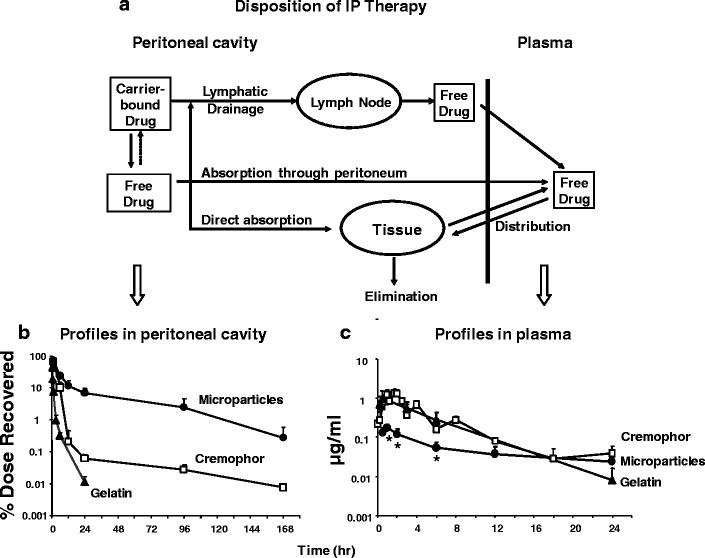
Pharmacokinetic model of disposition of IP therapy and effects of carrier. a A model of kinetic processes for IP therapy. b, c Tumor-free mice were given IP injections of paclitaxel solubilized in Cremophor EL/ethanol (white square), paclitaxel-loaded gelatin nanoparticles (black triangle), and paclitaxel-loaded polymeric microparticles (black circle), all at 10 mg/kg. Paclitaxel concentration-time profiles in peritoneal lavage samples (b) and plasma samples (c). Note the different time scales for panels b and c. Reprinted from (7) with permission
Spatial Drug Distribution after Intravenous and IP Administration
We compared the spatial distribution of 3H-paclitaxel in mice after IP or intravenous injection of the Cremophor micellar solution, using whole body autoradiography (7). Figure 2 shows the results. While IP therapy, as expected, yielded greater drug localization in the peritoneal cavity, most of the IP dose disappeared after 8 h, which is short relative to the typical tumor doubling time. In addition, analysis of the kinetics in liver and intestines suggests direct absorption of the IP dose into intestines, which explains the greater gastrointestinal toxicity of IP therapy. These findings led to the conclusions that fractionated dose presentation may reduce the intestinal toxicity and prolonging drug retention in the cavity may improve the efficacy of IP therapy.
Fig. 2.
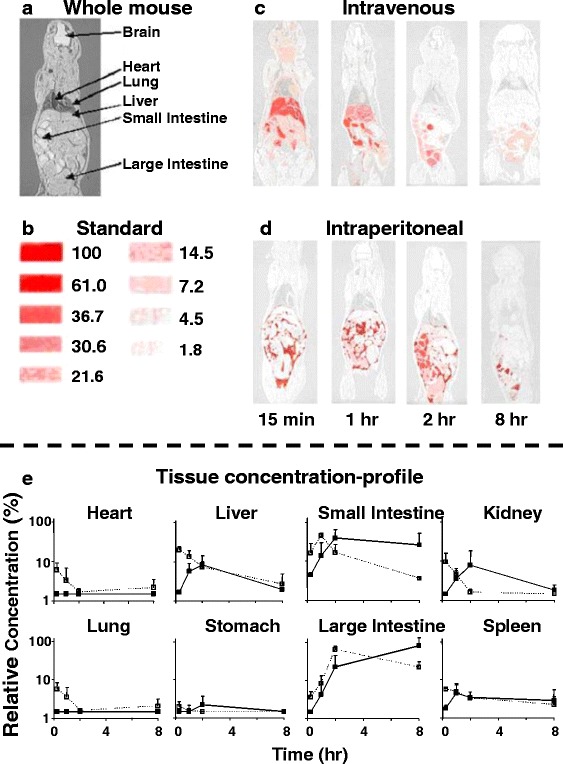
Spatial and tissue distribution of intravenous and IP injections of 3H-paclitaxel solubilized in Cremophor micelles. A mouse was given an IP or intravenous injection of the Cremophor formulation of paclitaxel (a mixture of radiolabeled and nonlabeled paclitaxel, equivalent to 10 mg/kg and 1 mCi/kg). a Whole body section of a mouse. b Densitometric signals of microscale tritium standards. The numbers correspond to the relative concentrations, with the highest level set at 100%. c Whole body autoradiographs at various time points after an intravenous dose. d Whole body autoradiographs after an IP dose. e Relative tissue concentration-time profiles, determined by digital videodensitometry, after an intravenous dose (open symbols, dotted lines) or an IP dose (closed symbols, solid lines). Reprinted from (7) with permission
Effects of Carriers on IP Pharmacokinetics
Multiple studies have shown that drug clearance from the peritoneal cavity and toxicity/activity of IP therapy are affected by drug carriers. For example, IP injection of sustained release cisplatin-loaded polymeric microparticles (50–200 μm) resulted in significantly higher drug concentrations in omental tumors and longer survival of tumor-bearing animals, as well as provided locoregional control in patients with malignant ascites (66,67). Likewise, sustained-release 5-fluorouracil-loaded PLGA microparticles yielded significantly higher drug concentrations in peritoneal tissues (omentum and mesentery) compared to systemic tissues (blood, lungs, and heart) (68,69). IP paclitaxel delivered in liposomes is better tolerated compared to paclitaxel dissolved in Cremophor (70).
In order to determine the relationship between carrier size, drug release rate, and pharmacokinetics of IP therapy, we compared three formulations of paclitaxel with different drug release rates and different particle sizes: Cremophor micellar solution, gelatin nanoparticles, and PLGA microparticles (7). The rank order of drug release was nanoparticles (100% release in 4 h under sink conditions) > Cremophor micelles (maintaining an equilibrium of about 10% free drug fraction until the entire drug load is released or until depletion of micelles) > microparticles (about 70% in 24 h under sink conditions). The rank order of particle size was microparticles (about 4–6 μm diameter) > nanoparticles (about 600 nm) > Cremophor micelles (13 nm). Another major difference is the drug uptake and release. For nanoparticles and microparticles, paclitaxel, once released, will not re-enter the particles. In contrast, paclitaxel partitions into and out of Cremophor micelles. Figure 1b, c compares the paclitaxel concentration-time profiles in peritoneal fluid and plasma; the results indicate that both particle size and drug release rate affect the drug residence in peritoneal cavity; retention is greatest for microparticles (with size at or exceeding that of lymphatic duct openings) and, among the two nanoparticles, Cremophor micelles with the slower drug release (i.e., short retention for nanoparticles due to the rapid clearance of paclitaxel after its complete release).
We next studied the effect of size on the spatial distribution of microparticles (4 and 30 μm, fluorescence-labeled) (6). The smaller particles were widely dispersed throughout the cavity including omentum, mesentery, diaphragm, and lower abdomen, whereas the larger particles were primarily localized in the lower abdomen near the injection site (Fig. 3a). Based on these data, we chose a particle size of 4–6 μm to simultaneously retard the lymphatic clearance and promote intra-cavity distribution.
Fig. 3.
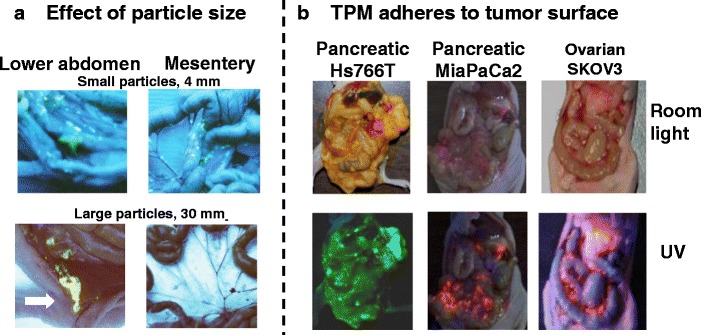
Intraperitoneal distribution of PLGA microparticles. a Effect of particle size. Tumor-free mice were given IP injections of acridine orange-labeled PLGA microparticles with average diameters of 4 or 30 μm. Acridine orange appears yellow under UV light. The smaller particles were dispersed throughout the cavity, and on mesenteric membrane and omentum that are common sites of local metastases of ovarian tumors. The larger particles were localized in lower abdomen near the injection site (indicated by an arrow) and were absent on mesenteric membrane and omentum. b TPM (4–6 μm) adheres to tumor surface. Mice were implanted with IP human xenograft tumors (pancreatic Hs766T, pancreatic MiaPaCa2, or ovarian SKOV3). After tumors were established (day 21, 28, and 42, respectively), mice were given an IP dose of FITC- or rhodamine-labeled blank TPM. Three days later, the animal was anesthetized and the abdominal cavity exposed. Green or red color under UV light indicated localization of FITC and rhodamine, respectively. Reprinted from (6) with permission
Choice of Polymers
Synthetic polymers have been used for decades in a variety of biomedical devices such as surgical sutures, implants, microspheres, and nanoparticles (71–77). PLGA breaks down to biocompatible and progressively smaller compounds, i.e., lactic acid and glycolic acid, which are further metabolized to carbon dioxide and water. Locoregional administration of PLGA is generally well tolerated in humans; intramuscular administration elicited mild tissue response followed by complete recovery (78,79). Another benefit is that these polymers have diverse properties and offer a wide range of drug release rates. We selected two PLGA copolymers to provide the desired range of rapid and slow release (from 1 to 70% in 1 day) and tumor selectivity (6). As shown below, the combination of rapid and slow drug release is required to induce tumor priming, to sustain drug retention in tumors, and to control tumors with diverse growth rates.
Tumor Selectivity
Figure 3b shows that TPM selectively adheres to tumor surface and not on the surface of peritoneum and other IP organs, in mice with metastatic IP Hs766T, MiaPaCa2, and SKOV3 tumors (4,6). The underlying mechanism of this tumor-selective biointerfacial interaction is unknown. But the consistent finding of preferential localization of TPM on tumors with different growth patterns and different disease stages (i.e., with or without carcinomatosis) indicates adherence to tumor surface is a general TPM property.
Getting into Tumors: Tumor Priming
In tumor priming, an apoptosis-inducing drug (e.g., paclitaxel, doxorubicin) is used to transiently expand the interstitial space (e.g., for 96 h) and thereby promotes drug/particulate penetration and dispersion in solid tumors (52–57,80). This effect is tumor selective due to the greater susceptibility of tumor cells to apoptosis relative to normal cells (56). The tumor priming concept is captured in the design of TPM; the rapid release component of TPM provides the initial tumor priming and promotes the penetration of remaining microparticles that continue to release the drug to maintain the antitumor effect and extend the period of effective tumor priming (6). Figure 4a shows that tumor priming is necessary for particle penetration into inner parts of peritoneal tumors. Figure 4b shows that TPM produced more extensive and more sustained tumor priming compared to the paclitaxel/Cremophor micellar solution, as indicated by the deeper penetration and wider dispersion of micron-size latex beads administered 10 days after IP TPM. Figure 4c shows the concentration-depth profiles in tumors; TPM shows 16 times higher CxT and deeper tumor penetration, compared to paclitaxel/Cremophor.
Fig. 4.
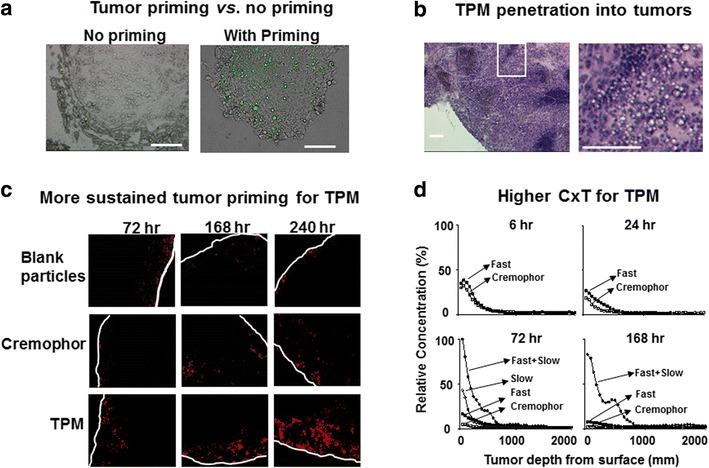
Tumor priming. a Tumor priming is essential for microparticle penetration into inner parts of tumors. Mice bearing IP SKOV3 tumors were given IP injections of acridine orange-loaded blank PLGA particles (no paclitaxel or no priming, left) or acridine orange and paclitaxel-loaded TPM at 10 mg/kg (right). Omental tumors were removed at 72 h after treatment, sectioned and observed under fluorescence microscope. Particles appear in green fluorescence. ×200. Bars, 100 μm. b TPM penetration into tumor interior. Mice bearing IP SKOV3 tumors were given IP injections of 2-component TPM (40 mg/kg, 1:1 fast/slow). Omental tumor was removed at 72 h after treatment, sectioned, and stained with hematoxylin and eosin. Left panel shows the areas with clusters of TPM (circumscribed with dotted lines). Right panel shows the enlarged picture of the white-boxed area in the left panel. c TPM produces more sustained tumor priming relative to paclitaxel/Cremophor micellar solution. Mice bearing IP SKOV3 tumors were given IP injections of a tumor priming treatment with either paclitaxel/Cremophor or the fast release component of TPM (40 mg/kg), followed by IP injections of fluorescent drug-free latex beads (2 μm diameter, red) given 48, 144, and 216 h later. Tumors were excised 24 h after bead injection, or 72, 168 and 240 h after the two priming treatments. Control group received blank, drug-free microparticles (i.e., no tumor priming pretreatment). Tumor sections were examined using fluorescence microscopy. White lines indicate the outer perimeter of tumor nodules. ×100 magnification. (d) TPM yields higher CxT and deeper tumor penetration. The spatial drug distribution in tumors was studied using autoradiography. Mice bearing SKOV3 tumors were treated with paclitaxel/Cremophor, fast release TPM, slow release TPM, or 2-component TPM (1:1 fast/slow). The total paclitaxel dose was 20 mg/kg for Cremophor and single component TPM groups and 40 mg/kg for the 2-component TPM group. All treatments consisted of a mixture of 3H-labeled and nonradiolabeled drug (1.6 mCi/20 mg paclitaxel). At predetermined time points, omental tumors were harvested, flash-frozen and cut into 20-μm sections. Comparison of drug penetration used tumor sections obtained at equal depths. Autoradioluminographic images were obtained and analyzed using standard curves established with commercially available, pre-calibrated microscale tritium autoradiography standards. Drug concentration as a function of distance from tumor periphery was quantified using computer-assisted densitometric analysis. Radioactivity was expressed as paclitaxel-equivalents, with the highest level set at 100%. Reprinted from (6) with permission
Preclinical Proof-of-Concept: Pharmacodynamics of IP TPM in Tumor-Bearing Mice
To improve the therapeutic index of IP therapy, the rate of drug presentation in peritoneal cavity should be optimized so that the drug level and residence in tumors is (a) high enough to provide adequate control of the disease but at the same time below the threshold for producing dose-limiting local toxicity and (b) sufficient to control tumors with diverse growth characteristics as is often found in human tumors. The two-component feature in TPM enables fractionated dose presentation to minimize the local toxicity and improve the control of rapidly and slowly growing tumors. The sustained release may eliminate the need of frequent treatments. These expectations have been confirmed by studies in tumor-bearing animals.
In anticipation of the heterogeneous disease presentation in human patients, we used three IP metastatic human xenograft tumor models with diverse growth characteristics and tumor structures, at early and late (i.e., ascites-producing) stages (Table II) (4,6). The ovarian SKOV3 model is the slowest growing and typically shows small solitary tumors on omentum (<50 mg). In comparison, the two pancreatic tumors, Hs766T and MiaPaCa2, grow more rapidly, with wide-spread nodules that are more bulky (ranging from about 300 mg to up to 1000 mg or about 5% of total body weight of a mouse), and routinely present with carcinomatosis. The median survival time of tumor-bearing mice ranges from 25 to 52 days for these three tumor models. Figure 5a compares the structures and morphologies of tumors located on the omentum, excised at times corresponding to about 80% of the median survival time of untreated animals; the results show substantial differences in tumor structures with respect to cell density (porous Hs766T vs. densely packed MiaPaCa2 and SKOV3) and stromal fraction (low fraction in Hs766 and MiaPaCa2 vs. high fraction in SKOV3). These three tumor models were used to compare the toxicity and efficacy of TPM and the intravenous Cremophor micellar solution. Drug treatments were administered by IP injection, in single or repeated doses. The equi-toxic doses of these two delivery systems are the milligarm paclitaxel-equivalents that produced similar body weight of 7–8% and their equi-effective doses are the milligarm paclitaxel-equivalents that produced the same survival benefits.
Table II.
Characteristics of IP Human Metastatic Xenograft Tumors
| Pancreatic Hs766T | Pancreatic MiaPaCa2 | Ovarian SKOV3 | |
|---|---|---|---|
| Median survival time of untreated animals | 25 days (n = 12) | 36 days (n = 6) | 52 days (n = 12) |
| Growth rate | Rapid | Moderate | Slow |
| Tumor location | Wide-spread throughout peritoneal cavity and retroperitoneal cavity | Multiple nodules on omentum, mesentery, lower abdomen, underneath liver and retroperitoneal cavity | Solitary tumors on omentum, occasionally in other locations, not in retroperitoneal cavity |
| Ascites formation | 100% (mucinous) on day 19 (range, 15–24) | 100% (not mucinous) on day 28 (range, 18–41) | 50% (not mucinous) |
| Average tumor Size | 1.05 g on day 17 | 0.32 g on day 22 | 0.014 g on day 45 |
Fig. 5.
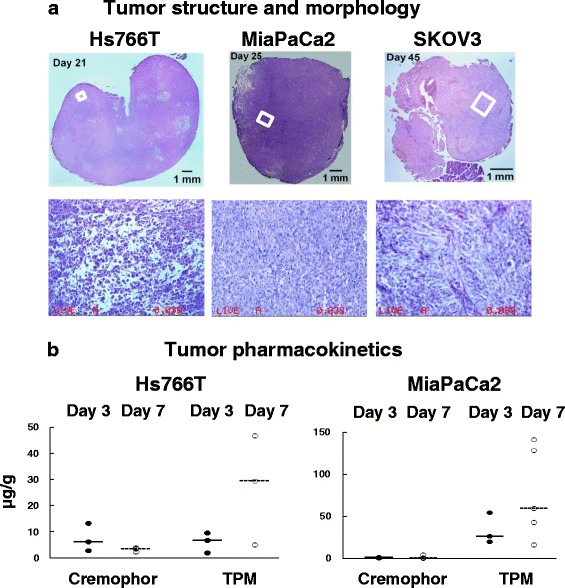
Effect of tumor structure and morphology and drug delivery system on tumor pharmacokinetics. Mice were implanted with IP human xenograft tumors (pancreatic Hs766T, pancreatic MiaPaCa2, or ovarian SKOV3). Omental tumors were excised on 21, 25, and 45 days or about 80% of the median survival time of untreated animals (25, 36, and 52 days, respectively) and stained with hematoxylin and eosin. a Tumor structure and morphology. Top: Whole tumor section (top panels). Note difference in size between the three tumors. Micrographs (×200 magnification) of areas marked in white boxes in the top panel pictures. b Tumor pharmacokinetics. Mice were treated with equi-toxic doses of paclitaxel/Cremophor (40 mg/kg) and TPM (120 mg/kg). Treatments were given when tumors were well established (i.e., 17 and 22 days post-tumor implantation for Hs766T and MiaPaCa2, respectively), and tumors were collected 3 and 7 days post-treatment. The respective tumor weight was 1054 ± 414 mg for Hs766T tumors, and 316 ± 150 mg for MiaPaCa-2 tumors. Note that the results are the sum of total paclitaxel concentration comprising free, protein-bound, cell-associated and particle-associated drug. Horizontal lines are median values. Solid circles and solid lines are for tumors removed 3 days post-treatment. Open circles and dashed horizontal lines are for tumors removed 7 days post-treatment. Data are normalized to 40 mg/kg dose. Note the different y-scales. Reprinted from (4) with permission
For toxicity, the equi-toxic dose of TPM (120 mg/kg paclitaxel-equivalent) is three times that of the Cremophor micellar solution (40 mg/kg). Conversely, the equi-effective dose of TPM (single dose of 120 mg/kg) produces less intestinal toxicity (toxicity was measured as reduction of proliferation index of intestinal crypt cells) compared to three daily doses of 40 mg/kg of the Cremophor formulation (Fig. 6a). In mice, TPM does not cause tissue adhesion (none observed in 26 mice) (6). In general, TPM is more efficacious on a milligarm paclitaxel-equivalent basis on reducing early death, prolonging overall survival time, and increasing the cure rate, and requires less frequent dosing (a single dose is equally effective as three to eight divided doses of the Cremophor micelles) (Fig. 6b, c) (4,6,7). The therapeutic benefits of TPM obtained in three tumors with diverse growth rates, structures, and disease stages indicate broad-spectrum activity and suggest potential utility for managing human tumors that typically are more heterogeneous with respect to pathological or physical attributes compared to experimental animal tumor models.
Fig. 6.
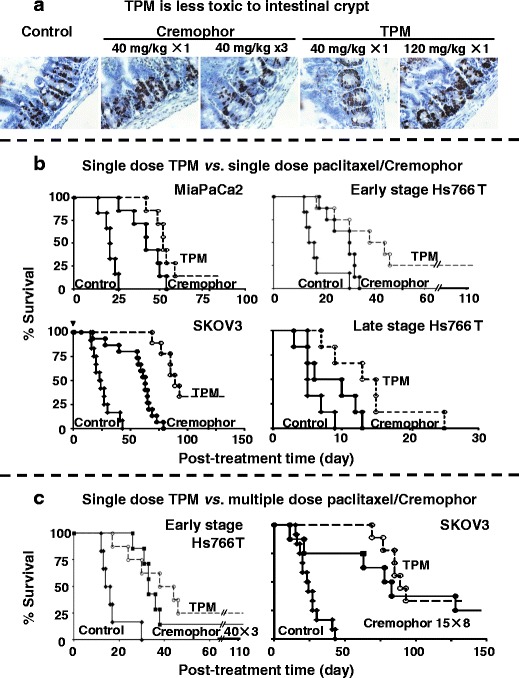
TPM is less toxic and more efficacious compared to paclitaxel/Cremophor. a TPM produces less intestinal toxicity. Mice were given IP injections of paclitaxel/Cremophor at 40 mg/kg (single dose or three daily doses over three consecutive days), Priming TPM at 40 mg/kg (single dose), or two-component TPM at 120 mg/kg (1:2 fast/slow, single dose). Control group received physiological saline. Mice in the single dose groups were euthanized at 24 h post-treatment and mice in the multiple dose group and two-component TPM were euthanized at 120 h after the initial treatment. Intestinal crypts were labeled by 5-bromodeoxyuridine (brown color). A lower labeling index indicates a higher inhibition of the crypt cell proliferation (i.e., higher toxicity). The group that received three doses of the Cremophor formulation had significantly lower labeling index compared to all other groups (p < 0.05). b Single-dose TPM is more efficacious compared to single-dose paclitaxel/Cremophor. Mice were implanted IP with Hs766T, MiaPaCa2, or SKOV3 tumor cells (20 × 106). For MiaPaCa2, SKOV3, and early-stage Hs766T, treatments were initiated at about 40% of the median survival time of untreated animals (10, 15, and 28 days post-implantation for early-stage Hs766T, MiaPaCa2, and SKOV3, respectively). For the late-stage Hs766T tumors, mice were monitored for the appearance of ascites and treatments were initiated when a mouse showed ≥20% weight gain plus abdominal extension. Mice were given IP injections of either blank microparticles or physiological saline (control, solid diamond), a single dose of 40 mg/kg paclitaxel/Cremophor (solid circles), or a single dose of 2-component TPM (120 mg/kg, 1:2 fast/slow, open circles). c Single-dose TPM is equally or more efficacious compared to multiple dose paclitaxel/Cremophor. For Hs766T, mice were treated with three doses of 40 mg/kg paclitaxel/Cremophor weekly on days 0, 7, and 14 (solid symbols, solid lines) or a single dose of 120 mg/kg TPM (1:2 fast/slow; open symbols, dotted lines). For SKOV3, mice were treated with eight doses of 15 mg/kg paclitaxel/Cremophor twice weekly (solid symbols, solid lines) or a single dose of 120 mg/kg TPM (1:2 fast/slow; open symbols, dotted lines). Day 0 represents the day of treatment initiation. Reprinted from (4,6,8) with permission
Correlation Between In Vitro Release and In Vivo Pharmacodynamics
The quantitative relationship between in vitro drug release from delivery systems and treatment outcome is rarely studied. For example, in spite of the repeated studies of IP particulate delivery systems, there is no data to indicate the threshold effective or toxic drug exposure in the peritoneal cavity, either in animals or humans. We investigated the relationship between paclitaxel release (extent and rate) and in vivo pharmacodynamics of TPM in tumor-bearing mice. A method was developed to simulate the dosing rate and cumulative dose released in the peritoneal cavity based on the in vitro release data. Briefly, we established a model of biphasic (rapid and slow) first-order drug release to fit the in vitro drug release data over 28 days; the best-fit model parameters (e.g., the drug release rate constants and dose fractions released over the rapid and slow release components) were used to simulate the dosing rate and the drug amount released from microparticles in the peritoneal cavity up to 76 days, the time when the last disease-related death occurred. For combinations of different microparticles, we assumed that each microparticle dose release is independent of each other and calculated the total amount of drug released as the sum of the individual components in the combinations. The simulated drug release in the peritoneal cavity linearly correlated with treatment efficacy in mice (r2 > 0.8, p < 0.001) (Fig. 7). TPM showed greater dose efficiency (defined as extent of survival benefit per administered dose) and lower cumulative toxicity, relative to the paclitaxel/Cremophor micellar solution. As the major differences between these two delivery systems are the slower clearance of TPM from peritoneal cavity and the slower drug release rate from TPM, the difference in their pharmacodynamics indicates a temporal component of drug presentation, in addition to the drug dose, that determines the treatment outcome (8).
Fig. 7.

Correlation of in vitro drug release and in vivo pharmacodynamics. a The dose amounts released in peritoneal cavity for the nine drug treatment groups were simulated based on the in vitro drug release profiles. Pac/Crem 40 × 1 refers to a single treatment of paclitaxel/Cremophor micellar solution at 40 mg/kg. Abbreviations for single type microparticles are as follows: MP (dose) (type of microparticles). Abbreviations for combination microparticles are as follows: MP (dose of first microparticles) (type of first microparticles) (dose of second microparticles) (type of second microparticles). Abbreviations for repeated treatments are as follows: Pac/Crem 40 × 3 is 3 weekly treatments of 40 mg/kg on days 0, 7, and 14 post-treatment (equivalent to 10, 17, and 24 days post tumor-implantation). MP40SF80SSx2 is two treatments of MP40SF plus MP80SS given on days 0 and 21 post-treatment (equivalent to 10 and 31 days post tumor-implantation). b In vivo biological activity. Day 0 represents the day of treatment initiation, which corresponded to 10 days post-tumor implantation (about 40% of the MST of controls). Survival over time is shown in Kaplan Meier curves. c Correlation of simulated drug release in peritoneal cavity and in vivo pharmacodynamics. Open circle paclitaxel/Cremophor; solid circle TPM. Treatment efficacy was expressed in median survival time (MST). The drug amount released in peritoneal cavity at MST for individual treatments were obtained from the data in panel b. The best-fit linearly regressed lines were MST = 0.39 × (released dose) + 19 for microparticles (r 2 = 0.86, p = 0.0003) and MST = 0.15 × (released dose) + 17 for paclitaxel/Cremophor (r 2 = 0.85, p = 0.1). The data points at 0 mg/kg correspond to the respective vehicle control for each group (saline and blank microspheres). Reprinted with permission (8)
Effects of Tumor Property and Drug Carriers on Drug Delivery and Residence in Peritoneal Tumors
We investigated how differences in tumor structures, disease presentation, and drug delivery systems may affect the drug delivery and residence (CxT) in peritoneal tumors (4). Comparison of tumor pharmacokinetics (on days 3 and 7) derived from IP injection of TPM and paclitaxel/Cremophor micellar solution, in two tumor models with different characteristics (Hs766T and MiaPaCa2, see Table II and Fig. 5a) shows up to 55-fold differences (Fig. 5b). Some of the differences were readily explained by the known differences in the nature of the drug delivery systems or transport barriers within tumors, whereas other observations contradicted the expectations. For example, the two delivery systems showed opposite tumor pharmacokinetic behaviors. The Cremophor group showed declining tumor concentrations over time whereas the TPM group showed increasing concentrations with time; these findings are consistent with the differences in drug release and clearance from peritoneal cavity, i.e., rapid release and rapid clearance of the Cremophor solution resulted in the more rapid drug removal vs. the slow release/clearance and continuing accumulation of TPM in tumors. With respect to transport, Hs766T tumor showed two to seven times higher concentrations relative to MiaPaCa2 tumors for the Cremophor solution; this finding is also as expected in view of more porous structure of Hs766T. However, the opposite was found for TPM; the finding of three to six times higher concentrations in MiaPaCa2 compared to Hs766T could not be explained by the difference in tumor structures, suggesting there are other unaccounted-for determinants and the need of further studies. In short, our collective findings indicate complex qualitative and quantitative relationships between tumor pharmacokinetics, tumor structure and morphology, disease stage, and drug delivery systems.
Summary
Through studying the pharmacokinetics and pharmacodynamics of IP therapy, we have identified several spatial and temporal determinants of treatment efficacy and toxicity, and were able to make use of the versatility of PLGA polymers to design TPM that yielded pharmacodynamically optimized drug delivery to achieve fractionated dosing, tumor priming, and enhanced particle penetration (greater depth and wider dispersion in peritoneal tumors) and intratumoral retention in mouse xenograft tumor models. Preclinical studies show that the first generation, paclitaxel-loaded TPM is more efficacious and less toxic and requires less frequent dosing, compared to the intravenous paclitaxel/Cremophor micellar solution that has been used off-label in previous IP studies. The broad-spectrum activity of paclitaxel-loaded TPM against several IP metastatic tumors with different characteristics (fast vs. slow growing, porous vs. densely packed structures, wide-spread vs. solitary tumors, early vs. late stage, with or without peritoneal carcinomatosis) has motivated the follow-up clinical development of TPM.
Part III. Academia-to-Industry Transition: IND-Enabling Studies
Transitioning from academic preclinical research to first-in-human investigation requires obtaining US FDA approval for the Investigational New Drug (IND) status. The requirements are provided in several well-articulated FDA Guidances (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM078933.pdf, http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm227351.pdf, http://www.fda.gov/downloads/Drugs/…/Guidances/ucm074980.pdf).
IND application requires information in three areas: (a) manufacturing to ensure product uniformity, (b) animal pharmacology and toxicology to ensure safety and to identify the starting dose in humans, and (c) clinical protocol and investigator brochure. The IND-enabling studies are relatively costly, require specialized skill sets, and are typically the bottleneck for transitioning academic discoveries to the clinic. We have been fortunate to obtain support through several funding mechanisms of the US National Institutes of Health, including research grants from the SBIR program of National Cancer Institute and the Bridging Interventional Development Gaps (BrIDGs) program of National Center for Advancing Translational Sciences. The BrIDGs program, in addition to providing the financial support for the Current Good Manufacturing Practice (CGMP) and toxicology studies, also provide significant expertise and know-how to chaperone these challenging IND-enabling studies.
The goal of manufacturing is to obtain clinical grade materials with well-defined product specifications and stability. Note the different FDA CGMP regulations and requirements for an investigational new drug at different clinical development phases, with generally lower requirements for the earlier phase trials (http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070273.pdf, http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211, http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=210). CGMP requires the following: (a) well-defined written procedures, (b) adequately controlled equipment and manufacturing environment, and (c) accurately and consistently recorded data from manufacturing (including testing). The FDA Guidances further provide specific, detailed information regarding the manufacturing control on (a) personnel, (b) QC function, (c) facility and equipment, (d) control of components, containers and closures, (e) manufacturing and records, (f) laboratory controls, (g) packaging, labeling and distributing, and (h) record keeping. Validation of the analytical methods is generally not required in the initial IND application, but is required upon progression to phase 2 and phase 3 stage (http://www.fda.gov/downloads/Drugs/…/Guidances/ucm074980.pdf, http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf, http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm386366.pdf). Stability data are required in all phases of the IND to demonstrate that the drug substance and drug product are within acceptable chemical and physical limits for the clinical study duration (http://www.fda.gov/downloads/Drugs/…/Guidances/ucm074980.pdf, http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073369.pdf). A common source of CGMP facilities are the contract manufacturing organizations. Selection of appropriate CMOs requires review of manufacturing facility and capability, expertise and experience, history of FDA inspection (e.g., Form 483 report), QA/QC system, regulatory and analytical support, and personnel training.
The purpose of the nonclinical toxicology study (performed under good laboratory practice) is to identify (a) the initial safe dose and dose escalation scheme in humans, (b) potential target organs subjected to toxicity, (c) testing methods and procedures to monitor clinical toxicity, and (d) patient eligibility criteria. Two mammalian species (one non-rodent and one rodent) are generally required; the relevant FDA Guidance is ICH M3(R2) (http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm073246.pdf). Specific to oncology products is the recently issued ICH S9 Guidance (http://www.fda.gov/downloads/Drugs/…/Guidances/ucm085389.pdf), describing the less stringent requirements for patients with advanced cancer, e.g., single dose instead of repeated dose toxicology study in certain circumstances and elimination of genotoxicity studies for the early phase trials.
The requirement of a clinical study protocol is to assess whether the initial clinical study will expose patients to unnecessary risks. The Investigator’s Brochure provides clinical investigators the pertinent information regarding the test item.
Part IV. Perspectives
In general, drug development is a high-cost and high-risk business, with an even lower success rate for cancer drugs. From 1996 to 2002, 209 candidates acting on the then-newly identified molecular targets (signaling and apoptosis cascades, angiogenesis, extracellular matrix proteins, growth factors) entered clinical evaluation. Among these 209 agents, only 12 (<6%) showed survival benefits in patients and solid tumors generally fared worse compared to liquid/blood cancers (81). A more recent 2014 review shows similarly low success rate of 6.7% (82). These data suggest development, instead of discovery, has become the bottleneck. We surmise the clinical development of TPM, in view of the confounding factors outlined below, requires a different approach.
As discussed in our reviews (5,83–86), tumors are highly heterogeneous with respect to size, vascularization, blood flow, growth rate, capillary permeability, extracellular protein contents, and tumor cell density. In addition, many of these properties are dynamic, dependent on the host (e.g., larger tumors in humans than in mice, higher stromal fraction in humans), patient dependent (e.g., location in relation to normal tissues, size), diverse in nature, and will change with time (e.g., tumor growth) or with treatments (e.g., apoptosis or necrosis due to chemotherapy or irradiation, changes in vasculature due to antiangiogenic treatment). In addition, changes in one property can affect other properties (e.g., increase in size will affect the vascularization). Such diverse and dynamic tumor properties create uncertainties on drug delivery to target sites. For example, how should one select treatments (dose intensity and dosing interval) in anticipation of intratumoral heterogeneity in the transport mechanisms (diffusion vs. convection) in different parts of a tumor? What are the margins of error if the treatment design/selection does not take into account these dynamic processes? In the case of particulate drug delivery systems, there are additional complications such as intra-cavity distribution, tumor penetration, and drug release rate. To our knowledge, there have been no or few studies to address these critical issues. For example, there have been multiple studies, including several from our group, on the pharmacokinetics of IP therapy (e.g., 3,7,8,15,25,87). But these studies primarily deal with the time scale and not the spatial scale. This is in part because the often-used pharmacokinetic methods, such as the compartmental and the physiological-based pharmacokinetic analyses, view a tissue as a homogeneous compartment (i.e., same concentration throughout). This practice contradicts the knowledge that solid tumors comprise compartments with distinct vascularization status (e.g., greater perfusion in the periphery vs. hypoxic region in the center) and consequently vastly different drug concentrations in these subcompartments. Another drawback is the inability to accommodate the well-established spatial-dependent drug response (e.g., hypoxic region displays greater chemoresistance). Finding the optimal treatment conditions (e.g., dose intensity and frequency) requires knowing the drug concentrations at different tumor subcompartments at any given times and the resulting pharmacodynamics. As very little is known in this area, we are exploring the use of computational approach and have developed multiscale spatiokinetic models that capture the various disposition and transport processes in peritoneal cavity, whole body, and tumor. These first generation models were successfully applied to mice given IP injection of paclitaxel/Cremophor solution, such that the model-predicted data agreed with the experimental results at early times (before treatment-induced changes in tumor structures occurred); the deviations were ≥60-fold lower compared to the observed inter-animal variations (Fig. 8). While the animal-to-human translation would require significantly more complex models to account for the unavoidable intersubject and intrasubject variabilities that would undoubtedly affect the treatment outcome, we are encouraged by these initial findings and are pursuing additional, clinically relevant models to assist the development of TPM.
Fig. 8.
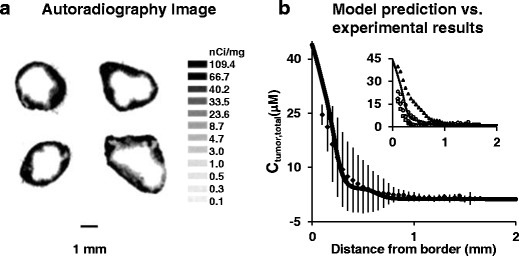
Comparison of experimentally-observed and model-simulated spatiokinetics in tumors. Mice bearing implanted IP SKOV3 tumors were treated with a mixture of 3H-paclitaxel and nonradiolabeled paclitaxel. At 6 h post-treatment, tumors were excised and processed for autoradiography. Drug concentrations were obtained by converting the grayscale values derived from densitometric analysis of autoradiograms. Results were normalized to a paclitaxel-equivalent dose of 10 mg/kg. a Autoradiograms of four tumors (one per mouse). b Concentration-tumor depth profile at 6 h. Solid line: model-simulated profile. Symbols and bars: mean (±1 SD). Inset: model-simulated (solid line) vs. profiles of individual tumors (symbols). Reprinted with permission (3)
Acknowledgments
The study was supported in part by research grants R43/R44CA103133 (Z. Lu) from the National Cancer Institute, R01GM100487 (J. Au) from the National Institute of General Medical Sciences, R01EB015253 (J. Au) from the National Institute of Biomedical Imaging and Bioengineering, X01NS069198 (J. Au) and X01TR000329 (J. Au) from the National Center for Advancing Translational Sciences, National Institutes of Health, Department of Human Health and Services.
Abbreviations
- CxT
Concentration-time product
- IP
Intraperitoneal
- MST
Median survival time
- PLGA
Poly(lactic-glycolic acid) copolymer
- TPM
Tumor-penetrating microparticles
- FDA
Food and Drug Administration
- IND
Investigational new drug
- CGMP
Current good manufacturing practice
References
- 1.Au JL, Wientjes MG. Intravesical chemotherapy of superficial bladder cancer: optimization and novel agents. In: Lerner S, Schoenberg MP, Sternberg CN, editors. Textbook of bladder cancer. Taylor&Francis; 2006. 341–52.
- 2.Shen Z, Shen T, Wientjes MG, O’Donnell MA, Au JL. Intravesical treatments of bladder cancer: review. Pharm Res. 2008;25:1500–10. doi: 10.1007/s11095-008-9566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Au JL, Guo P, Gao Y, Lu Z, Wientjes MG, Tsai M, et al. Multiscale tumor spatiokinetic model for intraperitoneal therapy. AAPS J. 2014;16:424–39. doi: 10.1208/s12248-014-9574-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu Z, Tsai M, Wang J, Cole DJ, Wientjes MG, Au JL. Activity of drug-loaded tumor-penetrating microparticles in peritoneal pancreatic tumors. Curr Cancer Drug Targets. 2013;14:70–8. doi: 10.2174/15680096113136660110. [DOI] [PubMed] [Google Scholar]
- 5.Lu Z, Wang J, Wientjes MG, Au JL. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010;6:1625–41. doi: 10.2217/fon.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Z, Tsai M, Lu D, Wang J, Wientjes MG, Au JL. Tumor penetrating microparticles for intraperitoneal therapy of ovarian cancer. J Pharmacol Exp Ther. 2008;327:673–82. doi: 10.1124/jpet.108.140095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai M, Lu Z, Wang J, Yeh TK, Wientjes MG, Au JL. Effects of carrier on disposition and antitumor activity of intraperitoneal Paclitaxel. Pharm Res. 2007;24:1691–701. doi: 10.1007/s11095-007-9298-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai M, Lu Z, Wientjes MG, Au JL. Paclitaxel-loaded polymeric microparticles: quantitative relationships between in vitro drug release rate and in vivo pharmacodynamics. J Control Release. 2013;172:737–44. doi: 10.1016/j.jconrel.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Lu Z, Yeung BZ, Wientjes MG, Cole DJ, Au JL. Tumor priming enhances siRNA delivery and transfection in intraperitoneal tumors. J Control Release. 2014;178:79–85. doi: 10.1016/j.jconrel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer facts and figures. American Cancer Society. 2013.
- 11.Coates G, Bush RS, Aspin N. A study of ascites using lymphoscintigraphy with 99m Tc-sulfur colloid. Radiology. 1973;107:577–83. doi: 10.1148/107.3.577. [DOI] [PubMed] [Google Scholar]
- 12.Feldman GB, Knapp RC, Order SE, Hellman S. The role of lymphatic obstruction in the formation of ascites in a murine ovarian carcinoma. Cancer Res. 1972;32:1663–6. [PubMed] [Google Scholar]
- 13.Amadori D, Sansoni E, Amadori A. Ovarian cancer: natural history and metastatic pattern. Front Biosci. 1997;2:g8–10. [PubMed] [Google Scholar]
- 14.del Castillo CF, Warshaw L. Peritoneal metastases in pancreatic carcinoma. Hepatogastroenterology. 1993;40:430–2. [PubMed] [Google Scholar]
- 15.Collins JM. Pharmacologic rationale for regional drug delivery. J Clin Oncol. 1984;2:498–504. doi: 10.1200/JCO.1984.2.5.498. [DOI] [PubMed] [Google Scholar]
- 16.Alberts DS, Liu PY, Hannigan EV, O’Toole R, Williams SD, Young JA, et al. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335:1950–5. doi: 10.1056/NEJM199612263352603. [DOI] [PubMed] [Google Scholar]
- 17.Verwaal VJ, van Ruth S, de Bree E, van Sloothen GW, van Tinteren H, Boot H, et al. Randomized trial of cytoreduction and hyperthermic intraperitoneal chemotherapy versus systemic chemotherapy and palliative surgery in patients with peritoneal carcinomatosis of colorectal cancer. J Clin Oncol. 2003;21:3737–43. doi: 10.1200/JCO.2003.04.187. [DOI] [PubMed] [Google Scholar]
- 18.Gadducci A, Carnino F, Chiara S, Brunetti I, Tanganelli L, Romanini A, et al. Intraperitoneal versus intravenous cisplatin in combination with intravenous cyclophosphamide and epidoxorubicin in optimally cytoreduced advanced epithelial ovarian cancer: a randomized trial of the Gruppo Oncologico Nord-Ovest. Gynecol Oncol. 2000;76:157–62. doi: 10.1006/gyno.1999.5677. [DOI] [PubMed] [Google Scholar]
- 19.Markman M, Bundy BN, Alberts DS, Fowler JM, Clark-Pearson DL, Carson LF, et al. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: an intergroup study of the gynecologic oncology group, southwestern oncology group, and eastern cooperative oncology group. J Clin Oncol. 2001;19:1001–7. doi: 10.1200/JCO.2001.19.4.1001. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 21.Nagel JD, Varossieau FJ, Dubbelman R, Bokkel Huinink WW, McVie JG. Clinical pharmacokinetics of mitoxantrone after intraperitoneal administration. Cancer Chemother Pharmacol. 1992;29:480–4. doi: 10.1007/BF00684852. [DOI] [PubMed] [Google Scholar]
- 22.Elferink F, van der Vijgh WJ, Klein I, Bokkel Huinink WW, Dubbelman R, McVie JG. Pharmacokinetics of carboplatin after intraperitoneal administration. Cancer Chemother Pharmacol. 1988;21:57–60. doi: 10.1007/BF00262740. [DOI] [PubMed] [Google Scholar]
- 23.Zimm S, Cleary SM, Lucas WE, Weiss RJ, Markman M, Andrews PA, et al. Phase I/pharmacokinetic study of intraperitoneal cisplatin and etoposide. Cancer Res. 1987;47:1712–6. [PubMed] [Google Scholar]
- 24.Speyer JL, Collins JM, Dedrick RL, Brennan MF, Buckpitt AR, Londer H, et al. Phase I and pharmacological studies of 5-fluorouracil administered intraperitoneally. Cancer Res. 1980;40:567–72. [PubMed] [Google Scholar]
- 25.Markman M, Rowinsky E, Hakes T, Reichman B, Jones W, Lewis JL, Jr, et al. Phase I trial of intraperitoneal taxol: a gynecologic oncology group study. J Clin Oncol. 1992;10:1485–91. doi: 10.1200/JCO.1992.10.9.1485. [DOI] [PubMed] [Google Scholar]
- 26.Markman M, Hakes T, Reichman B, Hoskins W, Rubin S, Jones W, et al. Intraperitoneal therapy in the management of ovarian carcinoma. Yale J Biol Med. 1989;62:393–403. [PMC free article] [PubMed] [Google Scholar]
- 27.Kerr DJ, Los G. Pharmacokinetic principles of locoregional chemotherapy. Cancer Surv. 1993;17:105–22. [PubMed] [Google Scholar]
- 28.Ajani JA, Mansfield PF, Lynch PM, Pisters PW, Feig B, Dumas P, et al. Enhanced staging and all chemotherapy preoperatively in patients with potentially resectable gastric carcinoma. J Clin Oncol. 1999;17:2403–11. doi: 10.1200/JCO.1999.17.8.2403. [DOI] [PubMed] [Google Scholar]
- 29.Yano M, Yasuda T, Fujiwara Y, Takiguchi S, Miyata H, Monden M. Preoperative intraperitoneal chemotherapy for patients with serosa-infiltrating gastric cancer. J Surg Oncol. 2004;88:39–43. doi: 10.1002/jso.20133. [DOI] [PubMed] [Google Scholar]
- 30.Esquivel J, Sticca R, Sugarbaker P, Levine E, Yan TD, Alexander R, et al. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in the management of peritoneal surface malignancies of colonic origin: a consensus statement. Soc Surg Oncol Ann Surg Oncol. 2007;14:128–33. doi: 10.1245/s10434-006-9185-7. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Trigo V, Stuart OA, Stephens AD, Hoover LD, Sugarbaker PH. Surgically directed chemotherapy: heated intraperitoneal lavage with mitomycin C. Cancer Treat Res. 1996;81:51–61. doi: 10.1007/978-1-4613-1245-1_6. [DOI] [PubMed] [Google Scholar]
- 32.Stephens AD, Belliveau JF, Sugarbaker PH. Intraoperative hyperthermic lavage with cisplatin for peritoneal carcinomatosis and sarcomatosis. Cancer Treat Res. 1996;81:15–30. doi: 10.1007/978-1-4613-1245-1_3. [DOI] [PubMed] [Google Scholar]
- 33.Witkamp AJ, de Bree E, Van Goethem R, Zoetmulder FA. Rationale and techniques of intra-operative hyperthermic intraperitoneal chemotherapy. Cancer Treat Rev. 2001;27:365–74. doi: 10.1053/ctrv.2001.0232. [DOI] [PubMed] [Google Scholar]
- 34.Sugarbaker PH, Graves T, DeBruijn EA, Cunliffe WJ, Mullins RE, Hull WE, et al. Early postoperative intraperitoneal chemotherapy as an adjuvant therapy to surgery for peritoneal carcinomatosis from gastrointestinal cancer: pharmacological studies. Cancer Res. 1990;50:5790–4. [PubMed] [Google Scholar]
- 35.Yu W, Whang I, Suh I, Averbach A, Chang D, Sugarbaker PH. Prospective randomized trial of early postoperative intraperitoneal chemotherapy as an adjuvant to resectable gastric cancer. Ann Surg. 1998;228:347–54. doi: 10.1097/00000658-199809000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugarbaker PH, Cunliffe WJ, Belliveau J, de Bruijn EA, Graves T, Mullins RE, et al. Rationale for integrating early postoperative intraperitoneal chemotherapy into the surgical treatment of gastrointestinal cancer. Semin Oncol. 1989;16:83–97. [PubMed] [Google Scholar]
- 37.Stewart JH, Levine EA, Shen P. The current role of hyperthermic intraperitoneal chemotherapy for peritoneal dissemination of appendiceal tumors. Curr Probl Cancer. 2009;33:142–53. doi: 10.1016/j.currproblcancer.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Jacquet P, Averbach A, Stephens AD, Stuart OA, Chang D, Sugarbaker PH. Heated intraoperative intraperitoneal mitomycin C and early postoperative intraperitoneal 5-fluorouracil: pharmacokinetic studies. Oncology. 1998;55:130–8. doi: 10.1159/000011847. [DOI] [PubMed] [Google Scholar]
- 39.de Bree E, Witkamp AJ, Zoetmulder FA. Peroperative hyperthermic intraperitoneal chemotherapy (HIPEC) for advanced gastric cancer. Eur J Surg Oncol. 2000;26:630–2. doi: 10.1053/ejso.2000.0964. [DOI] [PubMed] [Google Scholar]
- 40.Los G, Mutsaers PH, van der Vijgh WJ, Baldew GS, De Graaf PW, McVie JG. Direct diffusion of cis-diamminedichloroplatinum(II) in intraperitoneal rat tumors after intraperitoneal chemotherapy: a comparison with systemic chemotherapy. Cancer Res. 1989;49:3380–4. [PubMed] [Google Scholar]
- 41.Los G, Mutsaers PH, Lenglet WJ, Baldew GS, McVie JG. Platinum distribution in intraperitoneal tumors after intraperitoneal cisplatin treatment. Cancer Chemother Pharmacol. 1990;25:389–94. doi: 10.1007/BF00686048. [DOI] [PubMed] [Google Scholar]
- 42.Bokkel Huinink WW, Dubbelman R, Aartsen E, Franklin H, McVie JG. Experimental and clinical results with intraperitoneal cisplatin. Semin Oncol. 1985;12:43–6. [PubMed] [Google Scholar]
- 43.Markman M, Walker JL. Intraperitoneal chemotherapy of ovarian cancer: a review, with a focus on practical aspects of treatment. J Clin Oncol. 2006;24:988–94. doi: 10.1200/JCO.2005.05.2456. [DOI] [PubMed] [Google Scholar]
- 44.Ozols RF, Bookman MA, Young RC. Intraperitoneal chemotherapy for ovarian cancer. N Engl J Med. 2006;354:1641–3. doi: 10.1056/NEJMc060264. [DOI] [PubMed] [Google Scholar]
- 45.Wenzel LB, Huang HQ, Armstrong DK, Walker JL, Cella D. Health-related quality of life during and after intraperitoneal versus intravenous chemotherapy for optimally debulked ovarian cancer: a gynecologic oncology group study. J Clin Oncol. 2007;25:437–43. doi: 10.1200/JCO.2006.07.3494. [DOI] [PubMed] [Google Scholar]
- 46.Recio FO, Piver MS, Hempling RE, Driscoll DL. Five-year survival after second-line cisplatin-based intraperitoneal chemotherapy for advanced ovarian cancer. Gynecol Oncol. 1998;68:267–73. doi: 10.1006/gyno.1998.4940. [DOI] [PubMed] [Google Scholar]
- 47.Piver MS, Recio FO, Baker TR, Driscoll D. Evaluation of survival after second-line intraperitoneal cisplatin-based chemotherapy for advanced ovarian cancer. Cancer. 1994;73:1693–8. doi: 10.1002/1097-0142(19940315)73:6<1693::AID-CNCR2820730623>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 48.Barakat RR, Sabbatini P, Bhaskaran D, Revzin M, Smith A, Venkatraman E, et al. Intraperitoneal chemotherapy for ovarian carcinoma: results of long-term follow-up. J Clin Oncol. 2002;20:694–8. doi: 10.1200/JCO.20.3.694. [DOI] [PubMed] [Google Scholar]
- 49.Topuz E, Saip P, Aydmer A, Salihoglu Y, Berkman S, Bengisu E. Intraperitoneal cisplatin-mitoxantrone and intravenous ifosfamide combination as first-line treatment of ovarian cancer. Eur J Gynaecol Oncol. 1998;19:265–70. [PubMed] [Google Scholar]
- 50.Markman M, Reichman B, Hakes T, Rubin S, Lewis JL, Jr, Jones W, et al. Evidence supporting the superiority of intraperitoneal cisplatin compared to intraperitoneal carboplatin for salvage therapy of small-volume residual ovarian cancer. Gynecol Oncol. 1993;50:100–4. doi: 10.1006/gyno.1993.1171. [DOI] [PubMed] [Google Scholar]
- 51.Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs. 2010;2. [DOI] [PMC free article] [PubMed]
- 52.Zheng JH, Chen CT, Au JL, Wientjes MG. Time- and concentration-dependent penetration of doxorubicin in prostate tumors. AAPS Pharm Sci. 2001;3 doi: 10.1208/ps030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JL. Determinants of paclitaxel penetration and accumulation in human solid tumor. J Pharmacol Exp Ther. 1999;290:871–80. [PubMed] [Google Scholar]
- 54.Jang SH, Wientjes MG, Au JL. Enhancement of paclitaxel delivery to solid tumors by apoptosis-inducing pretreatment: effect of treatment schedule. J Pharmacol Exp Ther. 2001;296:1035–42. [PubMed] [Google Scholar]
- 55.Chen CT, Au JL, Wientjes MG. Pharmacodynamics of doxorubicin in human prostate tumors. Clin Cancer Res. 1998;4:277–82. [PubMed] [Google Scholar]
- 56.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther. 2007;322:80–8. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 57.Jang SH, Wientjes MG, Au JL. Determinants of paclitaxel uptake, accumulation and retention in solid tumors. Invest New Drugs. 2001;19:113–23. doi: 10.1023/A:1010662413174. [DOI] [PubMed] [Google Scholar]
- 58.Au JLS, Li D, Gan Y, Gao X, Johnson AL, Johnston J, et al. Pharmacodynamics of immediate and delayed effects of paclitaxel: role of slow apoptosis and intracellular drug retention. Cancer Res. 1998;58:2141–8. [PubMed] [Google Scholar]
- 59.Baron MA. Structure of the intestinal peritoneum in man. Am J Anat. 1941;69:439–97. doi: 10.1002/aja.1000690305. [DOI] [Google Scholar]
- 60.Flessner MF, Fenstermacher JD, Blasberg RG, Dedrick RL. Peritoneal absorption of macromolecules studied by quantitative autoradiography. Am J Physiol. 1985;248:26–32. doi: 10.1152/ajpheart.1985.248.1.H26. [DOI] [PubMed] [Google Scholar]
- 61.Hirano K, Hunt CA. Lymphatic transport of liposome-encapsulated agents: effects of liposome size following intraperitoneal administration. J Pharm Sci. 1985;74:915–21. doi: 10.1002/jps.2600740902. [DOI] [PubMed] [Google Scholar]
- 62.Mirahmadi N, Babaei MH, Vali AM, Dadashzadeh S. Effect of liposome size on peritoneal retention and organ distribution after intraperitoneal injection in mice. Int J Pharm. 2010;383:7–13. doi: 10.1016/j.ijpharm.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 63.Supersaxo A, Hein WR, Steffen H. Mixed micelles as a proliposomal, lymphotropic drug carrier. Pharm Res. 1991;8:1286–91. doi: 10.1023/A:1015807913934. [DOI] [PubMed] [Google Scholar]
- 64.Negrini D, Mukenge S, Del Fabbro M, Gonano C, Miserocchi G. Distribution of diaphragmatic lymphatic stomata. J Appl Physiol. 1991;70:1544–9. doi: 10.1152/jappl.1991.70.4.1544. [DOI] [PubMed] [Google Scholar]
- 65.Li JC, Yu SM. Study on the ultrastructure of the peritoneal stomata in humans. Acta Anat (Basel) 1991;141:26–30. doi: 10.1159/000147094. [DOI] [PubMed] [Google Scholar]
- 66.Natsugoe S, Tokuda K, Shimada M, Kumanohoso T, Baba M, Takao S, et al. Morphology of the designed biodegradable cisplatin microsphere. Anticancer Res. 1999;19:5163–7. [PubMed] [Google Scholar]
- 67.Sugiyama T, Kumagai S, Nishida T, Ushijima K, Matsuo T, Yakushiji M, et al. Experimental and clinical evaluation of cisplatin-containing microspheres as intraperitoneal chemotherapy for ovarian cancer. Anticancer Res. 1998;18:2837–42. [PubMed] [Google Scholar]
- 68.Hagiwara A, Takahashi T, Sawai K, Sakakura C, Tsujimoto H, Imanishi T, et al. Pharmacological effects of 5-fluorouracil microspheres on peritoneal carcinomatosis in animals. Br J Cancer. 1996;74:1392–996. doi: 10.1038/bjc.1996.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hagiwara A, Sakakura C, Tsujimoto H, Imanishi T, Ohgaki M, Yamasaki J, et al. Selective delivery of 5-fluorouracil (5-FU) to i.p. tissues using 5-FU microspheres in rats. Anticancer Drugs. 1997;8:182–8. doi: 10.1097/00001813-199702000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Sharma A, Sharma US, Straubinger RM. Paclitaxel-liposomes for intracavitary therapy of intraperitoneal P388 leukemia. Cancer Lett. 1996;107:265–72. doi: 10.1016/0304-3835(96)04380-7. [DOI] [PubMed] [Google Scholar]
- 71.Peppas NA, Langer R. New challenges in biomaterials. Science. 1994;263:1715–20. doi: 10.1126/science.8134835. [DOI] [PubMed] [Google Scholar]
- 72.Pillai O, Panchagnula R. Polymers in drug delivery. Curr Opin Chem Biol. 2001;5:447–51. doi: 10.1016/S1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 73.Okada H, Toguchi H. Biodegradable microspheres in drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12:1–99. doi: 10.1615/CritRevTherDrugCarrierSyst.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- 74.Schacht EH. Using biodegradable polymers in advanced drug delivery systems. Med Device Technol. 1990;1:15–21. [PubMed] [Google Scholar]
- 75.Eldridge JH, Staas JK, Tice TR, Gilley RM. Biodegradable poly(DL-lactide-co-glycolide) microspheres. Res Immunol. 1992;143:557–63. doi: 10.1016/0923-2494(92)80069-W. [DOI] [PubMed] [Google Scholar]
- 76.Heller J. Biodegradable polymers in controlled drug delivery. Crit Rev Ther Drug Carrier Syst. 1984;1:39–90. [PubMed] [Google Scholar]
- 77.Gilding DK, Reed AM. Biodegradable polymers for use in surgery-polyglycolic/poly(lactic acid) homo-and copolymers. Polymer. 1979;20:1459–84. doi: 10.1016/0032-3861(79)90009-0. [DOI] [Google Scholar]
- 78.Visscher GE, Robison RL, Maulding HV, Fong JW, Pearson JE, Argentieri GJ. Biodegradation of and tissue reaction to 50:50 poly(DL-lactide-co-glycolide) microcapsules. J Biomed Mater Res. 1985;19:349–65. doi: 10.1002/jbm.820190315. [DOI] [PubMed] [Google Scholar]
- 79.Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/S0169-409X(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 80.Griffon-Etienne G, Boucher Y, Brekken C. Suit Hd, Jain RK. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: clinical implications. Cancer Res. 1999;59:3776–82. [PubMed] [Google Scholar]
- 81.Nygren P, Larsson R. Overview of the clinical efficacy of investigational anticancer drugs. J Intern Med. 2003;253:46–75. doi: 10.1046/j.1365-2796.2003.01098.x. [DOI] [PubMed] [Google Scholar]
- 82.Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- 83.Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010;12:492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang J, Lu Z, Gao Y, Wientjes MG, Au JL. Improving delivery and efficacy of nanomedicines in solid tumors: role of tumor priming. Nanomedicine (Lond) 2011;6:1605–20. doi: 10.2217/nnm.11.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res. 2003;20:1337–50. doi: 10.1023/A:1025785505977. [DOI] [PubMed] [Google Scholar]
- 86.Au JL, Jang SH, Wientjes MG. Clinical aspects of drug delivery to tumors. J Control Release. 2002;78:81–95. doi: 10.1016/S0168-3659(01)00488-6. [DOI] [PubMed] [Google Scholar]
- 87.Armstrong DK, Fleming GF, Markman M, Bailey HH. A phase I trial of intraperitoneal sustained-release paclitaxel microspheres (Paclimer) in recurrent ovarian cancer: a gynecologic oncology group study. Gynecol Oncol. 2006;103:391–6. doi: 10.1016/j.ygyno.2006.02.029. [DOI] [PubMed] [Google Scholar]