

Abstract
Physiologically based absorption models can be an important tool in understanding product performance and hence implementation of Quality by Design (QbD) in drug product development. In this report, we show several case studies to demonstrate the potential application of absorption modeling in rational design of drug product under the QbD paradigm. The examples include application of absorption modeling—(1) prior to first-in-human studies to guide development of a formulation with minimal sensitivity to higher gastric pH and hence reduced interaction when co-administered with PPIs and/or H2RAs, (2) design of a controlled release formulation with optimal release rate to meet trough plasma concentrations and enable QD dosing, (3) understanding the impact of API particle size distribution on tablet bioavailability and guide formulation design in late-stage development, (4) assess impact of API phase change on product performance to guide specification setting, and (5) investigate the effect of dissolution rate changes on formulation bioperformance and enable appropriate specification setting. These case studies are meant to highlight the utility of physiologically based absorption modeling in gaining a thorough understanding of the product performance and the critical factors impacting performance to drive design of a robust drug product that would deliver the optimal benefit to the patients.
KEY WORDS: absorption modeling, PBPK, pharmacokinetics, Quality by Design (QbD), quality target product profile (QTPP)
INTRODUCTION
In recent years, there has been an increased attention in the application of Quality by Design (QbD) during the development of drug pharmaceutical products (1–3). Under QbD paradigm, the product critical quality attributes (CQAs), which may include attributes of the drug substance, the formulation composition, and/or the manufacturing process, are linked to the in vivo performance of the product, as defined by the quality target product profile (QTPP), and are controlled via a control strategy and appropriate clinically relevant specifications. Implementing QbD in drug development enables rational product design based on a thorough understanding of product and process performance and, most importantly, benefits the patient population. The role of biopharmaceutics, through the use of dissolution testing, modeling and simulation, preclinical and clinical pharmacokinetic studies, in creating this link between drug product characteristics and performance has been the subject of a few recent publications (3,4).
Drug product performance has been traditionally studied in a preclinical setting via dissolution testing, with the primary intent of providing a quality control for the product/manufacturing process consistency. However, dissolution testing can be a powerful tool to link CQAs to the QTPP and indeed several publications have demonstrated the use of dissolution testing in setting of clinically relevant specifications (5,6). In recent years, there has been an increased focus on biorelevant dissolution which is conducted in media intended to better simulate the gastrointestinal tract. Over the years, significant advances have been made in defining composition of such media or establishing new dissolution methodologies (7). However, the relationship between formulation properties and oral absorption in vivo is complex and cannot always be captured solely by dissolution testing. In these cases, additional tools need to be implemented to establish a link between a drug product (or its dissolution) and clinical performance as encouraged by the QbD paradigm.
Modeling and simulation can provide this link and can be an important tool in implementation of QbD in every stage of drug development. A variety of models can be developed depending on the stage of development and availability of data. Oftentimes, the link between product dissolution and in vivo performance is accomplished via the establishment of an in vitro–in vivo correlation (IVIVC) (8). Development of an IVIVC does not necessarily require complex dissolution methodologies, and there is a significant experience with its application both in development and regulatory settings at least for modified release (MR) products. However, the development of an IVIVC can be challenging especially for immediate-release (IR) products. Additionally, availability of clinical data on multiple formulations is a prerequisite, which limits its application especially in early development. Since the underlying principle of QbD is the understanding of in vivo formulation behavior, which is established throughout development, alternative approaches to IVIVC are sought. As a result, oral absorption physiologically based pharmacokinetic (PBPK) models are increasingly being used during formulation development to interrogate and understand formulation performance (9). Availability of commercial software such as GastroPlus (10), Simcyp (11), PK-Sim (12), or Intellipharm (13) has facilitated adoption of these tools during routine pharmaceutical development. Significant efforts across academia and industry are also being undertaken to further refine existing models, such as the efforts from the Oral Biopharmaceutics Tools (OrBiTo) IMI project (9). Given the ability of mechanistic absorption modeling to incorporate formulation properties in a PBPK model setting, it would seem appropriate that such modeling approaches could play a critical role in linking product CQAs to the QTPP. While several publications have presented the use of PBPK models to understand absorption, majority of publications focus on human dose predictions (14,15) or food effect (16,17), and relatively few have specifically focused in detail on studying formulation properties. Nevertheless, literature reports have demonstrated the potential applicability of PBPK modeling to study API properties (18–20) or understand the impact of dissolution differences during manufacturing changes (21–23), all areas that would be relevant to a QbD approach to formulation development. The use of modeling tools in implementing QbD has indeed been highlighted in two relevant publications (3,24).
In this manuscript, we present several case studies related to studying the impact of formulation properties on their in vivo performance via absorption modeling, and the use of this information to guide robust formulation development. The examples presented here span early and late development stages and include both IR and controlled-release (CR) dosage forms. Case studies are presented in a sequential fashion representing the timing when the modeling and simulation activities took place in the drug development continuum, from pre-first-in-human (FIH) to life cycle management. The modeling questions and approaches are framed in the context of the QTPP for each case study. The potential for utilizing the models to set clinically relevant specifications is also discussed in some of the case studies, where applicable.
MATERIALS AND METHODS
Software Used
GastroPlus™ (Simulations Plus, Lancaster, CA, USA) was primarily used for absorption modeling in all the case studies. Simcyp® (Certara, Sheffield, UK) was also used in case study 4. Human pharmacokinetic (PK) parameters were estimated by fitting the human PK profiles in WinNonLin® (Certara, Princeton, NJ, USA), except in case study 2 where human PK parameters were estimated in NONMEM® (Icon, Dublin, Ireland).
Case study 1: Developing Quality by Design in biopharmaceutics from early development stages—understanding the effects of gastric pH (a critical biopharmaceutics attribute) on absorption, relative to QTPP, prior to FIH studies
Merck Development Compound A Physicochemical Properties
Compound A is a BCS II/IV free base with pKa of 2.0, 5.4, and 6.5 and log D of 1.95 (pH 7). The following solubility values were obtained for the free base and were used in the simulations: 5 mg/mL at pH <5, 0.034 mg/mL at pH 6.1, and 0.01 mg/mL at pH 8.0. FaSSIF and FeSSIF solubilities were in agreement with the pH solubility profile indicating no significant bile micelle solubilization; therefore, simulations were conducted using the pH solubility profile alone. Permeability across MDCKII cells was estimated at 9 × 10−6 cm/s which would indicate moderate-high permeability. Precipitation time is difficult to a priori assess in the absence of clinical data. For these simulations, conducted prior to FIH study, two scenarios were simulated; one with the default precipitation time (900 s; i.e., 15 min, similar to gastric emptying time for solids) and one with prolonged precipitation time (20,000 s, i.e., ~5.5 h, exceeding small intestinal transit time). Simulations were run over a dose range of 50–300 mg to bracket the projected clinical doses.
Physiology
The default human fasted physiological model in GastroPlus™ (Opt logD SA/v6.1) was used in these simulations.
Simulation
Since simulations were conducted ahead of FIH studies and the goal was to understand absorption limitations, simulations focused on estimating fraction absorbed (Fa) rather than the area under the curve (AUC) which would be confounded by any errors on the human CL estimates. Since Fa was simulated in this case, pharmacokinetic parameter input is not required.
Case study 2: Identification of optimal drug release rate from a controlled-release (CR) tablet to guide the development of a QD formulation
Merck Development Compound B Physicochemical Properties
Compound B is a BCS I compound. The following key compound properties were used in building the model—molecular weight 382, log P 2.0, pKa 4.0, density 1.2 g/mL, particle size 2 μm, calculated human effective permeability (based on LLCPK1 data) was 3.6 × 10−4 cm/s, and diffusion coefficient was calculated based on molecular weight. Solubility was adjusted in GastroPlus™ to account for changes in solubility across the gastrointestinal (GI) tract based on bile salt concentration changes, using the measured SGF (3 mg/mL), FaSSIF (0.1 mg/mL), and FeSSIF (1.2 mg/mL) solubilities. The precipitation time was fixed at the default value of 900 s.
Dosage Form and Dissolution Data Input
The absorption model was built based on the FIH data generated using the IR capsule formulation at 3 mg dose. Subsequently, a priori prediction of PK profiles of the CR integral tablets at 6 mg dose was conducted using assumed target dissolution profiles of 80% drug release at 8, 12, and 16 h. These simulations were conducted before availability of any dissolution data with the aim to a priori guide formulation development. An assumption was also made that the in vitro release will be representative of in vivo. This assumption is reasonable in early development of CR formulations when the goal of the simulations is to identify optimal dissolution rates to meet pharmacokinetic targets; in this case, the C24 h plasma target of 0.041 μg/mL.
Physiology
The default human fasted physiological model in GastroPlus™ (Opt logD SA/v6.1) was used in these simulations, including the absorption scaling factors (ASF) for caecum and ascending colon assuming good colonic absorption in human based on the high colonic absorption of the compound observed in a dog colonic absorption study.
Pharmacokinetic (PK) Parameters
Human PK parameters were estimated from a one-compartment population PK model built in NONMEM using the single ascending dose oral data (Merck data on file). The mean PK parameters used in these simulations were CL/F = 2.67 L/h and V/F = 21.4 L. Based on the low dose and linear pharmacokinetics, it was assumed that F = 1 (i.e., CL/F and V/F were used directly to simulate systemic disposition)
Single and Population Simulations
Single simulations were conducted to predict the mean PK profiles and parameters. In addition, population simulations were conducted in 25 healthy subjects, randomly generated by GastroPlus™ using the Virtual Trials function, in a crossover design, to compare bioperformance of the CR tablets. The intent of these simulations was to investigate the effect of variations in population physiology (for e.g., GI transit times) on PK of the CR tablets. The default values and %CVs for all the parameters in GastroPlus™ were used in these simulations except for permeability (CV = 100%), V (CV = 49%), and CL (CV = 35%). The variability in these parameters was estimated in the population PK model. For the permeability, the variability was based on the estimated ka variability.
Case study 3: Assessment of the effect of API particle size and particle size distribution (PSD) of a low solubility molecule on tablet bioperformance during late-stage formulation development
Merck Development Compound C Physicochemical Properties
The following key compound properties were used in building the model—molecular weight 504, log D 2.1 (pH 7.0), pKa 6.9, density 1.2 g/mL, calculated human effective permeability (based on LLCPK1 data) was 2.5 × 10−4 cm/s, and the pH-solubility profile of 0.3 mg/mL (pH 2.1), 0.28 mg/mL (pH 4), 0.48 mg/mL (pH 6.9), 1.31 (pH 7.7). Compound C is a BCS II molecule. The precipitation time was fixed at 900 s. Four API lots with the following PSD were simulated to investigate the impact of PSD on tablet bioperformance—lot A (d10 = 9 μm, d50 = 39 μm, d90 = 96 μm), lot B (d10 = 3 μm, d50 = 17 μm, d90 = 62 μm), lot C (d10 = 13 μm, d50 = 77 μm, d90 = 198 μm), and lot D (d10 = 29 μm, d50 = 52 μm, d90 = 109 μm). In these cases, d10, d50, and d90 are defined as the diameter at which 10, 50, and 90%, respectively, of a sample’s distribution is comprised of smaller particles. The PSD was entered as radius vs. % cumulative, and it was assumed that the API radius was constant in each bin.
Dosage Form and Dissolution Data Input
The absorption model was built based on the phase I single ascending dose data generated using a suspension formulation across a 10- to 800-mg dose range. The API PSD (mv; mean diameter of the “volume distribution”) in the suspension was 35 ± 18 μm. Dissolution of the tablet formulations using the four different API lots was generated in FaSSIF (USP-II apparatus) and was used as input to simulate the impact of PSD. FaSSIF was selected as the dissolution media in order to mimic the dissolution of this weakly acidic compound in the small intestinal media. To allow for mechanistic modeling of the dissolution process in the gastrointestinal (GI) tract, instead of incorporation of the dissolution curve directly in the simulation, the in vitro dissolution data were fit in GastroPlus™ using the built-in Johnson dissolution model to obtain a representative diffusion coefficient value. The resulting diffusion coefficients (Deff) for tablets manufactured using different PSD lots were used in the subsequent simulations, and this represents a correction value which takes into account the contribution of formulation to the release rate over the simple drug particle-based dissolution. In order to do the dissolution fitting, a separate drug record was created, and the reference solubility and pH in the compound tab were adjusted as per the dissolution condition. In the physiology tab, the stomach pH was adjusted to the pH of the dissolution media, e.g., pH 6.5, and the volume was changed to 500 mL to mimic the FaSSIF volume used in the in vitro dissolution study. Subsequently, a simulation was run for 90 min to mimic the duration of the in vitro dissolution study, and the simulated amount dissolved was compared to the in vitro dissolution data, the diffusion coefficient was changed iteratively to match the simulated amount dissolved and the in vitro dissolution data.
Physiology
The default human fasted physiological model in GastroPlus™ (Opt logD SA/v6.1) was used in these simulations. No colonic absorption was assumed for these simulations. This assumption was based on deconvolution of the phase I data, which showed minimal absorption past 5 h post dose and was in line with our experience with the previous compounds in the series. Based on the comparison of the observed and predicted PK for the phase I studies, it was observed that the GastroPlus™ setting of 15% fluid volume in the small intestine best captured the PK profiles, especially Cmax at doses of 200 mg and above. While in our experience, this adjustment is not always needed, this change in the intestinal fluid volume has been previously reported in the literature (25) and facilitated a better simulation of compound C data. Based on these outcomes, the subsequent simulations of the impact of the API particle size were conducted using 15% intestinal fluid volume. While it is possible that other factors contribute to the deviations between simulated and observed data (e.g., differential permeability values in the small intestine), the intestinal volume model that directly affects the dissolution process was selected as the most sensitive around the assessment of dissolution rate input and impact on bioavailability.
Pharmacokinetic (PK) Parameters
The human PK parameters were estimated by simultaneously fitting the oral phase I data and IV data to a three-compartment model in WinNonLin (Merck data on file). The microdose IV data of this compound has been reported previously (26). In that publication, this compound was referred to as compound D. The simultaneous fitting was necessary to accurately capture the terminal phase rate constants. The mean final parameters and associated coefficient of variation (%CV) were as follows—V = 11.8 L (17% CV), k12 = 3.4 1/h (47% CV), k21 = 2.0 1/h (27% CV), k13 = 0.09 1/h (25% CV), and k31 = 0.08 1/h (45% CV). Liver first-pass extraction (41.4%) was calculated based on the clearance, liver blood flow, and blood/plasma ratio. The intestinal extraction (13%) was estimated as the ratio between F and Fa*Fh from the lowest dose, where the total dose would be in solution; hence, Fa was expected to be 100%.
Case study 4: Informing API phase product specifications: simulation of the impact of API phase (salt and free base) on bioavailability in Caucasian and Japanese populations
Pioglitazone Physicochemical Properties
Pioglitazone is a free base with a pKa of 4.8. It is dosed as a HCl salt in the clinic at doses up to 45 mg and results in high bioavailability (>80%) irrespective of stomach pH. The following solubility values were obtained for the free base and were used in the simulations: 0.38 mg/mL at pH 1.8, 0.03 mg/mL at pH 2.6, and 0.0003 mg/mL at pH >6. The HCl salt rapidly dissolves in acidic environment to ~1.3 mg/mL at pH 1.4. FaSSIF and FeSSIF values are in line with the pH solubility profile indicating no significant bile micelle solubilization of this compound; hence, simulations were conducted using the pH solubility profile alone. Human effective permeability was estimated to be 4 × 10−4 cm/s.
Pharmacokinetic (PK) Parameters
The following pharmacokinetic parameters were used in these simulations CL 2.4 L/h and V = 0.253 L/kg (27). The low CL results in negligible FPE (~3%).
Physiology/Simulations
Initial simulations on average plasma concentration profiles were conducted using the default human fasted physiological model in GastroPlus™ (Opt logD SA/v6.1). The initial model was qualified by simulating the performance of the HCl salt tablets tested in the clinic (30 mg dose) (28). A long precipitation time was required to describe the observed plasma concentration profiles. For subsequent simulations of mixed API forms, given the known high bioavailability of the HCl salt regardless of stomach pH, the HCl salt was simulated as a non-precipitating solution (set at 100,000 s) to represent best case scenario for bioavailability and thus most conservative scenario in terms of impact of free base. Free base absorption was simulated based on pH solubility curve. Outcome for different free base fractions was interpolated from combinations of these two simulations via the use of the mixed multiple dose module. To further understand the impact of free base content in a population setting, additional simulations were conducted in Simcyp® (v8) that allows for use of well-defined populations for absorption simulations based on literature information on population variability of key physiological parameters (e.g., in this case, stomach pH). The setup of the simulations was similar to that of GastroPlus™. Identical physicochemical, permeability, and compartmental PK parameters were used. The ADAM solid model was used for absorption simulation. For either the HCl salt or the free base, ten trials with 25 subjects were simulated (total 250 subjects); the default healthy volunteer population was used (ages 18–65, 34% females) for initial simulations, while the Japanese population was explored as a next step to simulate the potential differences in the stomach pH between the two populations. The different free base fractions were interpolated from the two simulations.
Case study 5: Understand the impact of dissolution rate differences on bioavailability of fixed dose combination (FDC) tablets of losartan
Losartan Physicochemical Properties
The following key compound properties were used in building the model—molecular weight 423, log P 6.1, density 1.2 g/mL, calculated human effective permeability of 1.15 × 10−4 cm/s, diffusion coefficient was calculated based on molecular weight, FaSSIF solubility profile of 1.6 mg/mL (pH 6.5), and mean API particle size of 40 μm. The precipitation time was fixed at 900 s.
Dissolution Data Input
The dissolution of losartan from three tablets (target tablet, slow tablet, and fast tablet) was generated in 250 mL of simulated gastric fluid (SGF) at 75 rpm and was used as input in the model as in vivo controlled release vs. time. The “target” tablet provided 80% losartan dissolution at 30 min, while the “fast” and “slow” tablet showed 100 and 82%, respectively. It was assumed that the in vitro and in vivo release profiles for these tablets will be similar for this highly soluble API. This assumption is in line with similar simulations to support biowaiver arguments in the literature (23,29). Losartan is present as a potassium salt in the tablet, which is expected to protonate in the low gastric pH and produce losartan acid. This free acid form will then undergo dissolution. The conversion to free acid and subsequent dissolution in the gastric fluid is expected to be the rate limiting step of the tablet dissolution; hence, SGF was selected as the dissolution media.
Physiology
The default human fasted physiological model in GastroPlus™ (Opt logD SA/v6.1) was used in these simulations.
Pharmacokinetic (PK) Parameters
Human PK parameters were estimated by fitting the human IV data in WinNonLin to a one-compartment model. The mean PK parameters used in these simulations were CL = 21.7 L/h and V = 28.7 L.
Population Simulations
Virtual trials were conducted at 50 mg losartan dose in 25 healthy subjects randomly selected by GastroPlus™ in a crossover design to compare bioavailability of the slow and fast tablets as compared to the target tablet formulation. The default population in GastroPlus™ was used in these simulations. However, the mean values and %CV for parameters specific to losartan such as dose, permeability, solubility, drug particle size, fraction unbound in plasma, and the PK parameters were changed to the measured values. These simulations allowed the assessment of the combined effects of variations in population physiology and formulation variables, thus enabling assessment of a potential bioequivalence study outcome from formulations of different dissolution rates.
RESULTS AND DISCUSSION
Case study 1: Developing Quality by Design in biopharmaceutics from early development stages, understanding the effects of gastric pH (a critical biopharmaceutics attribute) on absorption, relative to QTPP, prior to FIH studies
Quality by Design is often viewed as a late-stage formulation development activity, focused on CMC activities that allow for a reproducible manufacturing of the final drug product. However, for biopharmaceutics, Quality by Design (QbD) should be developed throughout the product development cycle. In a recent commentary on a proposed biopharmaceutics risk assessment roadmap, the authors emphasized how QbD activities related to building product knowledge commences as early as drug candidate selection and with defining the QTPP (4). The authors proposed a series of steps toward building the necessary biopharmaceutics knowledge, starting with the preclinical experiments to identify the appropriate formulation options. To that extent, we discuss in this case study the use of absorption modeling to investigate the sensitivity of stomach pH on the bioavailability for an early drug candidate compound and use of these models to design early preclinical PK experiments to guide formulation selection. Due to the pH solubility profile and the expected use of the compound in populations where the use of co-medications like proton pump inhibitors (PPIs) may be prevalent, lack of interactions with these agents is an important aspect of the QTPP.
The results of the simulations are presented in Fig. 1. Regardless of precipitation time, at stomach pH 5 and below exposures were expected to be similar to those of regular fasted conditions. Therefore, the risk to bioavailability due to co-administration of medications, like PPIs, that typically result at stomach pH values of 3–5, would appear to be minimal. On the other hand, the simulation at pH 6 indicates a significant risk of reduced exposures, with up to threefold reduction in bioavailability. To further qualitatively verify the outcome of these clinical simulations, a dog study was conducted where the compound was dosed at 2 mg/kg after either pentagastrin or famotidine pretreatment. After pentagastrin pretreatment exposures were found to be adequate (AUC of 2.0 ± 0.5 μM×h—covering the predicted clinical exposure targets), indicating that enough stomach solubilization was achieved to meet the required exposure (i.e., in vivo precipitation was not significantly limiting oral absorption). Famotidine pretreatment resulted in extremely low exposures of compound A, with most time points being below the LOQ. Famotidine pretreatment in dogs in our experience results in stomach pH ~7–7.5. This would explain the significant impact on compound A bioavailability given the extremely low solubility of the compound at that pH. Based on the simulation results and the confirmatory dog study, it was evident that to meet the QTPP of this drug candidate and to minimize interaction with H2 receptor antagonists (H2RAs), a formulation that mitigates the pH sensitivity would be required. Subsequently, an early formulation prototype with incorporation of acid in the composition was tested in dogs and indeed resulted in significant recovery of exposures, with less than twofold difference between pentagastrin and famotidine pretreatments.
Fig. 1.
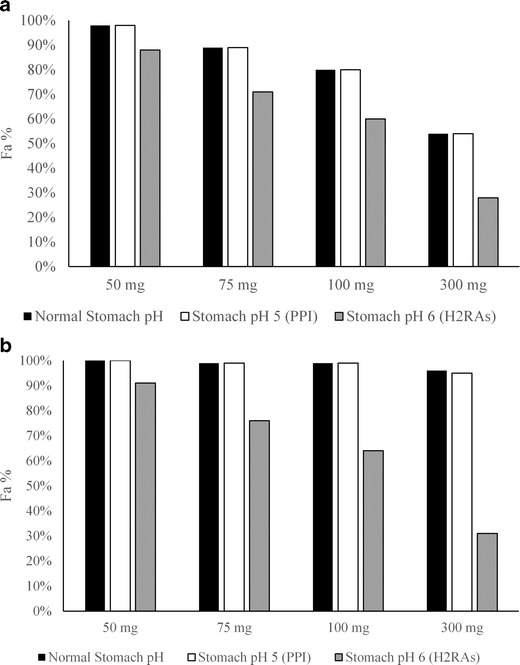
Projections for total absorption (Fa) for compound A as a function of stomach pH and dose under default precipitation (a) or prolonged precipitation (b) settings
The QTPP defines the quality characteristics of the drug product that will ensure the desired quality, taking into account safety and efficacy (1,2). Absorption modeling provides a versatile tool to formulator and biopharmaceutics scientists to very early on interrogate the biopharmaceutical performance of compounds and their ability to meet the QTPP. As demonstrated with the case study on compound A, such simulations can take place before introduction of the compound in the clinic. For compound A, the simulations allowed for an early evaluation of liabilities against PPI or H2RA co-administration. These simulations indicated that while no drug-drug interaction (DDI) would be expected with the former, a reduction of exposures would be anticipated with the latter. The model results led to a preclinical experiment that further confirmed the importance of stomach pH on the absorption of the compound and to the development an early formulation prototype designed to address the solubility limitation at higher pH. While many subsequent steps would be required that would lead to the development of a final drug product, this modeling and simulation work provides an early direction to the formulation development as suggested in the previously published biopharmaceutics risk assessment roadmap (4).
Case study 2: Identification of optimal drug release rate from a controlled release (CR) tablet to guide the development of a QD formulation
CR formulations are often employed to modulate the plasma concentration profile of orally administered drug compounds toward either more favorable dosing regimen (e.g., enable QD administration for a compound with short half-life) or mitigate potential Cmax related adverse events. For compound B, a QD administration was required based on the QTPP. While half-life was not necessarily short, the limitations with Cmax-related adverse events limited the utility of IR dosage form. An absorption model was thus developed to guide formulation development.
The dissolution profiles of the CR formulations used in these simulations are shown in Fig. 2a. As described in the “Methods” section, three distinct dissolution profiles were used in these simulations. Figure 2b shows a comparison of the observed and predicted PK profile at 3 mg IR dose. Based on this, it was concluded that the model was adequate for prediction of human PK and guide formulation development. The model predicted that even a 6-mg-dose IR formulation will not be able to meet the C24 h plasma target (0.041 μg/mL). Simulations predicted that a 3-mg BID dose would be able to meet the C24 h target. However, BID dosing was not acceptable from a target product profile (QTPP) perspective. In addition, in the FIH study, Cmax-related adverse events were observed; hence, there was a need to blunt the Cmax but still meet the target C24 h plasma levels. Due to these reasons, a CR formulation was deemed necessary to meet the requirements of the QTPP, and several simulations were conducted to guide the development of a formulation with adequate drug release rate. Based on prediction of mean PK profiles, it was predicted that in fasted state and at 6 mg dose, the CR formulations with target 80% drug release of 12 and 16 h would meet the C24 h target (Fig. 2b). The population simulations also predicted that the 12- and 16-h release CR tablets would meet the target plasma levels. However, the population simulations also highlighted the potential that these prolonged release rates may result in loss of bioavailability based on the anticipated population variability of colonic transit times; thus, there could be some portion of the population that might not meet the target (Table I). CR formulations with even slower drug release profiles were not simulated due to significantly higher complexities of developing such formulations and the increased risk that making the release rate too slow might compete with the GI transit time and result in excretion of tablet with unreleased drug. In a clinical PK study, clear differentiation in Tmax was seen for the three CR tablets, blunting of Cmax as compared to the IR formulation was observed, and it was concluded that the CR formulation with the T80% of 12-h release rate afforded the optimal PK profile (Merck data on file), as was predicted by the model.
Fig. 2.
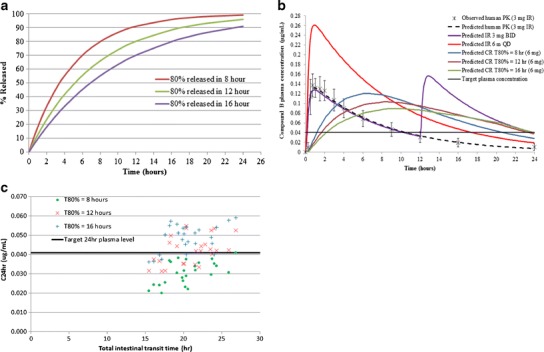
a Assumed target dissolution profiles of controlled-release formulation showing target 80% (T 80%) release of compound B at 8, 12, and 16 h. b Predicted PK profiles of controlled-release (CR) formulations (6 mg dose) and a comparison of the observed (mean ± standard deviation) and predicted PK profile of immediate-release (IR) formulation at 3 mg dose. c Plot of predicted individual plasma concentration at 24 h (C 24 h) as a function of total intestinal transit time for the three controlled-release formulations
Table I.
Predicted Mean, Minimum, and Maximum PK Parameters of Compound B from Population Simulations of Three Controlled-Release Formulations with Different Release Rates, Under Fasted Condition
| Formulation | AUC0–24 h
(min, max) (μg×h/mL) |
C
max
(min, max) (μg/mL) |
T
max
(min, max) (h) |
Fa (min, max) (%) |
C
24 h
(min, max) (μg/mL) |
|---|---|---|---|---|---|
| 80% released in 8 h | 1.74 (0.65, 3.65) |
0.135 (0.058, 0.242) |
7.0 (5.1, 10.3) |
90 (76, 93) |
0.031 (0.020, 0.041) |
| 80% released in 12 h | 1.67 (0.64, 3.51) |
0.099 (0.055, 0.210) |
9.1 (4.1, 13.3) |
89 (75, 91) |
0.042 (0.032, 0.055) |
| 80% released in 16 h | 1.52 (0.58, 3.20) |
0.086 (0.043, 0.123) |
10.2 (4.3, 15.7) |
81 (70, 95) |
0.049 (0.036, 0.059) |
AUC area under the curve, Fa fraction absorbed
The simulations conducted were instrumental in informing formulation direction and building an understanding of the anticipated interplay between formulation parameters (i.e., release rate) and physiological variability (i.e., intestinal transit time). As seen in Fig. 2c, the formulation release rate and the total intestinal transit time have an impact on the C24 h achieved in individual subjects, and it was clear that for the CR formulation with T80% = 8 h, a majority of the population might not meet the target plasma levels. Understanding this interplay is crucial in the context of QbD as optimal absorption in the lower GI dictates ability of the formulation to achieve the desired PK profile and deliver the intended therapeutic effect to the patients. In the context of the previously proposed biopharmaceutics risk assessment roadmap (4), the modeling exercise described here represents the initial evaluation step prior to the first clinical introduction of a CR formulation. It is worth noting that the utility of the model could be further expanded at later development stages in the context of IVIVC to guide the development of clinically relevant specifications.
Case study 3: Assessment of the effect of API particle size and particle size distribution (PSD) of a low-solubility molecule on tablet bioperformance during late-stage formulation development
API particle size is a commonly considered CQA for BCS II and IV compounds where dissolution rate may be limiting bioavailability. In this case, in order to accurately capture the effect of API PSD as well as formulation effect on tablet dissolution, the in vitro dissolution data were fit as described in the “Methods” section (Fig. 3a). The resulting diffusion coefficients (Deff) for tablets manufactured using different PSD lots were used in the subsequent simulations, and this represents a correction value which takes into account the contribution of formulation to the release rate over the simple drug particle-based dissolution. Slight changes were also made to the default fasted human physiology in GastroPlus™, as described in the “Methods” section, to accurately capture the phase I ascending dose PK data of compound C (Fig. 3b). Subsequently, this model was used to simulate the API PSD effect of this BCS II molecule on its tablet bioperformance. Simulations for compound C showed that at 200- and 400-mg doses, a minimal impact on bioperformance is predicted (~10% difference in AUC and Cmax) between the lots with mean particle sizes of 23 and 57 μm (Table II). However, a more pronounced impact on bioperformance is predicted on increasing the particle size to 85 μm, with >25% reduction in AUC and Cmax was observed as compared to the 23 μm particle size at 200 and 400 mg doses. At the 800-mg dose (upper end of the efficacious dose range), up to 20% reduction in AUC and Cmax is predicted when particle size is increased from 23 to 57 μm; also, a significant reduction in AUC (~20–40%) and Cmax (~30–40%) is predicted for 85-μm particle size as compared to the 23-μm particle size. In this case, BID dosing was acceptable per the QTPP; however, maintaining trough plasma concentration levels above the target was paramount for adequate efficacy of this product. Hence, particular attention was given to the C12 h predictions in the simulations. The predicted C12 h data suggested that C12 h is much less sensitive to the API particle size. Comparison to the phase I data showed that particle sizes of 23–57 μm were predicted to show similar C12 h as in phase I. The 85-μm API might show a lower C12 h as compared to the phase I data at 400 and 800 mg. However, this reduction in C12 h is only ~15% as compared to the phase I suspension data and hence can be considered acceptable if the significant impact on AUC and Cmax can be tolerated. As summarized in Table II, reduction in AUC and Cmax was predicted with an increase in PSD, especially at the higher doses as would be expected for a BCS II molecule. However, the impact on C12 h was predicted to be minimal at the particle sizes and doses simulated here. Hence, based on these data, it was concluded that API particle sizes of 23–57 μm can be suitable for formulation development at the doses simulated here.
Fig. 3.
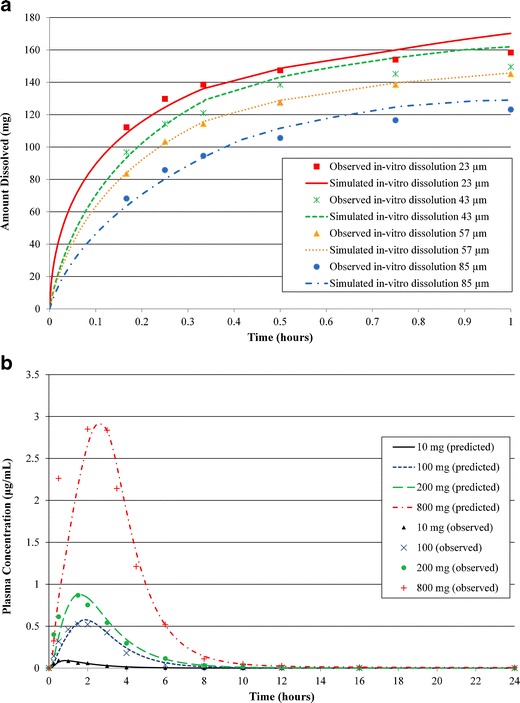
a Observed (mean) and fitted dissolution profiles of compound C tablets using API with four different particle size distributions. b Observed (mean) vs. model predicted plasma concentration vs. time profiles in fasted health volunteers at different doses
Table II.
Predicted Mean PK Parameters of Compound C from Tablets Manufactured Using API with Four Different Particle Size Distributions, Under Fasted Condition. The Ratio of the PK Parameters for Each Particle Size as Compared to the Phase I Suspension are Shown in Parenthesis
| 200 mg | 400 mg | 800 mg | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC (μg×h/mL) | C max (μg/mL) | C 12 h (μg/mL) | AUC (μg×h/mL) | C max (μg/mL) | C 12 h (μg/mL) | AUC (μg×h/mL) | C max (μg/mL) | C 12 h (μg/mL) | |
| Phase I suspension (35 μm) | 2.12 | 0.87 | 0.011 | 6.26 | 1.49 | 0.018 | 9.32 | 2.79 | 0.026 |
| 23 μm | 3.18 (1.5) |
0.98 (1.1) |
0.012 (1.1) |
6.39 (1.0) |
1.67 (1.1) |
0.019 (1.1) |
12.00 (1.3) |
2.75 (0.9) |
0.028 (1.1) |
| 43 μm | 2.91 (1.4) |
0.90 (1.0) |
0.011 (1.0) |
5.76 (0.9) |
1.54 (1.0) |
0.019 (1.1) |
10.40 (1.1) |
2.39 (0.9) |
0.028 (1.1) |
| 57 μm | 2.83 (1.3) |
0.88 (1.0) |
0.010 (0.9) |
5.62 (0.9) |
1.50 (1.0) |
0.018 (1.0) |
9.72 (1.0) |
2.21 (0.8) |
0.027 (1.0) |
| 85 μm | 2.32 (1.1) |
0.76 (0.9) |
0.010 (0.9) |
4.97 (0.8) |
1.25 (0.8) |
0.016 (0.9) |
7.29 (0.8) |
1.64 (0.6) |
0.025 (0.9) |
AUC area under the curve
API PSD-based absorption models have been previously reported in the literature (18,20). It is worth noting that while API PSD is often looked as the CQA, dissolution of formulated product is often different than that of dispersed API. Thus, when absorption modeling is used for assessment of particle size impact at late-stage formulation development, dissolution models should account for performance of final formulated product and not just rely on input of primary particle size. Otherwise, assessment of a CQA and/or specifications may be misleading. In the case of compound C, simulations of particle size were based on observed dissolution of the final formulations, which was incorporated in the model as a correction factor on the Deff parameter obtained by fitting the observed dissolution profiles of the tablets to their respective API PSD, as described in the methods. Alternative dissolution models, such as the Z-factor model, may also be useful in such cases.
Case study 4: Informing API phase product specifications: simulation of the impact of API phase (salt and free base) on bioavailability in Caucasian and Japanese populations
API form and phase selection is a critical step for optimizing bioavailability and stability of oral formulations. When the free (neutral) form of the API is used, oftentimes the most stable polymorph is selected. However, when the solubility of the free form is not sufficient to achieve sufficient bioavailability, alternative forms are considered. The use of either a salt that provides improved solubility/dissolution rate or of amorphous API form, oftentimes stabilized in the form of a solid dispersion, is commonplace during pharmaceutical development. If the higher solubility API form is critical to performance, maintaining that API form intact in the final product becomes a CQA and appropriate controls or specifications need to be put in place to ensure bioavailability (or stability) is not compromised due to API form conversion. Having an understanding of the potential impact of the alternate API form on bioavailability can further inform the specifications. In this case study, we discuss the use of absorption modeling in a population setting, rather than average profile, to investigate the potential impact of disproportionation of pioglitazone HCl salt to free base.
Figure 4a shows the predicted average plasma concentration profile and comparison to the observed profile. As seen from that figure, adequate agreement between observed and predicted data was observed when extremely slow precipitation was assumed, i.e., predicted plasma concentration profiles are within the error bars of the clinical data during the absorption phase. The terminal phase was underpredicted, perhaps because the PK parameters were estimated from an IV study in a different set of individuals. The likelihood of flip-flop kinetics not captured by the model explaining the underprediction of the C24 h was ruled out, as the drug solubility is extremely low and would not be able to sustain continuous absorption as also evident in the simulation with the default precipitation settings (Fig. 4a). However, the prediction of the terminal phase has no influence on the application of the models in these case studies as the focus here is on the impact on absorption. The slow precipitation is in line with the significant supersaturation previously reported in a novel dissolution system (30). It is acknowledged that other hypotheses (e.g., precipitation to a much higher solubility phase) may also result in an acceptable model fit. The intent of the simulation of the HCl salt behavior was in the context of the impact of another form (i.e., free base), that will dissolve as a function of pH, on bioavailability. Hence, the most conservative model was selected which attributes fast absorption to the HCl salt. Use of any other model for the HCl salt would be even more forgiving to the impact of free base on bioavailability. Figure 4b demonstrates the relative (to the HCl salt formulation) fraction absorbed (relative %Fa) as a function of pH, simulated in Simcyp® for the individual subject when a formulation with 100% free base is dosed. A clear relationship between relative %Fa and stomach pH was observed. Figure 4c summarizes the average impact of % free base as a function of stomach pH based on simulations in GastroPlus™. While in normal stomach pH on average, the presence of free base does not appear to affect bioavailability, the effect becomes more pronounced as stomach pH increases. Figure 4d shows the population simulation of the 20% free base in healthy Caucasian population vs. healthy Japanese population. The Japanese population exhibits a higher incidence of achlorhydria as evidenced by the distribution of pH values in Fig. 4d. These simulations clearly showed that there was a higher percentage of subjects in the latter population that may be at risk of reduced exposures due to the presence of free base in the formulation.
Fig. 4.
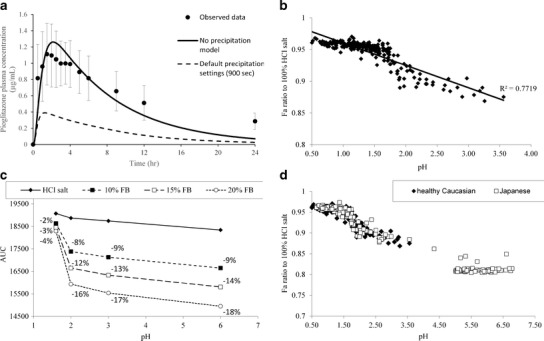
a Observed (mean ± standard deviation) vs. model predicted plasma concentration vs. time profile for pioglitazone HCl salt at 30 mg in fasted healthy volunteers. The default precipitation settings (900 s) model is also shown as a reference. b Model projected relationship between free base absorption (expressed as % relative Fa to the HCl salt simulation) and stomach pH. c Projected AUC values as a function of free base content and stomach pH. d Simulation of 80:20 HCl salt:free base mixture in healthy Caucasians or Japanese subjects. Exposure is presented as relative bioavailability to a 100% HCl simulation
Understanding the impact of API form changes, in the final product, on bioavailability is an important aspect of ensuring clinical performance according to the QTPP. For pioglitazone, the developed models reveal interplay between disproportionation of the HCl salt and stomach pH that dictate the impact on bioavailability (Fig. 4b and c). The complimentary use of average (Fig. 4a and c) and population (Fig. 4b and d) simulations further allows framing of this question relative to the QTPP. If the compound is intended mostly for use under normal fasted conditions, free base content may be less of a concern compared to products that are intended for use in populations with elevated stomach pH (e.g., the Japanese population simulated or patients on antacids). For pioglitazone, the simulations were subsequently applied to FDC tablets developed internally (Merck data in file) that were placed on accelerated stability conditions (40°C/60% RH). Based on the observed dissolution data, it was estimated that the tablet dissolution kinetics were similar to what would be expected by a 70:30 HCl:free base pioglitazone mixture. As seen in Fig. 4c and d, at 20% free base, a potentially meaningful reduction in absorption was expected. On average under normal gastric conditions (pH 1.6), the projections for the tablets on stability were 85% of AUC and 84% of Cmax. This indicated the need for control of the HCl salt conversion to free base at higher temperature and humidity, perhaps via appropriate packaging solutions. Based on the totality of the data, one may recommend a 15–20% free base content in the formulation as the acceptable limit, such that significant negative impact of bioavailability is not observed. However, in practice, additional testing may be required to further qualify the acceptable free base levels, perhaps by conducting an appropriate clinical study guided by the results of these simulations and by development of clinically relevant specifications for a dissolution test.
Case study 5: Understand the impact of dissolution rate differences on bioavailability of fixed-dose combination (FDC) tablets of losartan
In the absence of an in vitro–in vivo correlation, F2 similarity criteria are widely applied to assess the potential impact of dissolution changes and decide on the need for bioequivalence studies (31). It has been suggested before that for BCS III compounds, wider dissolution bounds may be acceptable (32). Therefore, an absorption model was developed for losartan, a BCS III compound, using the available in vitro dissolution data (Fig. 5) to better understand the impact of changes in dissolution on its in vivo behavior.
Fig. 5.
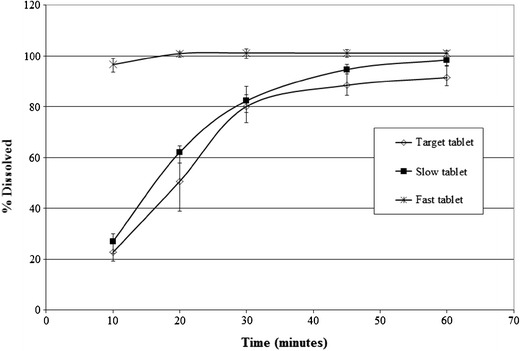
Dissolution profiles of 50 mg losartan from three tablet formulations in 250 mL of simulated gastric fluid (SGF)
The performance of the model for losartan was assessed against a previous bioequivalence study, and the model was deemed acceptable for prediction of the tablet bioperformance (data not shown). The results of the population simulations are summarized in Table III. Based on the current simulation results, it was predicted that the observed differences in the dissolution rates of the FDC formulations should not significantly impact the rate and extent of losartan absorption. This was primarily due to permeability limited absorption of the compound and hence the early differences in the dissolution rates for the slow and fast tablets were not expected to result in meaningful difference in PK as compared to the target tablet. However, the confidence in the Cmax prediction was low because previous clinical data had shown high variability in Cmax of losartan, which was not captured in this model. Nevertheless, based on the established PK/PD relationship for this compound, it was known that Cmax was not a critical parameter for efficacy (Merck data on file), and hence the focus of this modeling exercise was primarily to assess impact of dissolution on AUC. These predictions were corroborated by the PK study in human, which showed that the geometric mean ratio (GMR) of AUC and Cmax for slow tablet to target tablet was 0.97 and 0.89 and 1.01 and 1.05 for fast tablet to target tablet, respectively.
Table III.
Predicted Mean PK Parameters of Losartan from Population Simulations of Three Tablet Formulations, Under Fasted Condition at 50 mg Dose
| AUC0–24 h
(μg×hr/mL) |
C
max
(μg/mL) |
Relative AUC0–24h |
Relative C max |
|
|---|---|---|---|---|
| “Target” tablet | 0.681 | 0.183 | – | – |
| “Slow” tablet | 0.681 | 0.186 | 1.00 | 1.02 |
| “Fast” tablet | 0.682 | 0.185 | 1.00 | 1.01 |
AUC area under the curve
In the context of QbD, there is an increased emphasis on clinically relevant specifications, under this paradigm, the product release specifications would reflect clinical experience with the formulations. For losartan, while clear F2 dissolution differences were observed between different formulations, the formulations were found to have similar performance in vivo. The over-discrimination of the dissolution method is explainable based on the absorption characteristics and the BCS classification of the compound. While we acknowledge that for BCS III compounds the role of transporters and impact of formulation excipients on their bioavailability remains a question that may need to be addressed on a case by case basis, the use of modeling and simulation as presented here may provide additional information on understanding the associated risks with dissolution differences.
CONCLUSION
Understanding the in vivo performance of a dosage form is an integral part of the QbD paradigm. Recently, a biopharmaceutics risk assessment roadmap has been proposed to guide drug product development (4). In silico approaches such as PBPK modeling can play an integral part in advancing biopharmaceutics knowledge throughout development. While oftentimes PBPK modeling is thought of as a tool to project the outcome and thus potentially avoid conduct of clinical studies, in the context of QbD, the primary role of absorption modeling is to drive product performance understanding and potentially inform future experimentation. The case studies discussed in this manuscript demonstrate the potential application of absorption modeling in such context, from early development to late-stage bridging studies. Building the biopharmaceutical understanding starts as early as at the time a compound is selected for development, as demonstrated by case study 1, where simulations helped assess the impact of gastric pH on absorption for different co-medications, prior to conducting any experiments. As clinical data becomes available and focus shifts toward understanding specific formulation attributes that could be potential CQAs, absorption modeling can probe multiple “what if” scenarios to drive experimentation or optimization of performance. Case study 2, where the design of a CR formulation with optimal release rate to meet QTPP is described, and case study 3 on understanding the impact of API particle size on bioavailability are examples of such application. In both cases, the PBPK model allowed to identify optimal dissolution behavior of the formulations based on readily obtainable in vitro data. In late development, modeling can help understand the risk associated with specification settings as shown in case study 4 (free base content in formulation) and case study 5 (dissolution rate of a FDC tablet) and informs the need and the design of potential further clinical studies. The underlying principle of this approach should be that of iterative learning where in vitro, in silico, and clinical data are reviewed and combined, as the product development progresses, to drive design of the most optimal product. The biopharmaceutics knowledge gained through this approach maximizes the possibility of development of a final product that would deliver the optimal benefit to the patients via the intended in vivo performance as well as adequate manufacturing controls and release specifications.
ACKNOWLEDGMENTS
The authors would like to thank the following individuals for providing some of the key data used in the models—Becky Nofsinger for providing support for preclinical studies for the compound used in case study 1, Martin Johnson and Huub Kleijn for providing the human PK parameters for case study 2, Eric Sirota for providing the API PSD and Jen Zhao for the dissolution data used in case study 3, Mike Socki and Paul Harmon for the dissolution data used in case study 5.
Contributor Information
Filippos Kesisoglou, Email: filippos_kesisoglou@merck.com.
Amitava Mitra, Email: amitava_mitra@merck.com.
REFERENCES
- 1.U.S. Department of Health and Human Services, Food and Drug Administration, Guidance for Industry: Q8(R2) Pharmaceutical development. 2009.
- 2.Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–83. doi: 10.1208/s12248-014-9598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selen A, Cruañes MT, Müllertz A, Dickinson PA, Cook JA, Polli JE, et al. Meeting report: applied biopharmaceutics and quality by design for dissolution/release specification setting: product quality for patient benefit. AAPS J. 2010;12(3):465–72. doi: 10.1208/s12248-010-9206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selen A, Dickinson PA, Müllertz A, Crison JR, Mistry HB, Cruañes MT, et al. The biopharmaceutics risk assessment roadmap for optimizing clinical drug product performance. J Pharm Sci. 2014;103(11):3377–97. doi: 10.1002/jps.24162. [DOI] [PubMed] [Google Scholar]
- 5.Dickinson PA, Lee WW, Stott PW, Townsend AI, Smart JP, Ghahramani P, et al. Clinical relevance of dissolution testing in quality by design. AAPS J. 2008;10(2):380–90. doi: 10.1208/s12248-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong C, D'Souza SS, Parker JE, Mirza T. Commentary on AAPS Workshop: dissolution testing for the twenty-first century: linking critical quality attributes and critical process parameters to clinically relevant dissolution. Pharm Res. 2007;24(9):1603–7. doi: 10.1007/s11095-007-9280-x. [DOI] [PubMed] [Google Scholar]
- 7.Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S, et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:342–66. doi: 10.1016/j.ejps.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 8.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry: extended release oral dosage forms: development, evaluation, and application of in vitro/in vivo correlations. 1997.
- 9.Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:300–21. doi: 10.1016/j.ejps.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 11.Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11(2):225–37. doi: 10.1208/s12248-009-9099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thelen K, Coboeken K, Willmann S, Dressman JB, Lippert J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: extension to describe performance of solid dosage forms. J Pharm Sci. 2012;101(3):1267–80. doi: 10.1002/jps.22825. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KC. Dissolution and absorption modeling: model expansion to simulate the effects of precipitation, water absorption, longitudinally changing ntestinal permeability, and controlled release on drug absorption. Drug Dev Ind Pharm. 2003;29(8):833–42. doi: 10.1081/DDC-120024179. [DOI] [PubMed] [Google Scholar]
- 14.Gibson CR, Bergman A, Lu P, Kesisoglou F, Denney WS, Mulrooney E. Prediction of phase I single-dose pharmacokinetics using recombinant cytochromes P450 and physiologically based modelling. Xenobiotica. 2009;39(9):637–48. doi: 10.1080/00498250902954296. [DOI] [PubMed] [Google Scholar]
- 15.Heimbach T, Lakshminarayana SB, Hu W, He H. Practical anticipation of human efficacious doses and pharmacokinetics using in vitro and preclinical in vivo data. AAPS J. 2009;11(3):602–14. doi: 10.1208/s12248-009-9136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heimbach T, Xia B, Lin TH, He H. Case studies for practical food effect assessments across BCS/BDDCS class compounds using in silico, in vitro, and preclinical in vivo data. AAPS J. 2013;15(1):143–58. doi: 10.1208/s12248-012-9419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 2009;11(1):45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10(4):516–25. doi: 10.1208/s12248-008-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei H, Dalton C, Di Maso M, Kanfer I, Löbenberg R. Physicochemical characterization of five glyburide powders: a BCS based approach to predict oral absorption. Eur J Pharm Biopharm. 2008;69(3):1046–56. doi: 10.1016/j.ejpb.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 20.Parrott N, Hainzl D, Scheubel E, Krimmer S, Boetsch C, Guerini E, et al. Physiologically based absorption modelling to predict the impact of drug properties on pharmacokinetics of bitopertin. AAPS J. 2014;16(5):1077–84. doi: 10.1208/s12248-014-9639-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitra A, Kesisoglou F, Dogterom P. Application of absorption modeling to predict bioequivalence outcome of two batches of etoricoxib tablets. AAPS Pharm Sci Technol. 2014. [DOI] [PMC free article] [PubMed]
- 22.Honório Tda S, Pinto EC, Rocha HV, Esteves VS, dos Santos TC, Castro HC, et al. In vitro-in vivo correlation of efavirenz tablets using GastroPlus®. AAPS Pharm Sci Technol. 2013;14(3):1244–54. doi: 10.1208/s12249-013-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crison JR, Timmins P, Keung A, Upreti VV, Boulton DW, Scheer BJ. Biowaiver approach for biopharmaceutics classification system class 3 compound metformin hydrochloride using in silico modeling. J Pharm Sci. 2012;101(5):1773–82. doi: 10.1002/jps.23063. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing Quality by Design in drug development. AAPS J. 2011;13(1):59–71. doi: 10.1208/s12248-010-9250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutton SC. Role of physiological intestinal water in oral absorption. AAPS J. 2009;11(2):277–85. doi: 10.1208/s12248-009-9087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun L, Li H, Willson K, Breidinger S, Rizk ML, Wenning L, et al. Ultrasensitive liquid chromatography-tandem mass spectrometric methodologies for quantification of five HIV-1 integrase inhibitors in plasma for a microdose clinical trial. Anal Chem. 2012;84(20):8614–21. doi: 10.1021/ac301581h. [DOI] [PubMed] [Google Scholar]
- 27.Eckland DA, Danhof M. Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes. 2000;108:234–42. doi: 10.1055/s-2000-8525. [DOI] [Google Scholar]
- 28.Wittayalertpanya S, Chompootaweep S, Thaworn N. The pharmacokinetics of pioglitazone in Thai healthy subjects. J Med Assoc Thai. 2006;89(12):2116–22. [PubMed] [Google Scholar]
- 29.Kovacevi I, Parojci J, Tubi-Grozdanis M, Langguth P. An investigation into the importance of “very rapid dissolution” criteria for drug bioequivalence demonstration using gastrointestinal simulation technology. AAPS J. 2009;11(2):381–4. doi: 10.1208/s12248-009-9114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsume Y, Amidon GL, Takeuchi S. Dissolution effect of gastric and intestinal pH fora BCS class II drug, pioglitazone: new in vitro dissolution system to predict in vivo dissolution. J Bioequiv Availab. 2013;5(6):224–7. doi: 10.4172/jbb.1000162. [DOI] [Google Scholar]
- 31.Stevens RE, Gray V, Dorantes A, Gold L, Pham L. Scientific and regulatory standards for assessing product performance using the similarity factor, f2. AAPS J. 2015;17(2):301–6. doi: 10.1208/s12248-015-9723-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsume Y, Amidon GL. The biowaiver extension for BCS class III drugs: the effect of dissolution rate on the bioequivalence of BCS class III immediate-release drugs predicted by computer simulation. Mol Pharm. 2010;7(4):1235–43. doi: 10.1021/mp100053q. [DOI] [PubMed] [Google Scholar]