

Abstract
In contrast to considerable data demonstrating a decrease in cytochrome P450 (CYP) activity in inflammation and infection, clinically, traumatic brain injury (TBI) results in an increase in CYP and UDP glucuronosyltransferase (UGT) activity. The objective of this study was to determine the effects of TBI alone and with treatment with erythropoietin (EPO) or anakinra on the gene expression of hepatic inflammatory proteins, drug-metabolizing enzymes, and transporters in a cortical contusion impact (CCI) injury model. Microarray-based transcriptional profiling was used to determine the effect on gene expression at 24 h, 72 h, and 7 days post-CCI. Plasma cytokine and liver protein concentrations of CYP2D4, CYP3A1, EPHX1, and UGT2B7 were determined. There was no effect of TBI, TBI + EPO, or TBI + anakinra on gene expression of the inflammatory factors shown to be associated with decreased expression of hepatic metabolic enzymes in models of infection and inflammation. IL-6 plasma concentrations were increased in TBI animals and decreased with EPO and anakinra treatment. There was no significant effect of TBI and/or anakinra on gene expression of enzymes or transporters known to be involved in drug disposition. TBI + EPO treatment decreased the gene expression of Cyp2d4 at 72 h with a corresponding decrease in CYP2D4 protein at 72 h and 7 days. CYP3A1 protein was decreased at 24 h. In conclusion, EPO treatment may result in a significant decrease in the metabolism of Cyp-metabolized drugs. In contrast to clinical TBI, there was not a significant effect of experimental TBI on CYP or UGT metabolic enzymes.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9792-y) contains supplementary material, which is available to authorized users.
Keywords: anakinra, erythropoietin, hepatic metabolism, traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of acute and chronic disability. Although more individuals survive traumatic brain injury than in the past, the survivors endure residual physical, cognitive, emotional, and/or behavioral impairments from the cascade of neuropathological responses resulting from TBI. In spite of promising pre-clinical studies of potential treatments, clinical studies have been overwhelmingly disappointing (1,2). There have been many theories identifying the reasons for the lack of success in translation of compounds identified in pre-clinical studies to successful clinical treatment. Some of the pitfalls that have been proposed include the type of pre-clinical TBI models, a lack of understanding of the therapeutic window, inadequate or super-therapeutic concentrations of the parent and/or active metabolites, and the need for targeting multiple mechanisms of the secondary injury (2–4). The secondary cascade resulting from TBI is likely due to interrelated processes including mitochondrial energy failure, excessive generation of reactive oxygen species, activation of destructive enzymes, membrane disruption, neuronal death, thrombosis due to intravascular coagulation in small vessels, increased synaptic concentrations of excitatory amino acids, and activation of innate inflammatory responses (5–7). The activation of the inflammatory cascade by TBI results in the release of cytokines, the main regulators of the inflammatory response (8). An increase in both pro-inflammatory and anti-inflammatory cytokines occurs after injury in patients with TBI.
Pharmacologic treatment in TBI is also complicated by the effects of TBI on the pharmacokinetics of drugs. Increased hepatic metabolism and decreased plasma protein binding result in an increase in the clearance and decreased concentrations of both unbound and bound drugs (9,10). The increase in hepatic metabolism has been shown to be relatively non-specific, affecting drugs metabolized by a diverse group of hepatic enzymes including various cytochrome P450 (CYP) and UDP glucuronosyltransferase (UGT) isozymes. Increases in unbound clearance have been demonstrated for phenytoin (CYP2C9 and CYP2C19) (11) lorazepam (UGT2B15) (12), antipyrine (multiple Cyps) (12), and valproate (UGT1A6, UGT1A8, UGT2B7, β-oxidation, CYP2A6, CYP2B6, CYP2C9) (13), along with increased urinary excretion of 6β-hydroxycortisol and cortisol (CYP3A4) (14).
The increase in hepatic metabolism in TBI patients is in contrast to the considerable in vitro and in vivo data demonstrating a 20–30% decrease in CYP activity found in experimental models of infection and inflammation (15–17). The decrease in CYP activity has been correlated with circulating concentrations of inflammatory mediators, including tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), and interleukins IL-1α, IL-1β, IL-2, and IL-6, with IL-6 the most important mediator responsible for downregulation (15–17). The inflammatory mediators downregulate CYPs by blocking the action of the pregnane x receptor (PXR) via binding of the p65 subunit of NF-κB to the retinoid X receptor (RXR), resulting in suppression of transcription (17–19). The effect of inflammatory mediators on post-transcriptional regulation of Cyp appears to be due to the inflammatory induction of nitric oxide synthase (20).
Erythropoietin (EPO) and anakinra, inflammatory modulators, have been proposed as promising neuroprotective agents for treatment of TBI. The uses of immunomodulators in TBI that target the inflammatory cytokines or their receptors also have the potential to alter the activity of the drug-metabolizing enzymes (17). EPO is a hematopoietic growth factor which regulates red cell production. EPO has also been shown to have significant pleiotropic effects including the ability to protect nerve cells from hypoxia-induced glutamate toxicity, reduce the immune response and inflammatory reaction, enhance nerve recovery, play a role in neurogenesis, prevent neuronal apoptosis, inhibit nitric oxide formation, and prevent oxidative stress (21). Anakinra is a recombinant form of nonglycosylated human interleukin-1 receptor antagonist (IL-1ra). Elevation of IL-1ra after initiation of the inflammatory response is an important part of the auto-regulatory network controlling the inflammatory response (22). In patients with TBI, high concentrations of IL-1ra and a high ratio of IL-1ra/IL-1β were association with better outcome (23).
The overall aim of this study was to determine the effect of TBI, TBI + EPO, and TBI + anakinra on gene expression of the hepatic inflammatory proteins and drug metabolic enzymes and transporters following experimental TBI.
MATERIAL AND METHODS
Experimental Injury Model
Fifty male Sprague Dawley rats (Harlan, Indianapolis, IN) approximately 3 months of age (335 ± 28 g) were used in this study. All animal and surgical procedures were adhered to as described in the NIH Guide for the Care and Use of Laboratory Animals. The Southern Illinois University Institutional Animal Care and Use Committee (IACUC) and the University of Washington’s IACUC reviewed and approved all experimental procedures. Before and after injury, animals were housed in the university-maintained vivarium, with a 12-h light/dark schedule and a controlled environmental temperature of 22°C in standard housing cages with food and water available ad libitum. All surgeries were performed under aseptic conditions. The cortical contusion injury (CCI) model utilized in the present study was based on previous studies and produces a moderate injury (24–26). Animals were anesthetized using a mixture of isoflurane (2–4%) and oxygen (0.8 L/min). When the animal became unresponsive (no ocular or pedal reflexes), the head was shaved and scrubbed with 70% alcohol followed by betadine and placed into a stereotaxic device. A midline incision was made in the skin as well as through the underlying fascia. A circular craniotomy (5.0 mm) was centered 2.4 mm posterior to and 2.4 mm lateral (left) to the bregma. The moderate contusion injury was created with the Benchmark™ stereotaxic impactor with a 4.0-mm-diameter impactor tip (www.myneurolab.com, St. Louis, MO) and was induced with an impact speed of 3.0 m/s and an impact depth of 2.5 mm. The impact tip maintained contact with the brain tissue for 0.5 s before retraction. Normal body temperature (37°C) during surgery and recovery was maintained with a warm water recycling bed and pump system (EZ Anesthesia, Palmer, PA). Rats receiving sham surgeries underwent identical surgical preparation as the injured animals but did not receive craniotomies or injuries; skin was then sutured and the animal was allowed to recover.
Drug Administration
Animals were randomly assigned to one of four groups: (a) intact sham, (b) CCI-injured + EPO 2500 IU/kg (Procrit™, Amgen, Thousand Oaks, CA), (c) CCI-injured + anakinra 100 mg/kg (Kineret™, Amgen, Thousand Oaks, CA), and (d) CCI-injured + vehicle (saline). Based on the results of pharmacokinetic studies in healthy rats (27), the dosage regimen for EPO and anakinra was designed to obtain clinically relevant serum concentrations of 5000–10,000 mIU/mL and 15–25 μg/mL for EPO and anakinra, respectively. Peak concentrations occurred within 1 to 2 h by administering the initial dose by intraperitoneal injection and subsequent subcutaneous injections. Doses were administered 2 h, 12 h, and then every 12 h up to 72 h after the CCI injury or until the time of sacrifice. All animals were randomized to injury and treatment conditions, and investigators involved in sample collection were blinded to treatment.
Tissue Harvest
Five animals in each treatment group at specified time points post-CCI (24 h, 72 h, and 7 days) were overdosed with a mixture of CO2 (80%) and O2 (20%). The rats were then decapitated; a cardiac blood sample was collected, and brains and livers were rapidly extracted. To maintain quality control and to assure that all of the brains were injured, each brain was assigned a rating score (1 = no visual sign of trauma; 2 = bruised and swollen cortex; 3 = no remaining cortex or extensive damage) (26). Only livers from animals who had a score of 2 were used in the subsequent analyses. The five intact sham animals were sacrificed at 24 h. The median lobe of the liver was extracted and placed on ice. Six tissue punches were collected and placed into microcentrifuge tubes, snap frozen, and then stored at −80°C. Tissue and plasma samples were shipped by overnight carrier to the University of Washington on dry ice.
Gene Expression Analysis
Microarrays were scanned using the Affymetrix GeneChip® 3000 scanner, and the data were processed using various Bioconductor packages. Briefly, raw data were normalized using the robust multi-array average (RMA) algorithm implemented in the Bioconductor oligo package (28). Using the normalized data, we identified genes with significant evidence for differential expression using the Bioconductor limma package (29) by fitting a weighted analysis of variance (ANOVA) model and then computing empirical Bayes adjusted contrasts (30). Significance was defined as an unadjusted p value <0.05 and an absolute fold change >1.5. We used a combination of p value and fold-change criteria based on observations made by the MAQC consortium (31). When we report results of comparisons, the second sample group is always the reference (e.g., vehicle 24 h vs sham indicates the contrast was computed as the vehicle 24 h group relative to the sham group). Ingenuity Pathway Analysis (IPA) (Build 131235; Version 11904312; Database Status 02.20.2012; www.ingenuity.com) was used to analyze differentially expressed genes (>1.5-fold up- or downregulated, p < 0.05) using the Core Analysis feature. IPA is a commercial tool that is based on a proprietary database (http://www.ingenuity.com/) to facilitate the identification of biological themes and canonical pathways in microarray gene expression data. IPA uses a right-tailed Fisher’s exact test to calculate a p value determining the probability that each biological function, canonical pathway, or transcriptional network assigned to the data set is due to chance alone.
Validation of Data Obtained with Microarrays Using Fluorogenic 5′-Nuclease-Based Assay and Quantitative RT-PCR
Quantitative TaqMan-based RT-PCR (qPCR) analysis has a greater dynamic range for changes in gene expression levels compared to microarray-based analysis. Therefore, we used qPCR to validate expression changes of genes of interest that had been identified by microarray analysis. Briefly, reverse transcription was performed according to the manufacturer’s established protocol using total RNA and the SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA.). For gene expression measurements, 2 μL of cDNA was included in a PCR (12 μL final volume) that also consisted of the ABI TaqMan® Gene Expression Assays mix and the TaqMan Gene Expression Master Mix according to the manufacturer’s protocol (Applied Biosystems Inc., Foster City, CA). Amplification and detection of PCR amplicons were performed with the ABI PRISM 7900 system (Applied Biosystems Inc., Foster City, CA) with the following PCR profile: 1 cycle of 95°C for 10 min, 40 cycles of 95°C for 30 s, and 60°C for 1 min. GAPDH amplification plots derived from serial dilutions of an established reference sample were used to create a linear regression formula in order to calculate expression levels, and β-actin gene expression levels were utilized as an internal control to normalize the data.
CYP2D4, CYP3A1, EPHX1, and UGT2B7 Protein Assays
An estimated 30 to 50 mg aliquots of each liver sample were added to 200 μL ice-cold radio-immunoprecipitation assay (RIPA) lysis buffer (Millipore, Billerica, MA) containing one protease inhibitor cocktail tablet (Complete Mini, Roche, Indianapolis, IN) per 2.5 mL, vortexed, sonicated, and then rotated for 30 min at 4°C. The samples were then centrifuged at 16,000 G at 4°C for 30 min to remove debris, and the supernatant was transferred to several replicate aliquots in strip tubes. Total protein concentration was measured using the BCA Total Protein Assay (Thermo Fisher Scientific, Waltham, MA). The remaining aliquots were stored at −80°C until protein analysis. ELISA assay kits from Antibodies-online.com (Atlanta, GA) were used for the CYP2D4 and CYP3A1 assays. ELISA assay kits from MyBio Source (San Diego, CA) were used to determine the epoxide hydrolase 1 (EPHX1) and UGT2B7 protein concentrations. Small-scale test runs were conducted using nonessential liver tissue homogenates to determine optimal total protein loading concentrations for each assay, 20 μg per well for CYP2D4 and 1 μg per well for CYP3A1, EPHX1, and UGT2B7. Tissue homogenates were thawed on ice and diluted initially in RIPA buffer, then 1:100 in sample diluent to bring the total protein to the desired concentration. Total protein concentrations of the initial daily RIPA buffer dilutions were verified and used to normalize results from each ELISA assay. BCA Total Protein and ELISA plates were analyzed using a Molecular Devices Spectra MAX 190 Microplate Reader at 562 and 450 nm, respectively. Standard curves were constructed using linear regression of log concentrations and absorbance. Measurable concentrations ranged from 0.3 to 20 ng/mL (CYP2D4), 0.15 to 10 ng/mL (CYP3A1), 16 to 1000 pg/mL (EPHX1), and 20 to 1200 pg/mL (UGT2B7) from which we then estimated liver concentrations for each animal. One-way analysis of variance (ANOVA) and Tukey’s method of multiple comparisons were used to determine statistical significance comparing contrasts identical to those we made with the microarray data.
Quantitative Assessment of Cytokines in Plasma
Cytokine concentrations were determined in the plasma samples of the animals using the Proinflammatory Panel 1 (rat) V-PLEX Kit from Meso Scale Discovery (Meso Scale Discovery, Rockville, MD) according to the manufacturer’s recommended protocol; a 1:4 dilution of the plasma samples was used to perform the assays. This kit allows for measuring the analytes IFN-γ, IL-1β, IL-10, IL-13, IL-4, IL-5, IL-6, and TNF-α simultaneously. Data was generated using a MESO QuickPlex SQ 120 instrument (Meso Scale Discovery, Rockville, MD). Standard curves were constructed using linear regression of log concentrations and absorbance. Measurable concentrations ranged from 1.0 to 1000 pg/mL (IFN-γ), 10 to 10,000 pg/mL (IL-1β), 4 to 10,000 pg/mL (IL-10), 0.2 to 1000 pg/mL (IL-13), 0.9 to 1000 pg/mL (IL-4), 10 to 1000 pg/mL (IL-5), 5 to 10,000 pg/mL (IL-6), and 0.25 to 1000 pg/mL (TNF-α) from which we then estimated liver concentrations for each animal. ANOVA with Holm’s procedure was used to determine statistical significance comparing contrasts identical to those we made with the microarray data.
RESULTS
The microarray data passed all the standard and advanced quality control metrics. The number of differentially expressed genes (>1.5-fold change, p < 0.05) at 24 h, 72 h, and 7 days is presented in Table I. We have submitted both the normalized and raw data used in this manuscript to the NCBI GEO database (GSE64886). The vehicle to sham comparison reflects the effect of the TBI without treatment to sham controls without TBI. The EPO or anakinra (CCI animals that received treatment) to vehicle (CCI animals that received vehicle) comparison evaluates the effect of treatment on any gene expression changes attributable to CCI.
Table I.
Gene Expression Data. The Number of Differentially Expressed Gene Probes (>1.5-Fold Up or Down, p < 0.05)
| 24 h | 72 h | 7 days | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Down | Up | Total | Down | Up | Total | Down | Up | Total | |
| 111 | 63 | 174 | 58 | 33 | 91 | 22 | 44 | 66 | |
| 25 | 86 | 111 | 36 | 320 | 356 | 85 | 102 | 187 | |
| 16 | 27 | 43 | 20 | 36 | 56 | 65 | 62 | 127 | |
Effect of TBI on Gene Expression in Liver Tissue and Plasma Cytokines
In contrast to the differential expression of over 100 genes involved in the inflammatory response in the brain after TBI (Supplemental Table 1), there was a less pronounced effect of TBI on the expression of genes involved in inflammatory processes in the liver (Table II). IPA analysis was used to identify the genes involved in the inflammatory process (Supplemental Table 1). At 24 h, 72 h, and 7 days, there was no effect of TBI on gene expression on the inflammatory factors shown to be associated with the alteration in the expression of hepatic metabolic enzymes in models of infection and inflammation; Tnfa, Ifng, Il1a, Il1b, ll6, or inducible nitric oxide synthase (Nos2) at any of the time points. TNF-α, IFN-γ, IL-4, IL-6, IL-10, and IL-13 were measurable in the plasma at all time points (Fig. 1). IL1-β and IL-5 concentrations were below detectable concentrations. Plasma concentrations of IL-6 were slightly increased in the sham when compared to literature baseline values (32), suggesting an inflammatory response to the sham procedure itself. Compared to the sham animals, at 24 h post-TBI, there was a statistically significant increase in the plasma concentration of IL-6 in the vehicle-treated TBI animals. IL-6 remained elevated for 7 days, although the increase was not statistically significantly different than sham.
Table II.
Effect of TBI on Inflammatory Genes in the Liver. Genes Differentially Expressed (>1.5-Fold Change, p < 0.05)
| Affymetrix ID | Gene symbol | Genes | a |
|---|---|---|---|
| 24 h post-TBI | |||
| 10901166 | Angptl4 | Angiopoietin-like 4 | 0.58 |
| 10710028 | Arntl | Aryl hydrocarbon receptor nuclear translocator-like | 0.63 |
| 10907825 | Casp4 | Caspase 4, apoptosis-related cysteine peptidase | 0.66 |
| 10727260 | Ccnd1 | Cyclin D1 | 0.47 |
| 10828827 | Cdkn1a | Cyclin-dependent kinase inhibitor 1A | 0.52 |
| 10771660 | Cxcl9 | Chemokine (C-X-C motif) ligand 9 | 0.55 |
| 10738399 | G6pc | Glucose-6-phosphatase, catalytic subunit | 0.39 |
| 10862867 | Gadd45a | Growth arrest and DNA-damage-inducible, alpha | 1.65 |
| 10922826 | Il1r1 | Interleukin 1 receptor, type I | 1.83 |
| 10789857 | Il17rb | Interleukin 17 receptor B | 0.57 |
| 10787364 | Jak3 | Janus kinase 3 | 1.51 |
| 10841693 | Lbp | Lipopolysaccharide binding protein | 1.97 |
| 10829649 | Mif | Macrophage migration inhibitory factor | 0.57 |
| 10810867 | Nqo1 | NAD(P)H dehydrogenase, quinone 1 | 1.78 |
| 10828714 | Ppard | Peroxisome proliferator-activated receptor delta | 0.62 |
| 10857314 | Slc6a6 | Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 | 0.41 |
| 10749372 | Socs3 | Suppressor of cytokine signaling 3 | 1.62 |
| 10747506 | Stat3 | Signal transducer and activator of transcription 3 | 1.62 |
| 10887306 | Tnfaip2 | Tumor necrosis factor, alpha-induced protein 2 | 1.53 |
| 72 h post-TBI | |||
| 10744425 | Alox15 | Arachidonate 15-lipoxygenase | 2.67 |
| 10727260 | Ccnd1 | Cyclin D1 | 0.55 |
| 10828827 | Cdkn1a | Cyclin-dependent kinase inhibitor 1A | 0.65 |
| 10729791 | Ifit1 | Interferon-induced protein with tetratricopeptide repeats 1 | 1.89 |
| 10789857 | Il17rb | Interleukin 17 receptor B | 0.59 |
| 10747813 | Slc4a1 | Solute carrier family 4 (anion exchanger), member 1 | 1.84 |
| 10749372 | Socs3 | Suppressor of cytokine signaling 3 | 1.62 |
| 10777337 | Stx18 | Syntaxin 18 | 0.63 |
| 10887306 | Tnfaip2 | Tumor necrosis factor, alpha-induced protein 2 | 1.80 |
| 7 days post-TBI | |||
| 10901166 | Angptl4 | Angiopoietin-like 4 | 0.64 |
| 10727260 | Ccnd1 | Cyclin D1 | 0.62 |
aFold change in gene expression
Fig. 1.
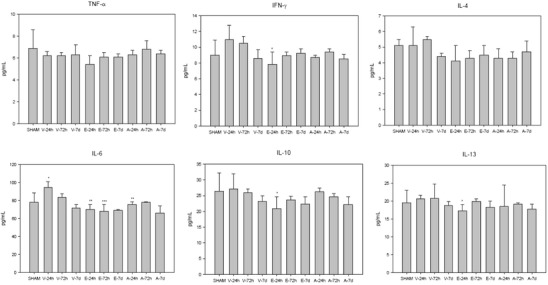
The effect of vehicle (V), erythropoietin (E), and anakinra (A) in TBI animals on cytokine plasma concentrations at 24 h, 72 h, and 7 days post-TBI. IFN-γ: *p = 0.01 compared to vehicle 24 h; IL-6: *p = 0.01 compared to sham, **p = 0.00 compared to vehicle 24 h, ***p = 0.03 compared to vehicle 72 h; IL-10: *p = 0.04 compared to vehicle 24 h; IL-13: *p = 0.03 compared to vehicle 24 h
There was no significant effect of TBI on gene expression of the Cyp1, Cyp2, and Cyp3 subfamily of enzymes known to be involved in the metabolism of exogenous compounds (Table I). Specifically, there was no effect of TBI on Cyp1a1, 2a1, 2a2, 2b1, 2C11, 2C13, 3a23/3, 2d2, 2d3, 2d4, 2d5, or 2e1. At 24 h, TBI significantly upregulated several members of the Cyp4f subfamily involved in the metabolism of the endogenous inflammatory substrates, i.e., leukotriene B4, prostaglandins, and arachidonic acid. TBI increased the expression of Cyp7a1 and Cyp51, metabolic enzymes involved in cholesterol biosynthesis and hydroxysteroid (17β) dehydrogenase 2, a metabolic enzyme involved in steroidogenesis.
There was a significant effect of TBI on the expression of the hepatic phase 2 enzymes involved in endogenous metabolism (Table III). The expressions of Ugt2B7, Ephx1, and glutathione S-transferase A2 (Gsta2) were downregulated 24 h post-injury compared to those of non-injured animals. In both rodents and humans, UGT2B7 is involved in the conjugation of many xenobiotics, including morphine (33). EPHX1 plays an important role in the activation and detoxification of exogenous chemicals (34); GSTA2 has glutathione peroxidase activity and protects cells from reactive oxygen species (35). At 7 days post-TBI, sulfotransferase 2A1 (Sult2a1) was upregulated. SULT2A1 is involved in the sulfate conjugation of primary and secondary alcohols including raloxifene, tibolone, estradiol, testosterone, and dehydroepiandrosterone (36).
Table III.
Effect of TBI on Liver Metabolizing Enzymes. Genes differentially expressed (>1.5-fold change, p < 0.05)
| Affymetrix ID | Symbol | Genes | a |
|---|---|---|---|
| 24 h post-TBI | |||
| 10716509 | Ces2c | Carboxylesterase 2C | 1.78 |
| 10871087 | Cyp4a1 | Cytochrome P450, family 4, subfamily a, polypeptide 1 | 1.63 |
| 10878780 | Cyp4a2 | Cytochrome P450, family 4, subfamily a, polypeptide 2 | 1.60 |
| 10878767 | Cyp4a8 | Cytochrome P450, family 4, subfamily a, polypeptide 8 | 1.65 |
| 10914411 | Cyp8b1 | Cytochrome P450, family 8, subfamily b, polypeptide 1 | 1.50 |
| 10853471 | Cyp51 | Cytochrome P450, family 51 | 1.59 |
| 10811341 | Hsd17b2 | Hydroxysteroid (17-beta) dehydrogenase 2 | 1.78 |
| 10769680 | Hsd17b7 | Hydroxysteroid (17-beta) dehydrogenase 7 | 1.88 |
| 10810867 | Nqo1 | NAD(P)H dehydrogenase, quinone 1 | 1.78 |
| 10868726 | Aldh1b1 | Aldehyde dehydrogenase 1 family, member B1 | 0.63 |
| 10807083 | Ces2g | Carboxylesterase 2G | 0.56 |
| 10770342 | Ephx1 | Epoxide hydrolase 1, microsomal | 0.63 |
| 10918822 | Gsta2 | Glutathione S-transferase A2 | 0.66 |
| 10863686 | Nat8 | N-Acetyltransferase 8 | 0.52 |
| 10771978 | Ugt2b7 | UDP glucuronosyltransferase 2 family, polypeptide B7 | 0.66 |
| 72 h post-TBI | |||
| 10863686 | Nat8 | N-Acetyltransferase 8 | 0.65 |
| 10875324 | Cyp7a1 | Cytochrome P450, family 7, subfamily a, polypeptide 1 | 1.94 |
| 10811341 | Hsd17b2 | Hydroxysteroid (17-beta) dehydrogenase 2 | 1.74 |
| 7 days post-TBI | |||
| 10719187 | Sult2a1 | Sulfotransferase family 2A, dehydroepiandrosterone (DHEA)-preferring, member 1 | 1.51 |
aFold change in gene expression
Although there was a significant effect of TBI on the expression of several solute carrier transporters 24 h post-injury (increased expression: Slc1a2, Slc4a1, Slc4a4, Slc13a5, Slc34a2; decreased expression: Slc2a5, Slc6a6, Slc28a2), 72 h post-injury (increased: Slc4a1, Slc34a2), and 7 days post-injury (increased: Slc28a2), there was no effect of TBI on the gene expression of the transporters shown to be involved in the transport of drug metabolism including Abcc1 (MRP1), Abcb1a (MDR1), Abcg2 (BCRP), Abce1 (OABP), Slc22a2 (OCT2), Slc22a6 (OAT1), Slc22a9 (OAT7), and Slco1b3 (OATP8).
At 24 h post-injury, the gene expressions of nuclear receptor 1D1 (Nr1d1) and Nr1d2 were upregulated (2.20- and 1.68-fold, respectively) and peroxisome proliferator-activated receptor delta (Ppard) was downregulated (0.62) in the TBI animals compared to sham. Hepatocyte nuclear factor 4, gamma (Hnf4g) was upregulated (1.57-fold) at 7 days post-injury in the TBI animals compared to the uninjured controls. There was no effect of TBI on the expression of Pxr, Rxr, or the constitutive androstane receptor (Car).
Effect of EPO and Anakinra on Hepatic Gene Expression and Plasma Cytokines
Overall, compared to vehicle, EPO treatment resulted in a significantly greater number of genes differentially expressed compared to anakinra (Table I). There was a significant effect of EPO on increased expression of genes involved in the inflammatory process in the liver at 24 and 72 h and both an increased and decreased expression of inflammatory genes 7 days post-injury in the presence of TBI (Table IV). Compared to the vehicle, EPO significantly decreased the plasma concentrations of IL-6 at 24 and 72 h post-injury and IL-10 and IL-13 at 24 h post-injury in the TBI animals (Fig. 1). The canonical pathways following EPO treatment identified by IPA at 24 h included several inflammatory signaling pathways (Table V, Supplemental Table 3). In contrast, the anakinra treatment only had a minimal effect at 72 h on the expression of genes involved in the inflammatory process (Table V, Supplemental Table 4). The anakinra treatment resulted in a small but significant decrease in the plasma concentrations of IL-6 at 24 h (Fig. 1). There was no effect of either EPO or anakinra on gene expression on any of the inflammatory factors shown to be associated with alteration in the expression of hepatic metabolic enzymes in models of infection and inflammation.
Table IV.
Effect of EPO and Anakinra on Inflammatory Genes in the Livers of TBI Animals
| Affymetrix ID | Gene symbol | Genes | a | a |
|---|---|---|---|---|
| 24 h post-TBI | ||||
| 10858408 | A2m | Alpha-2-macroglobulin | 2.27 | n.s. |
| 10744425 | Alox15 | Arachidonate 15-lipoxygenase | 2.86 | n.s. |
| 10894525 | Cry1 | Cryptochrome 1 (photolyase-like) | 1.54 | n.s. |
| 10711575 | Dmbt1 | Deleted in malignant brain tumors 1 | 1.55 | n.s. |
| 10732652 | Dusp1 | Dual specificity phosphatase 1 | 1.80 | n.s. |
| 10800919 | Egr1 | Early growth response 1 | 2.09 | n.s. |
| 10878112 | Jun | Jun oncogene | 2.13 | n.s. |
| 10742402 | Ltc4s | Leukotriene C4 synthase | 1.57 | n.s. |
| 10907913 | Mmp8 | Matrix metallopeptidase 8 | 1.67 | n.s. |
| 10747813 | Slc4a1 | Solute carrier family 4 (anion exchanger), member 1 | 1.93 | n.s. |
| 10857314 | Slc6a6 | Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 | 1.83 | n.s. |
| 72 h post-TBI | ||||
| 10745670 | Ccl6 | Chemokine (C-C motif) ligand 6 | 1.65 | n.s. |
| 10940473 | Cd36 | CD36 molecule | 2.97 | n.s. |
| 10777232 | Cd38 | CD38 molecule | 1.61 | n.s. |
| 10767388 | Cd55 | CD55 molecule | 1.78 | n.s. |
| 10858626 | Cd163 | CD163 molecule | 1.81 | n.s. |
| 10803110 | Cd226 | CD226 molecule | 1.59 | n.s. |
| 10765625 | Cd244 | CD244 molecule | 1.67 | n.s. |
| 10764069 | Chi3l1 | Chitinase 3-like 1 | 1.75 | n.s. |
| 10897428 | Csf2rb | Colony stimulating factor 2 receptor, beta, low-affinity (granulocyte-macrophage) | 1.60 | n.s. |
| 10763808 | Ctse | Cathepsin E | 1.83 | n.s. |
| 10924245 | Cxcr2 | Cxcr2 | 1.79 | n.s. |
| 10936899 | Cybb | Cytochrome b-245, beta polypeptide | 1.66 | n.s. |
| 10711575 | Dmbt1 | Deleted in malignant brain tumors 1 | 1.59 | n.s. |
| 10820586 | F2r | Coagulation factor II (thrombin) receptor | 1.57 | n.s. |
| 10812589 | F2rl2 | Coagulation factor II (thrombin) receptor-like 2 | 2.49 | n.s. |
| 10794734 | F13a1 | Coagulation factor XIII, A1 polypeptide | 1.72 | n.s. |
| 10813392 | Fyb | FYN binding protein (FYB-120/130) | 1.63 | n.s. |
| 10797527 | Gadd45g | Growth arrest and DNA-damage-inducible, gamma | n.s. | 1.67 |
| 10735317 | Gp1ba | Glycoprotein Ib (platelet), alpha polypeptide | 1.62 | n.s. |
| 10897805 | Grap2 | GRB2-related adaptor protein 2 | 2.38 | n.s. |
| 10923036 | Gulp1 | GULP, engulfment adaptor PTB domain containing 1 | 1.53 | n.s. |
| 10821370 | Gzma | Granzyme A | 1.61 | n.s. |
| 10860351 | Hgf | Hepatocyte growth factor | 1.60 | n.s. |
| 10806122 | Hmox1 | Heme oxygenase (decycling) 1 | 1.95 | n.s. |
| 10862842 | Hpgds | Hematopoietic prostaglandin D synthase | 1.78 | n.s. |
| 10729791 | Ifit1 | Interferon-induced protein with tetratricopeptide repeats 1 | 8.60 | n.s. |
| 10774141 | Igfbp1 | Insulin-like growth factor binding protein 1 | 2.08 | 1.92 |
| 10708015 | Isg20 | Interferon stimulated exonuclease gene 20 | 1.80 | n.s. |
| 10821851 | Il7r | Interleukin 7 receptor | 2.24 | n.s. |
| 10909874 | Il18 | Interleukin 18 | 1.55 | n.s. |
| 10832306 | Itgb2 | Integrin, beta 2 | 1.62 | n.s. |
| 10806585 | Junb | Jun B proto-oncogene | 1.75 | n.s. |
| 10844331 | Lcn2 | Lipocalin 2 | 3.51 | n.s. |
| 10779673 | Lgals3 | Lectin, galactoside-binding, soluble, 3 | 1.87 | n.s. |
| 10902547 | Lyz2 | Lysozyme 2 | 1.69 | n.s. |
| 10930693 | Marco | Macrophage receptor with collagenous structure | 1.63 | n.s. |
| 10907913 | Mmp8 | Matrix metallopeptidase 8 | 4.99 | n.s. |
| 10796476 | Mrc1 | Mannose receptor, C type 1 | 1.52 | n.s. |
| 10855506 | Npy | Neuropeptide Y | 2.20 | n.s. |
| 10775914 | Pf4 | Platelet factor 4 | 1.54 | n.s. |
| 10873341 | Pla2g2a | Phospholipase A2, group IIA (platelets, synovial fluid) | 1.60 | n.s. |
| 10768376 | Pla2g4a | Phospholipase A2, group IVA (cytosolic, calcium-dependent) | 1.63 | n.s. |
| 10771406 | Plac8 | Placenta-specific 8 | 1.86 | n.s. |
| 10778558 | Plek | Pleckstrin | 2.06 | n.s. |
| 10775918 | Ppbp | Pro-platelet basic protein (chemokine (C-X-C motif) ligand 7) | 3.56 | n.s. |
| 10810018 | Prdx2 | Peroxiredoxin 2 | 2.00 | n.s. |
| 10837381 | Prg2 | Proteoglycan 2, bone marrow | 1.72 | n.s. |
| 10768138 | Ptprc | Protein tyrosine phosphatase, receptor type, C | 1.63 | n.s. |
| 10742194 | Pttg1 | Pituitary tumor-transforming 1 | 1.75 | n.s. |
| 10817071 | S100a8 | S100 calcium binding protein A8 | 1.77 | n.s. |
| 10824695 | S100a9 | S100 calcium binding protein A9 | 2.78 | n.s. |
| 10752744 | Samsn1 | SAM domain, SH3 domain and nuclear localization signals, 1 | 1.64 | n.s. |
| 10765186 | Sell | Selectin L | 1.90 | n.s. |
| 10759173 | Selplg | Selectin P ligand | 1.51 | n.s. |
| 10763367 | Serpinb2 | Serpin peptidase inhibitor, clade B (ovalbumin),member 2 | 1.83 | n.s. |
| 10747813 | Slc4a1 | Solute carrier family 4 (anion exchanger), member 1 | 6.09 | n.s. |
| 10862820 | Snca | Synuclein, alpha (non A4 component of amyloid precursor) | 2.87 | n.s. |
| 10901996 | Socs2 | Suppressor of cytokine signaling 2 | n.s. | 0.37 |
| 10833180 | Srgn | Serglycin | 1.65 | n.s. |
| 10856525 | Tacr1 | Tachykinin receptor 1 | 2.41 | n.s. |
| 10746976 | Top2a | Topoisomerase (DNA) II alpha | 1.91 | n.s. |
| 10709093 | Ucp2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 2.65 | n.s. |
| 7 days post-TBI | ||||
| 10901166 | Angptl4 | Angiopoietin-like 4 | n.s. | 0.55 |
| 10751931 | Bcl6 | B cell CLL/lymphoma 6 | n.s. | 2.30 |
| 10727260 | Ccnd1 | Cyclin D1 | 0.59 | n.s. |
| 10940473 | Cd36 | CD36 molecule (thrombospondin receptor) | 1.98 | n.s. |
| 10828827 | Cdkn1a | Cyclin-dependent kinase inhibitor 1A | 0.62 | n.s. |
| 10732652 | Dusp1 | Dual specificity phosphatase 1 | n.s. | 0.54 |
| 10769825 | Fcer1g | Fc fragment of IgE, high affinity I, receptor for; gamma polypeptide | n.s. | 0.65 |
| 10729791 | Ifit1 | Interferon-induced protein with tetratricopeptide repeats 1 | 9.90 | n.s. |
| 10804463 | Lox | Lysyl oxidase | 0.43 | n.s. |
| 10855506 | Npy | Neuropeptide Y | n.s. | 0.59 |
| 10775914 | Pf4 | Platelet factor 4 | 1.50 | n.s. |
| 10775918 | Ppbp | Pro-platelet basic protein (chemokine (C-X-C motif) ligand 7) | 1.81 | n.s. |
| 10901996 | Socs2 | Suppressor of cytokine signaling 2 | n.s. | 0.25 |
| 10709093 | Ucp2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 2.46 | n.s. |
aFold change in gene expression
n.s. not significant
Table V.
Erythropoietin (EPO) and Anakinra Ingenuity Canonical Pathway Analysis
| Pathways | p value | Genes |
|---|---|---|
| 24 h post-TBI: EPO | ||
| Circadian rhythm signaling | 9.55E−05 | PER3, ARNTL, CRY1 |
| IL-17A signaling in fibroblasts | 4.07E−03 | JUN, LCN2 |
| IL-6 signaling | 4.17E−03 | IL-18, JUN, A2M |
| IL-12 signaling and production in macrophages | 4.27E−03 | ALOX15, IL18, JUN |
| Glucocorticoid receptor signaling | 4.90E−03 | JUN, DUSP1, CDKN1A, A2M |
| Acute phase response signaling | 1.05E−02 | ALOX15, IL18, JUN |
| Eicosanoid signaling | 1.07E−02 | ALOX15, LTC4S |
| ATM signaling | 1.12E−02 | JUN, CDKN1A |
| RAR activation | 1.12E−02 | CSF2RB, JUN, DUSP1 |
| IL-10 signaling | 1.58E−02 | IL-18, JUN |
| Crosstalk between dendritic cells and natural killer cells | 1.62E−02 | CSF2RB, IL-18 |
| IL-3 signaling | 1.66E−02 | CSF2RB, JUN |
| Leukotriene biosynthesis | 2.57E−02 | LTC4S |
| PPAR signaling | 2.82E−02 | IL-18, JUN |
| GADD45 signaling | 4.79E−02 | CDKN1A |
| Aryl hydrocarbon receptor signaling | 5.01E−02 | JUN, CDKN1A |
| 72 h post-TBI: EPO | ||
| Eicosanoid signaling | 1.62E−03 | ALOX15, PLA2G4A, PLA2G2A, HPGDS, DPEP2 |
| Fc epsilon RI signaling | 5.50E−03 | PLA2G4A, RAC2, GRAP2, FCER1G, MAP2K3, PLA2G2A |
| Phospholipases | 8.71E−03 | HMOX1, PLA2G4A, PLA2G2A, PLA1A |
| Oncostatin M signaling | 1.51E−02 | TIMP3, OSMR, CHI3L1 |
| GADD45 signaling | 2.95E−02 | PCNA, ADD45A |
| Leukocyte extravasation signaling | 5.75E−02 | ITGB2, RAC2, TIMP3, MMP8, CYBB, SELPLG |
| IL-8 signaling | 1.28E−01 | ITGB2, HMOX1, RAC2, CXCR2, CYBB |
| 72 h post-TBI: anakinra | ||
| PXR/RXR activation | 0.0002 | NR1l3, IGFBP1, CYP2B6 |
| GADD45 signaling | 0.0005 | GADD45A, GADD45G |
| VDR/RXR activation | 0.0098 | GADD45A, IGFBP1 |
| Acute phase response signaling | 0.0389 | IL1RN, SOCS2 |
| 7 days post-TBI: EPO | ||
| GADD45 signaling | 6.76E−03 | CDKN1A, CND1 |
| TR/RXR activation | 1.82E−02 | UCP2, CYP7A1, NCOA4 |
Abbreviations: A2M alpha-2-macroglobulin; ALOX15 arachidonate 15-lipoxygenase; ARNTL aryl hydrocarbon receptor nuclear translocator-like; CCND1 cyclin D1; CDKN1A cyclin-dependent kinase inhibitor 1A; CHI3L1 chitinase 3-like 1; CRY1 cryptochrome 1 (photolyase-like); CSF2RB colony stimulating factor 2 receptor, beta, low-affinity; CXCR2 chemokine (C-X-C motif) receptor 2; CYP2B6 cytochrome P450, family 2, subfamily b, polypeptide 3; CYP7A1 cytochrome P450, family 7, subfamily a, polypeptide 1; CYPP cytochrome b-245, beta polypeptide; DUSP1 dual specificity phosphatase 1; DPEP2 dipeptidase 2; FCER1G Fc fragment of IgE, high affinity I, receptor for; gamma polypeptide; GRAP2 GRB2-related adaptor protein 2; GADD45A growth arrest and DNA-damage-inducible, alpha; GADD45G growth arrest and DNA-damage-inducible, gamma; IGFBP1 insulin-like growth factor 2 mRNA binding protein 1; HMOX1 heme oxygenase (decycling) 1; HPGDS hematopoietic prostaglandin D synthase; IL1RN interleukin 1 receptor antagonist; IL-18 interleukin 18, nintegrin, beta 2; JUN Jun oncogene; LCN2 lipocalin 2; LTC4S leukotriene C4 synthase; MAP2K3 mitogen-activated protein kinase kinase 3; MMP8 matrix metallopeptidase 8; NCOA4 nuclear receptor coactivator 4; Nr1i3 nuclear receptor subfamily 1, group I, member 3; OSMR oncostatin M receptor; PER3 period homolog 3; PLA1A phospholipase A1 member A; PLA2G2A phospholipase A2, group IIA; PLA2G4A phospholipase A2, group IVA; RAC2 ras-related C3 botulinum toxin substrate 2 (rho family, small GTP binding protein Rac2); SELPLG selectin P ligand; TIMP3 TIMP metallopeptidase inhibitor 3; SOCS2 suppressor of cytokine signaling 2; UCP2 uncoupling protein 2
Treatment with EPO or anakinra in CCI animals resulted in minimal effects on the expression of genes of the hepatic metabolizing enzymes (Table VI). Injured animals treated with EPO had a decreased expression of Cyp2d4 at 72 h, the only CYP2D present in the brain, but also present in the liver (37). Anakinra decreased the expression of Sult2a1 at 72 h compared to sham controls. There was no effect of either EPO or anakinra on the gene expression of any of the major transporters involved in endogenous drug transports.
TABLE VI.
Effect of Erythropoietin and Anakinra on Hepatic Metabolizing Enzymes in TBI Animals. Genes Differentially Expressed (>1.5-fold change, p < 0.05)
| Affymetrix ID | Gene symbol | Genes | a | a |
|---|---|---|---|---|
| 24 h post-TBI | ||||
| 10875324 | Cyp7a1 | Cytochrome P450, family 7, subfamily a, polypeptide 1 | n.s. | 1.93 |
| 10863686 | Nat8 | N-Acetyltransferase 8 | 1.87 | 1.51 |
| 72 h post-TBI | ||||
| 10905721 | Cyp2d4 | Cytochrome P450, family 2, subfamily d, polypeptide 4 | 0.65 | n.s. |
| 10914411 | Cyp8b1 | Cytochrome P450, family 8, subfamily b, polypeptide 1 | 0.62 | n.s. |
| 10719187 | Sult2a1 | Sulfotransferase family 2A, dehydroepiandrosterone (DHEA)-preferring, member 1 | n.s. | 0.66 |
| 7 days post-TBI | ||||
| 10930766 | Cyp2c12 | Cytochrome P450, family 2, subfamily c, polypeptide 12 | n.s. | 1.51 |
| 10875324 | Cyp7a1 | Cytochrome P450, family 7, subfamily a, polypeptide 1 | 2.11 | 2.55 |
| 10730599 | Cyp17a1 | Cytochrome P450, family 17, subfamily a, polypeptide 1 | 0.40 | n.s. |
| 10769385 | Fmo3 | Flavin containing monooxygenase 3 | 0.61 | n.s. |
| 10855008 | Gstk1 | Glutathione S-transferase kappa 1 | n.s. | 0.66 |
| 10771936 | Ugt2a3 | UDP glucuronosyltransferase 2 family, polypeptide A3 | 0.63 | n.s. |
aFold change in gene expression
n.s. not significant
Effect of TBI, EPO, and Anakinra on Protein Expression of CYP2D6, CYP3A1, EPHX1, or UGT2B7
As shown in Fig. 2, CYP2D4 protein levels were significantly decreased by EPO treatment at 72 h and 7 days post-TBI compared to the 24-h time point. CYP3A1 was only significantly decreased at the 24-h time point in the EPO-treated animals compared to the 72-h and 7-day time points. There was no effect of TBI or anakinra treatment on levels of CYP2D4, CYP3A1, EPHX1, or UGT2B7.
Fig. 2.
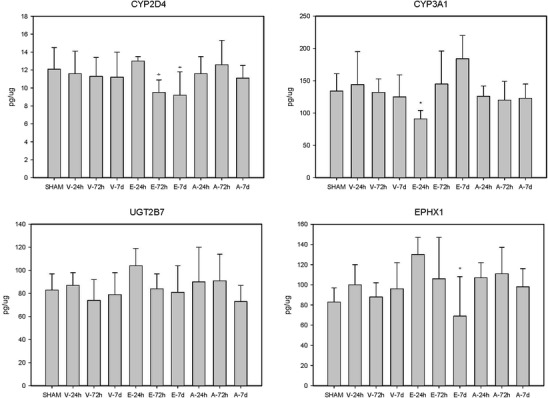
The effect of vehicle (V), erythropoietin (E), and anakinra (A) in animals on liver protein expression. Abbreviations CYP2D4 cytochrome P450 2D6, CYP3A1 cytochrome P450 3A1, EPHX1 epoxide hydrolase 1, UGT2B7 UDP glucuronosyltransferases 2B7. CYP2D4: *p < 0.01 compared to EPO 24 h; CYP3A1: *p < 0.02 compared to EPO 7 days; EPHX1: *p < 0.05 compared to EPO 24 h
Validation of Microarray Data
Based on differential expression, as assessed by microarray data analysis, 11 genes (Aldh1b1, Cyp4a1, Cyp4a2, Cyp51, Cyp7a1, Ephx1, Fmo3, Il1b, Nat8, Sult2a1, Ugt2b7) were chosen for q-RT-PCR validation. The array data indicated that these genes were differentially expressed only in certain but not all contrast as described in the manuscript. q-RT-PCR analysis confirmed differential expression observed with microarray analysis. Figure 3 shows the gene expression results of these 11 genes in the liver generated by microarray and qPCR (normalized to beta-actin). The qPCR findings were highly correlated with the microarray data (Pearson’s correlation = 0.77). Supplemental Figure 1 shows in more detail the differential expression of these 11 genes for all contrasts for both the microarray and q-RT-PCR data.
Fig. 3.
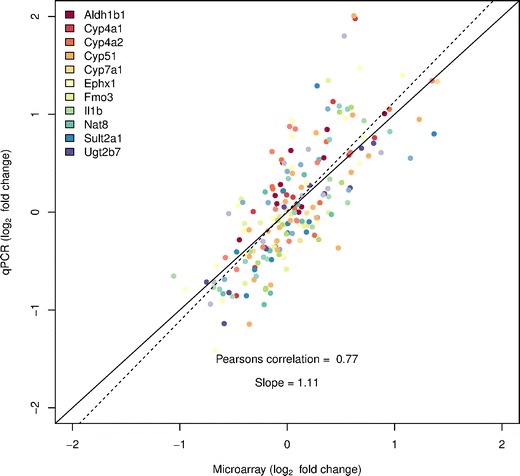
TaqMan-based RT-PCR validation of the microarray data for the selected genes: Aldh1b1 (aldehyde dehydrogenase 1 family, member B1), Cyp4a1 (cytochrome P450, family 4, subfamily a, polypeptide 1), Cyp4a2 (cytochrome P450, family 4, subfamily a, polypeptide 2), Cyp51 (cytochrome P450, family 51), Cyp7a1 (cytochrome P450, family7, subfamily a, polypeptide 1), Ephx1 (epoxide hydrolase 1), Fmo3 (flavin containing monooxygenase 1), Il1b (interleukin 1 beta), Nat8 (N-acetyltransferase 8), Sult2a1 (sulfotransferase family 2A, dehydroepiandrosterone (DHEA)-preferring, member 1), Ugt2b7 (UDP glucuronosyltransferase 2 family, polypeptide B7). The RT-PCR data shown in the figure was normalized to the housekeeping gene β-actin
DISCUSSION
TNF-α, IL-1β, and IL-6, the inflammatory mediators shown to be correlated with a decrease in CYP activity with infection and inflammation, are present in the cerebral spinal fluid (CSF) and serum of patients after a TBI (8). IL-6 serum concentrations were significantly lower and decreased significantly more rapidly in patients with mild/moderate TBI (Glasgow coma scale (GCS > 8)) compared to those in patients with more severe TBI (38). Shiozaki et al. compared patients with isolated TBI to those with TBI plus other injuries (39). Serum concentrations of TNF-α, IL-1β, and IL-10 at 6 h post-injury were significantly less than CSF concentrations in patients with isolated TBI. In contrast, in patients with TBI plus multiple injuries, serum concentrations exceeded CSF concentrations. Seekeamp et al. (40) also found that IL-6, IL-8, and IL-10 serum concentrations were significantly higher in patients with multiple injuries compared to TBI alone when measured out to 7 days post-injury. IL-6 was increased to ~150 pg/mL in days 1 and 2 in the isolated TBI patients and only stayed elevated in patients with multiple injuries, with an average IL-6 concentration of ~350 pg/mL. IL-8 and IL-10 serum concentrations were significantly elevated in day 1 after injury only in patients with TBI plus multiple injuries and stayed at low concentrations for at least 7 days.
In experimental rodent models of TBI, brain concentrations of TNF-α, IL-1β, and IL-6 increase rapidly after TBI and returned to below the detection limit within 24 h post-injury (41–43). Maegele et al. compared plasma concentrations in rodents with a severe TBI alone, peripheral bone fracture alone, or TBI plus fracture (32). TNF-α peak plasma concentrations were detectable within 30 min post-injury and returned to baseline by 6 h for TBI alone and TBI plus fracture. IL-1β plasma concentrations peaked at 6 and 48 h with TBI plus fracture and TBI alone, respectively. IL-6 plasma concentrations remained elevated for at least 7 days with significantly higher concentrations at 6 h post-injury with TBI plus fracture. IL-10 serum concentrations were only significantly elevated with TBI plus fracture and remained elevated for 7 days. Therefore, in rodents, the inflammatory response to experimentally induced TBI with and without multiple injuries does appear to mimic the inflammatory response found in humans with TBI. There is increased systemic exposure to inflammatory cytokines in humans and animals with TBI. The magnitude of exposure depends upon the severity of the TBI and the presence or absence of other injuries.
Clinically, the significant increase in CYP- and UGT-dependent metabolism occurs within 2 days after the TBI, peaking at 2–3 weeks, and is still increased at 30 days depending on whether the patient had an isolated TBI or had concurrent non-TBI (13). In patients receiving valproate, the severity of the TBI, the presence of non-TBI, the presence of ethanol at the time of injury, age, and whether or not the patient had a neurosurgical procedure (craniotomy/craniectomy) affected the magnitude and time course of the increased unbound clearance (Clu) (13). The severity of the TBI based on GCS was correlated with Clu, with the more severe TBI having a higher increase in Clu. If patients had a TBI in combination with other injuries, the average Clu was 51% higher at week 4 compared to patients with isolated TBI. Patients with age ≥40 years had a maximum increase in Clu 37% greater at 3 weeks after TBI than was found in the younger patients. The presence of ethanol at the time of TBI increased Clu by an average of 14% for the first 3 weeks post-injury.
Although plasma concentrations of IL-6 were increased in our CCI model, the increase was significantly less than found clinically in TBI patients, especially those with TBI plus multiple injuries. In both animal models and in patients, TBI plus multiple injury patients have higher IL-6 concentrations and, clinically, valproate metabolism was greater in patients with multiple injuries compared to isolated TBI (13). Conversely, in patients receiving a bone marrow transplant, serum IL-6 increased on average to 212 pg/mL and was associated with a threefold decrease in Cyp3A4-dependent cyclosporine metabolism (44). This further suggests that IL-6 does not play a role in the TBI-associated increased metabolism. All of the patient factors associated with increasing the magnitude and duration of Clu have been reported to cause a shift to the anti-inflammatory mediators, IL-4 and IL-10, from pro-inflammatory mediators in experimental models (40,45–47). Therefore, the increase in hepatic metabolism after TBI may be due to the increased presence of anti-inflammatory mediators in contrast to the inhibition effect of the pro-inflammatory mediators in non-TBI inflammation and infection. Although the administration of IL-10 (8 μg/kg for 6 days) to healthy subjects resulted in a slight decrease in the activity of CYP3A4 (midazolam) and no effect on CYP1A2 (caffeine), CYP2C9 (tolbutamide), or CYP2D6 (dextromethorphan) on days 4 and 5 (48), the estimated IL-10 serum concentrations after exogenous administration would have been 1000-fold higher (ng/mL) (49) than exogenously produced in TBI patients (pg/mL).
Clinically, there is also evidence of a selective effect on CYP isozymes in general trauma patients; 20 of 23 did not have a TBI (50). Probe substrates were administered during weeks 1 to 4 after injury. The metabolism of flurbiprofen (CYP2C9) and dapsone (CYP3A4, CYP2C9) was increased. In contrast, the metabolism of mephenytoin (CYP2C19) and chlorzoxazone (CYP2E1) was decreased.
In contrast to the increase in CYP and UGT activity found in patients after TBI, in the CCI model, there was no effect on the gene expression of any of the Cyp1, Cyp2, and Cyp3 subfamily of enzymes or transporters and a decrease in expression of phase II metabolism genes. Animals were only evaluated at 24 h, 72 h, and 7 days post-injury which is a limitation, as both early (<24 h) and delayed effects (>7 days) were not evaluated. There is limited evidence that TBI may alter CYP in the CCI model. Toler et al. (51) found no change in total CYP content and CYP2C11 and CYP3A protein amount or activity at 24 h after injury, although mRNA levels of both were decreased. Kalsotra et al. (52) found a decrease in total CYP450 content at 24 h and an increase at 2 weeks. CYP3A protein and activity were not altered at 24 h and were significantly increased at 2 weeks. Conversely, CYP1A was decreased at both time points and there was no effect on CYP2B, CYP2D, or CYP4F sub-families. Poloyac et al. (53) reported a decrease in Cyp2E1 activity 24 and 48 h following TBI.
EPO treatment also resulted in significant effects on the expression of genes involved in the inflammatory process similar to the effect found in the brain (27). IL-6 plasma concentrations were decreased at 24 and 72 h. EPO increased the expression of IL-18 at 72 h. IL-18 has been described as an IFN-γ-inducing factor (54). IL-18 binds to the IL-18 receptor and induces cell-mediated immunity, eventually resulting in the release of IFN-γ. IFN-γ has been associated with decreased expression of hepatic metabolic enzymes (55). EPO treatment decreased the gene expression of Cyp2d4 at 72 h with a corresponding decrease in CYP2D4 protein levels at 72 h and 7 days. CYP3A1 concentration was also decreased at 24 h compared to that at 7 days. Although there was not a statistically significant difference between vehicle treated and EPO treated at the corresponding time points, possibly due to the larger variability in the vehicle-treated animals, the concentrations are consistent with a decrease due to EPO treatment. The lack of effect of anakinra on hepatic metabolic enzymes was also consistent with the minimal effect on the expression of the gene involved in the inflammatory process in the liver. EPO and anakinra treatments were only evaluated in TBI animals which is a limitation.
CONCLUSIONS
IL-6 has been shown to be the mediator most responsible for downregulating Cyp activity in infection and inflammation. Although plasma concentrations of IL-6 were increased in our CCI model, the increase was significantly less than found clinically in TBI patients and there was no effect of TBI on the expression of Cyp and Ugt. This is in contrast to the large increase in Cyp and UGT metabolism that occurs in patients after a TBI, with the increased metabolism greatest in patients with the more severe injuries and highest IL-6 concentrations. In our CCI model, EPO treatment decreased plasma concentrations of IL-6 with a corresponding decrease in Cyp expression, opposite the effect found in models of infection and inflammation. Therefore, the pro-inflammatory cytokines do not appear to play a major role in the regulation of Cyp activity after TBI.
Electronic Supplementary Material
Barplots showing the consistency between microarray and qPCR data. Each individual plot shows data for a given contrast, indicated in the header for the plot. The horizontal axis separates the data by gene, and the vertical axis presents the log fold change between the two sample types, which can be inferred by the plot header (e.g., EPO_24 hrs vs SHAM indicates the comparison is EPO 24 hours - SHAM, so a positive fold change indicates up-regulation in EPO treated samples, and a negative fold change indicates down-regulation in EPO treated samples). Asterisks indicate significance - a single asterisk indicates p < 0.05 and two asterisks indicate p < 0.005. (PDF 38.5 kb)
Effect of TBI on Brain Inflammatory Genes: Genes differentially expressed (>1.5 fold change, p<0.05) (PDF 42 kb)
Ingenuity Canonical Pathways: Vehicle Sham Contrast (PDF 65 kb)
Ingenuity Canonical Pathways: EPO Sham Contrast (PDF 64 kb)
Ingenuity Canonical Pathways: Anakinra Vehicle Contrast (PDF 53 kb)
Acknowledgments
The research was supported by a grant from the National Institutes of Health/National Institute of Child, Health and Development (R01 HD061944-01) and by the NIEHS Center for Ecogenetics & Environmental Health (P30ES007033).
References
- 1.Statler KD, Jenkins LW, Dixon CE, Clark RS, Marion DW, Kochanek PM. The simple model versus the super model: translating experimental traumatic brain injury research to the bedside. J Neurotrauma. 2001;18(11):1195–206. doi: 10.1089/089771501317095232. [DOI] [PubMed] [Google Scholar]
- 2.Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002;19(5):503–57. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bullock MR, Lyeth BG, Muizelaar JP. Current status of neuroprotection trials for traumatic brain injury: lessons from animal models and clinical studies. Neurosurgery. 1999;45(2):207–17. doi: 10.1097/00006123-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Galanopoulou AS, Buckmaster PS, Staley KJ, Moshe SL, Perucca E, Engel J, Jr, et al. Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia. 2012;53(3):571–82. doi: 10.1111/j.1528-1167.2011.03391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang KK, Larner SF, Robinson G, Hayes RL. Neuroprotection targets after traumatic brain injury. Curr Opin Neurol. 2006;19(6):514–9. doi: 10.1097/WCO.0b013e3280102b10. [DOI] [PubMed] [Google Scholar]
- 6.Jennings JS, Gerber AM, Vallano ML. Pharmacological strategies for neuroprotection in traumatic brain injury. Mini Rev Med Chem. 2008;8(7):689–701. doi: 10.2174/138955708784567377. [DOI] [PubMed] [Google Scholar]
- 7.McConeghy KW, Hatton J, Hughes L, Cook AM. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs. 2012;26(7):613–36. doi: 10.2165/11634020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boucher BA, Hanes SD. Pharmacokinetic alterations after severe head injury. Clin Pharmacokinet. 1998;35:209–21. doi: 10.2165/00003088-199835030-00004. [DOI] [PubMed] [Google Scholar]
- 10.Anderson GD, Gidal BE, Hendryx RJ, Awan AB, Temkin NR, WA J. Decreased plasma protein binding of valproate in patients with acute head trauma. Br J Clin Pharmacol. 1994;37:559–62. doi: 10.1111/j.1365-2125.1994.tb04304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boucher B, Rodman J, Jaresko G, Rasmussen S, Watridge C, Fabian T. Phenytoin pharmacokinetics in critically ill trauma patients. Clin Pharmacol Ther. 1988;44:675–83. doi: 10.1038/clpt.1988.211. [DOI] [PubMed] [Google Scholar]
- 12.Boucher B, Kuhl D, Fabian T, Robertson J. The effect of neurotrauma on hepatic drug clearance. Clin Pharmacol Ther. 1991;50:487–97. doi: 10.1038/clpt.1991.173. [DOI] [PubMed] [Google Scholar]
- 13.Anderson GD, Temkin NR, Awan AB, Winn RH. Effect of time, injury, age and ethanol on interpatient variability in valproic acid pharmacokinetics after traumatic brain injury. Clin Pharmacokinet. 2007;46(4):307–18. doi: 10.2165/00003088-200746040-00004. [DOI] [PubMed] [Google Scholar]
- 14.Anderson GD, Awan A, Adams C, Temkin N, Winn H. Increases in metabolism of valproate and excretion of 6b-hydroxycortisol in patients with traumatic brain injury. Br J Clin Pharmacol. 1998;45:101–95. doi: 10.1046/j.1365-2125.1998.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46:123–49. doi: 10.1146/annurev.pharmtox.46.120604.141059. [DOI] [PubMed] [Google Scholar]
- 16.Renton KW. Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr Drug Metab. 2004;5(3):235–43. doi: 10.2174/1389200043335559. [DOI] [PubMed] [Google Scholar]
- 17.Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85(4):434–8. doi: 10.1038/clpt.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu X, Ke S, Liu D, Sheng T, Thomas PE, Rabson AB, et al. Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J Biol Chem. 2006;281(26):17882–9. doi: 10.1074/jbc.M601302200. [DOI] [PubMed] [Google Scholar]
- 19.Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A, et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116(8):2280–9. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khatsenko OG, Gross SS, Rifkind AB, Vane JR. Nitric oxide is a mediator of the decrease in cytochrome P450-dependent metabolism caused by immunostimulants. Proc Natl Acad Sci U S A. 1993;90(23):11147–51. doi: 10.1073/pnas.90.23.11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boogaerts M. Pleiotropic effects of erythropoietin in neuronal and vascular systems. Curr Med Res Opin. 2006;22(Suppl.4):S15–S22. doi: 10.1185/030079906X159588. [DOI] [Google Scholar]
- 22.Bartfai T, Sanchez-Alavez M, Andell-Jonsson S, Schultzberg M, Vezzani A, Danielsson E, et al. Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Ann N Y Acad Sci. 2007;1113:173–7. doi: 10.1196/annals.1391.022. [DOI] [PubMed] [Google Scholar]
- 23.Hutchinson PJ, O’Connell MT, Rothwell NJ, Hopkins SJ, Nortje J, Carpenter KL, et al. Inflammation in human brain injury: intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. J Neurotrauma. 2007;24(10):1545–57. doi: 10.1089/neu.2007.0295. [DOI] [PubMed] [Google Scholar]
- 24.Quigley A, Tan AA, Hoane MR. The effects of hypertonic saline and nicotinamide on sensorimotor and cognitive function following cortical contusion injury in the rat. Brain Res. 2009;1304:138–48. doi: 10.1016/j.brainres.2009.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goffus AM, Anderson GD, Hoane M. Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid Med Cell Longev. 2010;3(2):145–52. doi: 10.4161/oxim.3.2.11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson GD, Farin FM, Bammler TK, Beyer RP, Swan AA, Wilkerson HW, et al. The effect of progesterone dose on gene expression after traumatic brain injury. J Neurotrauma. 2011;28(9):1827–43. doi: 10.1089/neu.2011.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson GD, Peterson TC, Vonder Haar C, Kantor ED, Farin FM, Bammler TK, et al. Comparison of the effects of erythropoietin and anakinra on functional recovery and gene expression in a traumatic brain injury model. Front Pharmacol. 2013;4:129. doi: 10.3389/fphar.2013.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26(19):2363–7. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, et al. Empirical array quality weights in the analysis of microarray data. BMC Bioinform. 2006;7:261. doi: 10.1186/1471-2105-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Consortium M, Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24(9):1151–61. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maegele M, Sauerland S, Bouillon B, Schafer U, Trubel H, Riess P, et al. Differential immunoresponses following experimental traumatic brain injury, bone fracture and “two-hit”-combined neurotrauma. Inflamm Res. 2007;56(8):318–23. doi: 10.1007/s00011-007-6141-3. [DOI] [PubMed] [Google Scholar]
- 33.Ishii Y, Iida N, Miyauchi Y, Mackenzie PI, Yamada H. Inhibition of morphine glucuronidation in the liver microsomes of rats and humans by monoterpenoid alcohols. Biol Pharm Bull. 2012;35(10):1811–7. doi: 10.1248/bpb.b12-00568. [DOI] [PubMed] [Google Scholar]
- 34.Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene. 2013;526(2):61–74. doi: 10.1016/j.gene.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coles BF, Kadlubar FF. Human alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Methods Enzymol. 2005;401:9–42. doi: 10.1016/S0076-6879(05)01002-5. [DOI] [PubMed] [Google Scholar]
- 36.Falany CN, Comer KA, Dooley TP, Glatt H. Human dehydroepiandrosterone sulfotransferase. Purification, molecular cloning, and characterization. Ann N Y Acad Sci. 1995;774:59–72. doi: 10.1111/j.1749-6632.1995.tb17372.x. [DOI] [PubMed] [Google Scholar]
- 37.Hiroi T, Imaoka S, Chow T, Funae Y. Tissue distributions of CYP2D1, 2D2, 2D3 and 2D4 mRNA in rats detected by RT-PCR. Biochim Biophys Acta. 1998;1380(3):305–12. doi: 10.1016/S0304-4165(97)00157-8. [DOI] [PubMed] [Google Scholar]
- 38.McClain C, Cohen D, Phillips R, Ott L, Young B. Increased plasma and ventricular fluid interleukin-6 levels in patients with head injury. J Lab Clin Med. 1991;118:225–31. [PubMed] [Google Scholar]
- 39.Shiozaki T, Hayakata T, Tasaki O, Hosotubo H, Fuijita K, Mouri T, et al. Cerebrospinal fluid concentrations of anti-inflammatory mediators in early-phase severe traumatic brain injury. Shock. 2005;23(5):406–10. doi: 10.1097/01.shk.0000161385.62758.24. [DOI] [PubMed] [Google Scholar]
- 40.Seekamp A, van Griensven M, Lehmann U, Molituris U, Hildebrandt F, Pohlemann T. Serum IL-6, IL-8 and IL-10 levels in multiple trauma compared to traumatic brain injury and combined trauma. Eur J Trauma. 2002;28:83–9. doi: 10.1007/s00068-002-1134-y. [DOI] [Google Scholar]
- 41.Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72(2):169–77. doi: 10.1016/S0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- 42.Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42(2):177–85. doi: 10.1016/0165-5728(93)90008-M. [DOI] [PubMed] [Google Scholar]
- 43.Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, McIntosh TK. Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res Mol Brain Res. 1995;30(1):125–30. doi: 10.1016/0169-328X(94)00287-O. [DOI] [PubMed] [Google Scholar]
- 44.Chen YL, Le Vraux V, Leneveu A, Dreyfus F, Stheneur A, Florentin I, et al. Acute-phase response, interleukin-6, and alteration of cyclosporine pharmacokinetics. Clin Pharmacol Ther. 1994;55(6):649–60. doi: 10.1038/clpt.1994.82. [DOI] [PubMed] [Google Scholar]
- 45.Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S, et al. Cytokines and alcohol. Alcohol Clin Exp Res. 2006;30(4):720–30. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- 46.Kovacs EJ, Duffner LA, Plackett TP. Immunosuppression after injury in aged mice is associated with a TH1-TH2 shift, which can be restored by estrogen treatment. Mech Ageing Dev. 2004;125(2):121–3. doi: 10.1016/j.mad.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 47.Asadullah K, Woiciechowsky C, Docke WD, Liebenthal C, Wauer H, Kox W, et al. Immunodepression following neurosurgical procedures. Crit Care Med. 1995;23(12):1976–83. doi: 10.1097/00003246-199512000-00006. [DOI] [PubMed] [Google Scholar]
- 48.Gorski JC, Hall SD, Becker P, Affrime MB, Cutler DL, Haehner-Daniels B. In vivo effects of interleukin-10 on human cytochrome P450 activity. Clin Pharmacol Ther. 2000;67(1):32–43. doi: 10.1067/mcp.2000.103860. [DOI] [PubMed] [Google Scholar]
- 49.Huhn RD, Radwanski E, O’Connell SM, Sturgill MG, Clarke L, Cody RP, et al. Pharmacokinetics and immunomodulatory properties of intravenously administered recombinant human interleukin-10 in healthy volunteers. Blood. 1996;87(2):699–705. [PubMed] [Google Scholar]
- 50.Harbrecht BG, Frye RF, Zenati MS, Branch RA, Peitzman AB. Cytochrome P-450 activity is differentially altered in severely injured patients. Crit Care Med. 2005;33(3):541–6. doi: 10.1097/01.CCM.0000155989.54344.E0. [DOI] [PubMed] [Google Scholar]
- 51.Toler SM, Young AB, McClain CJ, Shedlofsky SI, Bandyopadhyay AM, Blouin RA. Head injury and cytochrome P-450 enzymes. Differential effect on mRNA and protein expression in the Fischer-344 rat. Drug Metab Dispos. 1993;21(6):1064–9. [PubMed] [Google Scholar]
- 52.Kalsotra A, Turman CM, Dash PK, Strobel HW. Differential effects of traumatic brain injury on the cytochrome p450 system: a perspective into hepatic and renal drug metabolism. J Neurotrauma. 2003;20(12):1339–50. doi: 10.1089/089771503322686139. [DOI] [PubMed] [Google Scholar]
- 53.Poloyac SM, Perez A, Scheff S, Blouin RA. Tissue-specific alterations in the 6-hydroxylation of chlorzoxazone following traumatic brain injury in the rat. Drug Metab Dispos. 2001;29(3):296–8. [PubMed] [Google Scholar]
- 54.Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin-18, more than a Th1 cytokine. Semin Immunol. 2013;25(6):439–48. doi: 10.1016/j.smim.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 55.Renton KW, Mannering GJ. Depression of hepatic cytochrome P-450-dependent monooxygenase systems with administered interferon inducing agents. Biochem Biophys Res Commun. 1976;73(2):343–8. doi: 10.1016/0006-291X(76)90713-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Barplots showing the consistency between microarray and qPCR data. Each individual plot shows data for a given contrast, indicated in the header for the plot. The horizontal axis separates the data by gene, and the vertical axis presents the log fold change between the two sample types, which can be inferred by the plot header (e.g., EPO_24 hrs vs SHAM indicates the comparison is EPO 24 hours - SHAM, so a positive fold change indicates up-regulation in EPO treated samples, and a negative fold change indicates down-regulation in EPO treated samples). Asterisks indicate significance - a single asterisk indicates p < 0.05 and two asterisks indicate p < 0.005. (PDF 38.5 kb)
Effect of TBI on Brain Inflammatory Genes: Genes differentially expressed (>1.5 fold change, p<0.05) (PDF 42 kb)
Ingenuity Canonical Pathways: Vehicle Sham Contrast (PDF 65 kb)
Ingenuity Canonical Pathways: EPO Sham Contrast (PDF 64 kb)
Ingenuity Canonical Pathways: Anakinra Vehicle Contrast (PDF 53 kb)