

Abstract
Allosteric modulators of G protein-coupled receptors (GPCRs), which target at allosteric sites, have significant advantages against the corresponding orthosteric compounds including higher selectivity, improved chemical tractability or physicochemical properties, and reduced risk of receptor oversensitization. Bitopic ligands of GPCRs target both orthosteric and allosteric sites. Bitopic ligands can improve binding affinity, enhance subtype selectivity, stabilize receptors, and reduce side effects. Discovering allosteric modulators or bitopic ligands for GPCRs has become an emerging research area, in which the design of allosteric modulators is a key step in the detection of bitopic ligands. Radioligand binding and functional assays ([35S]GTPγS and ERK1/2 phosphorylation) are used to test the effects for potential modulators or bitopic ligands. High-throughput screening (HTS) in combination with disulfide trapping and fragment-based screening are used to aid the discovery of the allosteric modulators or bitopic ligands of GPCRs. When used alone, these methods are costly and can often result in too many potential drug targets, including false positives. Alternatively, low-cost and efficient computational approaches are useful in drug discovery of novel allosteric modulators and bitopic ligands to help refine the number of targets and reduce the false-positive rates. This review summarizes the state-of-the-art computational methods for the discovery of modulators and bitopic ligands. The challenges and opportunities for future drug discovery are also discussed.
Key words: allosteric modulators, bitopic ligands, computational approaches, drug discovery, drug target discovery, G protein-coupled receptors
INTRODUCTION
G protein-coupled receptors (GPCRs), also known as 7-transmembrane (7TM) receptors, are the largest family of cell surface receptors in the human genome (1). Receptors of four major classes of human GPCRs (class A, B, C, and F) play key roles in many essential physiological processes including neurotransmission, immune defense, cell growth, cellular metabolism, secretion, and differentiation. GPCRs are targeted by more than 40% of marketed drugs worldwide (2–5), providing treatments for cancer, central nervous system (CNS) disorders, cardiac dysfunction, diabetes, obesity, inflammation, and pain.
Each GPCR has a distinctive binding pocket/site for its endogenous ligands. This so-called orthosteric binding pocket/site (Fig. 1a) can be bound with both native or synthetic ligands termed orthosteric ligands (6,7). Early drug development focused on the orthosteric site (8). Orthosteric ligands (Fig. 1(a)) have the advantages of better lipid solubility, fewer interactions with other proteins in the CNS, and no metabolism by CYP450 (9). However, most orthosteric ligands have poor efficacy, subtype selectivity, and/or resistance (1,6,10–14).
Fig. 1.

Docking results of bitopic ligand-SB269652 at dopamine 2 receptor (D2R). a The binding mode of the orthosteric fragment 1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (THIQ7C) at the orthosteric binding site, b the binding mode of the allosteric fragment at the allosteric binding site (TM1-2-7 region), c the binding mode of the whole structure of SB269652
In addition to the orthosteric binding pockets/sites, GPCRs, kinases, ion channels, caspases, and phospholipases have been recently reported to possess allosteric binding sites (Fig. 1(b)) (1,6,10–14). Each class contains many therapeutically relevant targets, and the detailed statistics data can be found in publication of Huang et al. (15) (or their website http://mdl.shsmu.edu.cn/ASD/). Briefly, allosteric binding sites functionally differ from orthosteric ones, allowing additional receptor–ligand interactions and signaling phenomena. To our knowledge, allosteric ligands (Fig. 1b) of GPCRs can be divided into three types according to their pharmacological effects. Positive allosteric modulators (PAMs) can potentiate agonist-mediated receptor response, while negative allosteric modulators (NAMs) can noncompetitively decrease receptor activity. Silent/neutral allosteric modulators (SAMs) could target allosteric binding sites, which can block the activity of either PAMs or NAMs instead of mediating orthosteric ligand responses (1,6,10–14).
The clinical successes of benzodiazepines and barbiturates, selective PAMs of neurotransmitter γ-aminobutyric acid type A (GABAA) (11,16), made allosteric regulation an attractive field for drug research. The application of allosteric drugs can enhance GABAA receptor activity in the treatment of CNS disorder and overcome shortcomings of traditional orthosteric compounds. The increasing number of both publications and new modulators reflects a general trend of developing allosteric modulators (7,17). Allosteric modulators are found to possess better receptor subtype selectivity when compared with orthosteric ligands (9,11,18,19).
A new class of ligands (20–25), termed bitopic or dualsteric ligands (Fig. 1(c)), have been reported to target at both orthosteric and allosteric sites simultaneously. The development of bitopic ligands is based on the combination of high affinity (via orthosteric sites) and high selectivity (via allosteric sites) (20). Moreover, some recent reports (26,27) showed that bitopic ligands possess either orthosteric or allosteric properties under different conditions. Both allosteric modulators and bitopic ligands of GPCRs possess advantages over orthosteric ones, so the discovery of allosteric modulators or bitopic ligands of GPCRs has become a new strategy in drug design.
Radioligand binding and functional assays ([35S]GTPγS and ERK1/2 phosphorylation) are used to characterize the effects for potential modulators or bitopic ligands (28–31). Kruse et al. (30) showed that LY2119620 (PAM) enhanced the affinity and potency of iperoxo (agonist for M2 muscarinic acetylcholine receptor (M2mAChR)) while activated M2mAChR signaling is measured directly by [35S]GTPγS and ERK1/2 phosphorylation. Lane et al. (29), Shore et al. (31), and Ahn et al. (28) explored the functional features of some allosteric modulators and bitopic ligands by testing for activity of the modulators of known-compound-induced ERK1/2 phosphorylation in GPCRs. Some biochemical experiments (32–34) have been carried out in the discovery of allosteric modulators or bitopic ligands of GPCRs, including high-throughput screening (HTS), disulfide trapping, and fragment-based screening (32–34).
However, experimental methods are costly and time-consuming, and can result in a large number of potential targets, including many false positives. Alternatively, many low-cost and efficient in silico methods based on sequence, structure, and dynamics are recognized as valuable tools to predict the allosteric site. The pharmacophore model and virtual docking screening that are successfully utilized in the discovery of orthosteric ligand are powerful tools for developing the allosteric/bitopic ligands as well. All-atoms molecular dynamics (MD) simulations are also successfully used to probe the detailed interactions of allosteric/bitopic ligands. This work will review some classical strategies of computational approaches and state-of-the-art computational methods to discover allosteric modulators or bitopic ligands of GPCRs.
RECENT ADVANCES OF ALLOSTERIC MODULATORS AND BITOPIC LIGANDS
Allosteric ligands are considerable potential sources of drugs due to clear therapeutic advantages and higher selectivity compared to traditional orthosteric ligands. Two GPCR allosteric modulators have already appeared on the market. Cinacalcet (Sensipar, Fig. 2a), conducted by Amgen, is a PAM that acts as a calcimimetic at the calcium-sensing receptor (CaSR), which was designed using a homology model of GPCR. Cinacalcet (35) (Fig. 2a) was approved by the FDA in 2004 as a treatment for hyperparathyroidism due to the function of CaSR in calcium homeostasis regulation, renal calcium resorption, and the maintenance of intracellular inositol triphosphate levels. Maraviroc (36) (Selzentry), designed by Pfizer Inc., is a NAM of chemokine CC-motif receptor 5 (CCR5) according to the result of HTS, and approved by the FDA in 2007 to treat patients infected with HIV.
Fig. 2.

Chemical structures of allosteric modulators and bitopic ligands discussed in the present work. a The structures of allosteric modulators and b the structures of bitopic ligands
Some GPCR allosteric modulators have entered clinical stages in recent years. Reparixin (37), a NAM for chemokine (C–X–C motif) receptor 1/2 (CXCR1/2), has shown a significant effect in the treatment of reperfusion injury to lung/kidney transplantation, and has entered into clinical trial II/III for further studies. Some PAMs or NAMs for metabotropic glutamate receptor 5 (mGluR5), which were well-studied and hotly debated, have already been carried out in clinical researches. For instance, ADX10059, an mGluR5 NAM, was recognized as the first compound to improve clinical symptoms in gastroesophageal reflux disease (GERD) patients (38). ADX10059 can also be used as an acute treatment for migraine and anxiety (39). When given in combination with levodopa, AFQ056, an mGluR5 NAM, has been shown to reduce the frequency of involuntary motor movements in patients with Parkinson’s disease levodopa-induced dyskinesia (PD-LID) in a phase II clinical trial (40). In addition, ADX48621 (41) (phase II) and other mGluR5 NAMs are also recognized as possible potential treatments for PD-LID. In a phase II clinical trial with AFQ056 (42) and fenobam (43), mGluR5 NAMs have also shown beneficial therapeutic effects in treating fragile X syndrome, which is an inherited cause of mental retardation and autism. Recently, a novel mGluR2 PAM has also entered into the phase II clinical trial testing for anxiety, depression, and schizophrenia (44,45) with promising results. Additional allosteric modulators, including mGluR5 PAMs for schizophrenia (46) and mGluR4 PAMs for Parkinson’s disease (47), are in late discovery or pre-clinical development showing similar clinical potential.
As a developing field, the discovery of bitopic ligands expands the scope of GPCR ligands design. Although no drug is yet on the market, some bitopic ligands show potential therapeutic effects. For instance, the compound SB269652, originally discovered as an antagonist of dopamine D2 receptor (D2R) and dopamine D3 receptor (D3R) (48), was proposed by Silvano et al. (49) as a bitopic ligand. The interaction between SB269652 and D3R was supported by work published by Maggoi et al. (27). Lane et al. (26) also provided evidence to confirm that SB269652 acted as a bitopic ligand at D2R. Their docking results of some representative D3R bitopic ligands were consistent with their previous studies (29): SB269652 shared same charge–charge interaction between the basic amino nitrogen and the carboxyl of Asp114 within the orthosteric pocket. Importantly, SB269652 also formed strong hydrogen bonding with Glu952.65 in the allosteric pocket (TM1-2-7 region).
Muscarinic acetylcholine receptors (mAChRs) are fruitful research objects for the development of bitopic ligands. Tahtaoui et al. (50) synthesized and used several fluorescent derivations of BPDIPY-label pirenzipine (an M1mAChR antagonist) to study ligand–receptor interactions. These analogs might interact with both orthosteric and allosteric binding domains of M1mAChR, and therefore may act as potential bitopic ligands. An M2mAChR partial agonist, McN-A-343, was considered as a bitopic ligand as well. Christopoulos et al. (51) found the reduction of receptor function after mutation of the allosteric site among McN-A-343 derivatives. Their molecular modeling and docking study indicates that the 3-chlorophenylcarbamate moiety of McN-A-343 tended to prolong to extracellular loop 2 (ECL2) region. ECL2 contains the allosteric site of M2mAChR, potentially explaining the previous results (51). Recently, Jo et al. (24) reported a novel bitopic antagonist (SPM-242) for the sphingosine 1-phosphate 3 (S1P3) receptor. They found that SPM-242 in the S1P3 receptor occupied both orthosteric and allosteric sites, which was consistent with the binding site of their novel allosteric agonist CYM-5541. More bitopic ligands with potential therapeutic effects can be found in Fig. 2b.
ALLOSTERIC BINDING SITE PREDICTIONS
Identification of the allosteric binding site is the first step to discover allosteric modulators or bitopic ligands. This section mainly introduces computational approaches for predicting allosteric binding pockets/sites, including sequence-based methods, structure-based approaches, conformational dynamics-based approaches, normal-mode-analysis-based (NMA) approaches, combinations of conformational dynamics-based and NMA-based methods, and other allosteric-related approaches (Table I). Some of these state-of-the-art computational approaches have been reviewed by Lu et al. (52) in their recent paper.
Table I.
The Computational Methods Discussed in the Present Work
| Categories | Name | Basic principle | Merit | Demerit | Representative example |
|---|---|---|---|---|---|
| Sequence-based approaches | Comparisons of sequence conservation (traditional) | Sequence conservation of pocket residues in one protein | Indicate the rearrangements of receptors | Not very accurate | Fenalti et al. Molecular control of δ-opioid receptor signaling. Nature 2014; 506: 191–196 |
| Statistical coupling analysis (traditional) | Multiple sequence alignments (MSA) Correlation information for residues |
Well-established Define effective covariance matrix directly contracted at sequence data level |
Inconsistent results caused by ignoring physical mechanism of energetic coupling | Lockless et al. Evolutionarily conserved pathways of energetic connectivity in protein families. Science, 1999; 286: 295–299 | |
| Structure-based approach | Allosite (state-of-the-art) | Pocket-based analysis Support vector machine (SVM) classifiers |
High accuracy and specificities | Imbalance problem in SVM classification | Huang et al. Allosite: a method for predicting allosteric sites. Bioinformatics 2013; 29: 2357–2359 |
| Conformational dynamics-based approaches | Molecular dynamics (MD) simulation (traditional) | Pocket-based analysis Virtual screening Fragment-based molecular dynamics |
Generate enhanced conformational diversity Show different topographies more representative of GPCR dynamics |
Incomplete conformation sampling for systems of same size | Matosin et al. Metabotropic glutamate receptor mGluR2/3 and mGluR5 binding in the anterior cingulate cortex in psychotic and nonpsychotic depression, bipolar disorder and schizophrenia: implications for novel mGluR-based therapeutics. J. Psychiatr. Neurosci. 2014; 39: 407 |
| Two-state Gō model (state-of-the-art) | Ability to reshape the energy landscape after binding to a ligand | Application on insufficient sampling models | Time-consuming Necessity of bound and unbound structures with difference in the residue–residue contact level |
Qi et al. Identifying allosteric binding sites in proteins with a two-state Gō model for novel allosteric effector discovery. J. Chem. Theory Comput. 2012; 8: 2962–2971 | |
| Normal model analysis-based approach | Protein allosteric and regulatory sites (PARS) (state-of-the-art) | Protein flexibility Structural conservation |
Simple and fast Ability of measuring all structure conservation of each pocket |
Phenomenological variety Accuracy in initial step |
Panjkovich et al. PARS: a web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics 2014; 30: 1314–1315 |
| Combination of conformational dynamics-based and NMA-based approach | SPACER (state-of-the-art) | Thermodynamic view of allostery | Simple and unique Application in a wide range of proteins |
Lowers chance of finding non-catalytic binding sites in proteins | Goncearenco et al. SPACER: server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 2013; 41: W266–W272 |
| Other allosteric-related approach | Ligand binding specificity analysis (LIBSA) (state-of-the- art) | Novel filtering algorithms Signal-to-noise ratios |
Potentially more accurate | Too expensive to be used for virtual screening a large sum of compounds and receptors | Hocker et al. LIBSA—a method for the determination of ligand-binding preference to allosteric sites on receptor ensembles. J. Chem. inf. Model. 2014; 54: 530 |
| Pharmacophore model | Pharmacophore-based virtual screening (traditional) | Built pharmacophore model based on known compounds, and performs virtual docking screening | High accuracy and fast | Not easy to build the pharmacophore model when the known compound is different | Castelli et al. Characterization of COR627 and COR628, two novel positive allosteric modulators of the GABAB receptor. J. Pharmacol. Exp.Ther. 2012; 340: 529–538 |
| Ligand-based similarity search (traditional) | Performed similarity search for the queries | Fast and simple | Hard to find novel compounds | Renner et al. New allosteric modulators of metabotropic glutamate receptor 5 (mGluR5) found by ligand-based virtual screening. ChemBioChem 2005; 6: 620–625 | |
| Support vector machine (SVM) filter (state-of-the-art) | Defined a general profile to filter general screening libraries | Reduces the size of the filtered chemical library and enhances the proportion of active compounds | Time-consuming | Hamon et al. 2P2IHUNTER: a tool for filtering orthosteric protein–protein interaction modulators via a dedicated support vector machine. J. Roy.l Soc. Interface 2013; 11 | |
| Structure-based virtual screening | Crystal structure-based method (state-of-the-art) | Target protein should have the crystal structure | High accuracy and specificities | High resolution-dependent | Chien et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010; 330: 1091–1095 |
| Homology model-based method (traditional) | Built the homology model for the target protein | Moderate accuracy | Not very accurate | Katritch et al. Ligand-guided receptor optimization. Meth. Mol. Biol. 2012; 857: 189–205 | |
| Computational fragment-based drug design for bitopic ligands | Fragment-based method (state-of-the-art) | Design orthosteric features and the allosteric pharmacophore separately | Easy to design and conform the orthosteric feature based on the known orthosteric compounds | Compound may be inactive when combing the active orthosteric and allosteric pharmacophore | Oguievetska et al. Computational fragment-based drug design to explore the hydrophobic sub-pocket of the mitotic kinesin Eg5 allosteric binding site. J. Comput.-Aid. Mol. Des. 2009; 23: 571–582 |
Sequence-Based Approaches
The conservation of amino acid sequences is often used to indicate biological importance. Sequence alignment (comparing sequence conservation) is a traditional way of arranging different sequences (including DNA, RNA, or protein) to identify regions of similarity that may reflect functional, structural, or evolutionary relationships among the sequences. This method is useful to identify similar/identical domains, especially highly conserved, functionally significant residues/binding pockets among proteins (especially within protein families and superfamilies). Moreover, this method is simple and traditional but not very accurate. The accuracy of sequence-based methods mainly relies on the similarity and inferred homology among sequences, which makes it unnecessary to know the 3D structure of the predicted protein.
Recently, Liu et al. (53) reengineered the human A2A adenosine receptor (A2AAR) and identified the precise allosteric site of sodium ion. Based on the conservation of this sodium binding pocket in known GPCR structures, Fenalti et al. (54) presented the 1.8-Å high-resolution crystal structure of the human δ opioid receptor (δ-OR), revealing the fundamental role of the sodium ion for mediating allosteric regulation of receptor subtype selectivity. Xie’s lab also combined the consideration of sequence comparison and residual mutations to confirm this allosteric binding site in cannabinoid receptor 2 (CB2) (5).
Statistical coupling analysis (SCA) (55) is a sequence-based technique of using multiple sequence alignments (MSA) to identify networks of coevolving residues and statistical interactions between amino acid positions in a protein family, providing indications of allosteric communication between functional and allosteric sites (56,57). This assumption was supported by a number of computational predictions of allosteric signaling for thermodynamically linked residues in PDZ domains (58). Recently, Reynolds et al. (59) confirmed this algorithm using model systems of the metabolic enzyme dihydrofolate reductase (DHFR) and a protein interaction module (the PDZ domain). Statistical cutoffs that contributed to the coevolving residues were detected by the SCA for an alignment of 418 phylogenetically diverse DHFR sequences. The results suggest that SCA selected a connection that has impact on the initial step of allosteric regulation between sectors and surface sites. However, analyzing relationships by calculating the covariance as in equations might lead to inconsistent results, which is caused by the characteristic of not revealing the physical mechanism of energetic coupling.
Structure-Based Methods
Structure-based methods rely greatly on the 3D structures of the target proteins obtained through methods such as X-ray crystallography or NMR spectroscopy. If there is no available structure for the target, it may be possible to build a 3D homology model. Computational software or algorithm (60) will be used to find the “allosteric binding sites” from receptor cavities or from the “known allosteric binding sites” of other target proteins. Comparing with the sequence-based approaches, structure-based methods are more accurate. The accuracy of the structure-based methods relies on the quality of the 3D structure, which means that the 3D structure of the predicted protein is required.
Based on the allosteric binding sites in certain receptors, Huang et al. (61) used 90 crystal structures of non-redundant allosteric proteins with a resolution better than 3 Å that were extracted from the allosteric database to develop a server-based automatic tool named Allosite (http://mdl.shsmu.edu.cn/AST/). This highly efficient model uses different algorithms to predict the locations of allosteric sites in proteins, such as pocket-based analysis (analysis based on the “druggable” cavities referring to target proteins where small drug-like molecules have been shown to bind) and support vector machine (SVM) classifiers. Twenty-one site descriptors best suited to delineate the characteristics of typical allosteric sites were selected by a discriminated feature selection method (61). Several software packages, including a novel geometry-based pocket-based algorithm named Fpocket (60), were chosen as an initial pocket to detect the cavities and covered all known allosteric sites. Then, SVM classifiers, a supervised learning model that employs learning algorithms for the classification of both linear and nonlinear data, was trained and tested to identify allosteric sites in proteins of interest as the final model in the web server of Allosite. They further validated the reliability of the server by predicting potential allosteric sites for five proteins collected from Allosteric Database (ASD) (15). Their results showed that the allosite server is able to identify potential allosteric sites with high accuracy.
Conformational Dynamics-Based Approaches
Molecular dynamic (MD) simulations are applied to show structural changes in a target protein at the molecular level, which can accurately reproduce local and large-scale conformational changes of proteins (62). It is frequently used to refine the 3D structure of a target protein. It is also used to examine the dynamics of atomic-level phenomena that cannot be observed directly. Therefore, this method was proved particularly useful in unraveling potential allosteric sites. However, accurate MD simulation is time-consuming, and long MD simulations are mathematically ill-conditioned, generating cumulative errors in numerical integration. Dror et al. (63) recently utilized large-scale unbiased MD simulation to locate the allosteric site for the M2mAChR. Their results showed that the allosteric modulator bound to the extracellular vestibule of the receptor, a region approximately 15 Å from the orthosteric site. Meanwhile, Matosin et al. (64) conducted a 20-ns MD simulation using the AMBER force field to predict the allosteric sites, and selected 31 cavities from 201 unique cavities. After assessing their energy distribution similarities, 21 pockets were considered to be potential allosteric sites. Druggable target binding sites were detected while correlations among the binding sites’ ionic locks were identified with the help of the CAVITY algorithm (65), which is an approach used in detecting the binding cavities of proteins and quantitatively calculate the ligandability of a cavity based on a de novo drug design program LigBuilder. Lately, Ivetac et al. (66) applied the FTMAP algorithm (67), a computational analog of experimental approaches, to identify affinities of “hot spots” with a large number of organic probe molecules corresponding to drug fragments. This method was composed of virtual screening and a fragment-based molecular dynamics approach. The authors mapped the surfaces of the β1 (β1AR) and β2 (β2AR) adrenergic receptor structures to detect potentially druggable allosteric sites. Their results revealed that distinct pockets formed at both solvent-exposed and lipid-exposed cavities, suggesting that a combined approach of fragment-based and virtual screening methods to detect novel targets for GPCR drug discovery may be effective. Compared to other methods, MD simulations generate enhanced conformational diversity and show different topographies that are more representative of GPCR dynamics. The method might detect some sites that are not apparent in static experimental structures due to the transient nature of some pockets. However, with advances in computer hardware, conventional MD simulations often suffer from incomplete conformation space sampling for systems with the same size. An increasing number of state-of-the-art computational methods have been applied in the generation of more diverse structure ensembles.
Another method, the coarse-grained two-state Gō model, can be used to identify allosteric sites in proteins based on the assumption that the allosteric site is able to reshape the energy landscape after binding to a ligand. Briefly, the model consists of two conventional single-state Gō potentials and generates an ensemble of two different functional states for the under study protein. With the help of the model, the combination of the two functional states of a protein and the allosteric coupling in the drug exporter were conducted. Perturbations are later added to a potential binding site to sample the distribution of populations of protein nearby in the state space. The binding site is predicted as a potential allosteric site if a population redistribution occurs in the system. Recently, Qi et al. (68) applied the approach and successfully identified all the known allosteric sites in a number of test proteins. They also selected Escherichia coli phosphoglycerate dehydrogenase (PGDH) as a case study and then predicted that the allosteric sites might have a larger size than known sites and provided new possibilities for novel ligand discoveries. Based on the conclusions, either the inhibitor, the corresponding cavity, or both could lead to the application of regulation of the E. coli amino acid synthesis pathway and metabolic network. One advantage of the two-state Gō is that it uses a measure of uncertainty to rule out sites that are unlikely to be important. Moreover, the two-state Gō model incorporates the binding of the ligand by changing the interactions within the binding site, which does not require background information of the protein–ligand structure. However, both the bound and unbound structures must be known with difference in the residue–residue contact level when using the approach. Calculation of the shape and energy landscape parameters is a time-consuming task and requires a large number of trials to provide sufficient sampling for the construction of a proper two-state ensemble.
Normal Model Analysis-Based Approaches
Normal mode analysis (NMA) possesses the ability to obtain the global modes that bear functional significance. Out of the all the methods discussed so far, NMA characterizes the largest number of functional motions of quaternary structures. NMA also unearths important sites that mediate or propagate allosteric signals (69). Recently, NMA was used to predict the allosteric sites of target proteins (70).
Protein allosteric and regulatory sites (PARS) (http://bioinf.uab.cat/pars) is a web server to predict the location of allosteric sites based on the alteration of protein flexibility upon ligand binding described by NMA. Briefly, after uploading protein-structure files (PDB format), the LIGSITEcsc program (70), an extension algorithm that can automatically and efficiently detect potential ligand-binding sites in proteins, is utilized to predict suspended ligand binding sites. NMA (69) is performed for the unbound structure. The binding site is marked as a potential allosteric site if a significant difference (Wilcoxon–Mann–Whitney test p value ≤0.05) is found between the unbound structure and the ligand bound protein flexibility. Panjkovich and Daura (70) marked PARS using 102 allosteric proteins. The result was a ranking of 44% proteins derived from the first position, and a total of 73% proteins were found in the top three positions. The approach starts with the test of whether the protein structure is available, followed either by protein sequence identification and homology modeling or ligand-binding sites prediction directly. The PARS constructs potential connections among changes in allosteric effects and protein flexibly according to the rule of structure conservation. The degree of conservation of the structure of each pocket is measured if enough structural data is provided for the protein family. However, with the limitations from both phenomenological variety and the dependence of accuracy in detecting allosteric binding sites in the initial steps, there is still room for improvement of this method using benchmarked datasets.
Combination of Conformational Dynamics-Based and NMA-Based Approaches
The SPACER web server (http://allostery.bii.a-star.edu.sg/) was developed by Goncearenco et al. (71) based on the recent advances in thermodynamic views of allostery. The server hosts a tool for predicting more potential allosteric sites as well as describing allosteric communications among the regulatory and functional sites. First, substrate binding sites and coupling to determine regulatory sites are characterized. Thus, users are able to detect communications between the allosteric and catalytic sites. The method and related theoretical support include local closeness (LC) (72) as a comprehensive quantitative criterion for the prediction of a ligand-binding site. Binding leverage (72) is used as a measurement of coupling between a binding site and its intrinsic motions of a protein, and leverage coupling (73) provides a measure to analyze communications between regulatory and catalytic sites. The results are based on generic allosteric models instead of size and degree of oligomerization or function of the protein. On the server, allosteric communication can be calculated for up to four sites. The online tutorial (http://allostery.bii.a-star.edu.sg/tutorial/) explains the features and detailed process by the case of the phosphofructokinase (PFK) homotetramer. SPACER has proven to be a simple and unique approach that can be applied in a wide range of proteins to provide meaningful links between structural dynamics and small molecules. However, the score measured local network used has an unclear empirical based to the model, and uses residue weighting in its original form, which lowers the chance of finding non-catalytic binding sites in proteins.
Other Allosteric-Related Approach
Ligand binding specificity analysis (LIBSA) was developed by Hocker et al. (74) to analyze multiple docking poses and provides a metric to describe the binding to a specific region. Instead of detecting binding affinity, the quantitative protocol uses novel filtering algorithms and signal-to-noise ratios (SNR) to calculate the relative binding frequency at different allosteric pockets. To clarify, the novel filtering favors binding consistency over affinity to reduce noise by valuing occurrence of the docking score over the magnitude of the score. SNR quantifies the binding preference and identifies the binding site of the protein. LIBSA provides a standard to identify binding sites and to rank ligands according to the consistency when binding to the correct sites. Hocker et al. (74) demonstrated the reliability of the approach by applying it on a diverse set of known receptors and corresponding ligands docked into Ras conformers with multiple binding sites. LIBSA correctly identified known allosteric sites for the ligands and predicted possible sites for determinate ligands. Although potentially more accurate, the approach is too computationally expensive to be used for virtual screening of large numbers of compounds and receptors. The combination of LIBSA and MD simulations may achieve balance between accuracy and efficiency.
PHARMACOPHORIC MODELS/FILTERS/DESCRIPTORS FOR DISCOVERY OF ALLOSTERIC MODULATORS
Despite the development of computer and computational technique, virtual docking screening with an unfiltered compound library often causes a waste of time and resources, which impedes later compound selection. Therefore, it is advisable to filter compounds from a library before performing virtual docking screening. A pharmacophore is a description of molecular features which are necessary for molecular recognition of a ligand by a biological macromolecule. The pharmacophoric model/filter/descriptor is a useful tool for enriching a library with compounds that satisfies specific geometric and/or physicochemical constraints. The process for developing a pharmacophoric model generally involves five steps: (1) choose structurally diverse molecules, (2) generate a set of conformations for the compounds with low energy, (3) superimpose all combinations of the low-energy conformations of the molecules, (4) transform the superimposed molecules into an abstract representation, and (5) validation for different biological activities of related receptors. By using pharmacophoric model for filtering, a big database (more than 200,000 compounds) can be efficiently narrowed down to a small dataset with several hundred or thousand compounds. However, some novel structures may be overlooked by using this method.
Pharmacophore-Based Virtual Screening
Castelli et al. (75) reported two novel PAMs (COR627 and COR628) in 2011. They selected six metabotropic γ-aminobutyric acidB (GABAB) receptor PAMs with diverse structures from literatures (76–79), and generated a five-features pharmacophore model, including three hydrophobic sites (H1–H3), one aromatic group (R), and one hydrogen-bond donor (D). They used this pharmacophore model to perform virtual screening of two databases (ASINEX Gold Collection and ChemBridge EXPRESS-Pick Collection), and identified molecules sharing equivalent steric and electronic features located at an appropriate distance from each other. COR627 was one of the fittest compounds with respect to the common features pharmacophore identified by query. COR628, a simplified analog of COR627, was produced by in-house chemical synthesis. These two compounds showed potential therapeutic effects on GABAB receptors according to in vivo assay.
In 2013, Kubas et al. (80) reported 2-aminoquinazoline derivatives as a novel class of mGluR5 NAMs. They aligned 19 mGluR5 active in-house compounds and reported compounds to build 30 different pharmacophore models. Each pharmacophore model possessed four to seven pharmacophoric features. Then, these pharmacophore models were analyzed against their in-house evolution databases (generated from a virtual screening compound library comprising 6.5 million unique commercially available compounds) to determine the ability of each model for distinguishing between decoys and actives using ROC–AUC and early enrichment factors (EF=1% and EF=2.5%). The top eight pharmacophore models were further tested and had at least a tenfold selectivity against mGluR1 activity (80). Finally, two best pharmacophore models with four pharmacophoric features were selected, to perform screening for a library of commercially available compounds. A total of 336 virtual hits were obtained, purchased, and tested for their in vitro activities on mGluR1 and mGluR5, respectively. The pharmacological screening yielded eight novel scaffolds with sufficient activity and selectivity for mGluR5.
Ligand-Based Similarity Search
In 2006, Renner et al. (81) performed a ligand-based similarity search (LBSS) to discover new allosteric modulators for mGluR5 without the crystal structure of mGluR5. The flexible-alignment module in MOE (an application for flexibly aligning small molecules) was used to align seven known compounds to detect the receptor-bound 3D conformations. A prescreening filter for a commercially available compound library with 194,563 molecules was applied to select 20,000 most “drug-like” compounds on the basis of their distance to “optimal” variable values, in which these variable values are obtained by principal component analysis. Finally, 27 top-scoring and most independent molecules were selected for experimental testing according to separate similarity-based searches (an independent similarity search was carried out for each molecule). Some compounds showed high affinity within 1 μM at mGluR5.
Noeske et al. (82) reported the application of alignment-free topological pharmacophore descriptors (CATs) for virtual screening of selective allosteric mGluR1 antagonists. CATs are based on the 2D structure of a molecule and circumvent problems associated with conformational flexibility. They used six known mGluR1 (allosteric) antagonists as queries to perform the similarity searching in a commercially available compound library (Gold Collection of Asinex Ltd) that includes 201,304 entries. For each seed compound, the CAT program generated 100 most similar hits. Noeske et al. (82) first selected the top-five scoring compounds for each seed compound. They then selected the hits that met at least three of the six criteria in the lists, irrespective of their rank. Finally, 23 compounds tested empirically. One compound was found to be highly active (<1 μM) at mGluR1, and five compounds were moderately active with IC50 values between 1 and 15 μM in functional assays (82).
Support Vector Machine Filter
Véronique Hamon et al. (83) analyzed the properties of 40 non-redundant small molecules presented in the 2P2I database (http://2p2idb.cnrs-mrs.fr/) to define a general profile of orthosteric inhibitors and proposed an original support vector machine (SVM) protocol for filtering general screening libraries. SVM modeling (implemented in the statistical software package R) was used for model training. The original data was mapped through a kernel function onto a higher dimensional space where the two sets (training and test sets) are more easily separable with a linear classifier. The positive or negative activity of the training set was recorded as a factor variable to specify the classification mode in problems (83). As a preprocessing treatment of data, each descriptor is centered on a mean of zero and scaled to a variance of 1. With a random selection of the training and test sets, the training is performed through a fivefold cross-validation procedure, which is repeated 30 times. They selected the final optimal model according to the average best performance of the ROC AUC (area under curve) statistical parameters. The filtering protocol has been validated using external datasets from PubChem BioAssay and in-house screening results, demonstrating that the SVM model can reduce the size of the filtered chemical library by eliminating up to 96% of compounds and enhancing the proportion of active compounds (83)..
STRUCTURE-BASED VIRTUAL SCREENING FOR LIGAND DISCOVERY TARGETING AT ALLOSTERIC SITE OF GPCRS
Structure-based virtual screening involves docking of candidate ligands into a target protein followed by applying a scoring function to estimate the likelihood that the ligand will bind to the protein with high affinity. The accuracy of this method relies on the 3D structure of the biological target. This method is able to obtain faster and better results if the compound library is first filtered by pharmacophore model. Recently, with advances in GPCR X-ray crystallography, new opportunities came to attention for developing novel GPCR allosteric modulators as drugs (84). This section summarizes different cases of developing allosteric modulators using structure-based computational approaches as shown in Fig. 3.
Fig. 3.
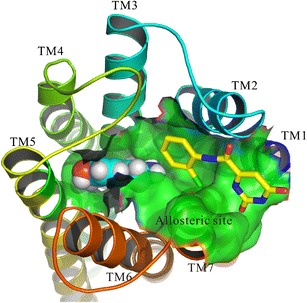
Structure-based approach for the virtual docking screening of allosteric modulators. Dopamine-bound model of D3R (PDB entry: 3PBL) for allosteric compound screening proposed by Lane and co-workers (Lane et al. Mol. Pharmacol. 2013). Dopamine is shown in sphere. The allosteric site was used for the virtual docking screening of allosteric compounds
Crystal Structure-Based Ligand Discovery
The X-ray crystal structure of D3R bound with antagonist eticlopride was reported by Chien et al. (85) in 2010. The structural basis of the binding pocket of D3R indicated a second potential allosteric binding site that extended toward ECL2, formed by TM1, TM2, and TM7 (Fig. 3, allosteric site highlighted in green). Lane et al. (29) reported their work on large-scale virtual screening for orthosteric and allosteric ligands of D3R, employing two optimized crystal-structure-based D3R models: one represented an apo state, D3RAPO, and another one bound with dopamine, D3RDopa (Fig. 3). At the beginning, they applied the LiBERO ((86), an algorithm that exploits ligand information for selecting the best performing protein models from an ensemble) optimization for the two models based on the D3R crystal structure (85). Conformers of the D3R crystal structure were generated by introducing minor variations (~0.1 Å) in the protein backbone, using energy-based side chain sampling with known antagonist sets, in which the binding energy of antagonists at the protein may be caused by different conformations of side chains. The established conformers of the binding pocket were assessed by virtual ligand screening (VLS) performance, which can separate D3R ligands from decoys. Second, the virtual screening of 4.1 million compounds prepared from the Molsoft Screen Pub database (Molsoft, LLC, San Diego, CA) was carried out by employing the VLS procedure. The binding energy of antagonists at the protein may be caused by different conformations of side chains implemented in the ICM-Pro molecular modeling software (87). Their screening work was performed independently for VLS models of D3RAPO and D3RDopa. They defined a large rectangular box that contains all the residues from orthosteric pocket, extended pocket, and ECLs for their docking procedure. Based on the docking results, they selected 300 compounds in each model hit list clustered by chemical similarity with 0.3 Tanimoto distance cutoff. The Tanimoto distance was calculated using dynamic linear fingerprints of 1536 bits with chains up to six atoms long extended with the atom/bond counts as performed in ICM-Pro v.3.7-2a (Molsoft, LLC). They found that the top 25 candidates in the D3RDopa model screening list did not have a positively charged amine forming a conserved salt bridge to Asp1103.32, which is contrary to the D3RAPO but has interactions with helices I, II, III, and VII as well as ECL1 and ECL2, with some larger compounds locating ECL3 residues. In addition, these hits also reach dopamine and Asp1103.32 at the end of the orthosteric pocket. They predicted that some motifs have polar interactions with Tyr3657.35 hydroxyl and/or the aromatic ring, polar interactions with Ser3667.36 and Glu902.65 side chains, and hydrogen bonding to Ser1925.42 in the backbone amides, Cys181 and Ile183 in the ECL2. Finally, they also further validated their predicted novel allosteric ligands by experiments. These allosteric modulators of the D3R receptor showed distinct functional profiles on dopamine-signaling efficiency to provide a starting platform for further optimization of selectivity.
Homology Model-Based Ligand Discovery
Recently, Radestock et al. (88) investigated the combination of two reported techniques for the improvement of homology model-based virtual screening for mGluR5 NAMs. They first utilized ligand-supported homology modeling to construct the receptor models that were congruent with the mutagenesis data and ligand structure–activity relationship (SAR) (88), which means that the selection of a set of ligands and related predefined interactions. Specially, side chains of a protein were manually optimized, and a database of drug-like compounds was docked against the validated receptor. Using ligand–receptor interaction fingerprint-based similarity (IFS), they scored and ranked chemical compounds from a virtual library. IFS is a method for similarity search, including three steps: (1) information about the patterns of interactions between the reference compounds and the binding site residues was used to rank order of the docking solutions, (2) predicted patterns of interactions of the docking solutions were compared with the patterns of interactions of the reference ligands, and (3) patterns of interactions were encoded in binary, and the similarity between two fingerprints was determined via the Tanimoto distance coefficient. Their method was evaluated in retrospective virtual screening experiments for mGluR5 NAMs (88). Comparing with the results obtained by conventional scoring functions, including PMF-score, Gold-score, FlexX-Score, Dock-Score, and ChemScore, the approach leads to significantly higher enrichment rates related to the competing scoring functions.
MOLECULAR DYNAMICS SIMULATION FOR EXPLORING THE DETAILED INTERACTIONS OF ALLOSTERIC MODULATORS
Although thousands of allosteric modulators are developed for more than 90 GPCRs, the accurate allosteric binding pocket and the detailed interactions between GPCRs and modulators are rarely reported. The breakthrough work of large-scale unbiased MD simulation to locate the allosteric site for M2mAChR, accomplished by Dror and co-workers (63), indicated that MD simulation is a valuable approach for exploring detailed interactions of modulators. Therefore, all-atoms MD simulation with calculation of the docking energy of both modulator and ligand is considered as a useful tool for predicting the exploring of the detailed interactions of allosteric modulators and GPCRs.
Recently, Shore et al. (31) reported the binding pocket and detailed interactions of ORG27569 in cannabinoid receptor 1 (CB1). The binding site of ORG27569 is located in the TM3-6-7 region of the CB1 receptor with extending to the extracellular side. They also performed 22.5 ns MD simulation with ~35,000 atoms for ORG27569 docked in the CB1-CP55940 complex and of CP55940 alone at the CB1 receptor. They used the OPLS2005 force field with a distance-dependent dielectric (coefficient of 2.0 to match the docking studies). The extended non-bonded treatment was employed. The dynamics module of Macromodel 9.1 (31) was invoked, using stochastic dynamics at 300 K with the use of SHAKE constraints for bonds to hydrogen allowing a 1.5-fs time step. The models were first minimized for 500 steps with restraints on all the heavy atoms The molecular dynamics was then initialized to 300 K, and an initial 100 ps of molecular dynamics was run (31). They found that the interactions between ORG27569’s peperidine nitrogen and Lys1923.28 kept stable during the MD simulation, with the distance of 3.19 Å. In the presence of ORG27569, the hydrogen bond between CP55940 and Lys1923.28 extended to 6.80 Å. However, the results of CP55940 along the docket at the CB1 receptor showed that the interactions between CP55940 and Lys1923.28 also kept stable at 3.19 Å. Importantly, by calculating the interaction energies, they found that ORG27569 did not influence the binding affinity of CP55940 for CB1 receptor.
Xie’s group (89) carried out the 100-ns MD simulation for the mGluR5 (residue 26-832, formed by Venus Flytrap domain, a so-called cysteine-rich domain, and 7TMD) bound with (a) antagonist-LY-344545 (an epimer of LY-341495, mGluR2/3 selective antagonist) and NAM-mavoglurant and (b) agonist-glutamate and PAM-VU0405386. Their MD simulations were carried out using the NAMD package (version 2.9b1) with CHARMM27 force field. Electrostatics were calculated using the Particle Mesh Ewald10 (PME) method with a 12-Å non-bonded cutoff and a grid spacing of 1 Å per grid point in each dimension. The van der Waals energies between protein and molecule were calculated using a smooth cutoff value (switching radius 10 Å, cutoff radius 12 Å). The temperature was maintained at 310 K using a Langevin thermostat and the pressure was maintained at 1 atm using the Langevin barostat, respectively. The time step of MD simulations was set to 1 fs. Their system included ~70,000 atoms with a 60 × 70 × 160 Å3 water–lipid box. Their results showed that the conformations of both mavoglurant (NAM) and VU0405386 (PAM) after stable 100-ns MD simulation results, when comparing with their conformations before MD simulation. However, the “ionic lock” in 7TMD of mGluR5 differed greatly depending on the ligand. The distance between Lys6653.50 and Glu7706.35 after 100-ns MD simulation remained within 3.0 Å. However, for mGluR5 bound with VU0405386 (PAM), the observed distance between Lys6653.50 and Glu7706.35 after 100-ns MD simulation extended to 7.1 Å.
BITOPIC LIGANDS
In recent years, bioactive compounds were designed to bind simultaneously to GPCR orthosteric and allosteric binding areas, which is derived from the “message-address” concept of Schwyzer (90). In principle, these molecules consist of two parts connected by a chemical linker, including the message part, effecting signal transduction responsible for the activation of the receptor, and the address part, located adjacent to the message, which provides additional binding affinity by guiding the ligand to the receptor. More specifically, the “message” may interact with the highly conserved orthosteric site and the “address” binds to the less-conserved allosteric binding pocket, contributing to selectivity. By combining these two considerations, ligand design efforts can incorporate selectivity as well as binding affinity as design criteria.
Bitopic ligands (also named as dualsteric ligands) consist of two pharmacophoric units connected via a linker. The pharmacophores can be identical (i.e. “homobivalent”) or different (i.e. “heterobivalent”). These pharmacophoric units (two or three) could bind to the orthosteric site and an allosteric site in the same receptor as well as bind to two orthosteric binding sites located on two different receptors.
Generally, the large size and molecular weight of bivalent ligands limit their in vivo use due to the impaired bioavailability. However, few synthesized bivalent ligands are not hindered by these limitations and have shown potential in vivo pharmacological activities.
Portoghese et al. (91) obtained MDAN-21, which could pass through the blood–brain barrier to induce antinociception by connecting oxymorphone, a μ opioid receptor agonist, with naltrindole, a δ opioid receptor antagonist.
Halazy et al. (92) have successfully conjugated two identical sumatriptans (5-HT1B agonist) together to obtain an orally active drug that could induce a stronger hypothermia effect than sumatriptan itself. It is surprising that such a drug was able to cross the blood–brain barrier in spite of its elevated molecular weight, polar surface area, and number of hydrogen-bond donors, indicating that an active transport is probably involved.
Bitopic–allosteric/orthosteric GPCR agonists were first discovered at M2mAChR, by molecular–mechanistic investigations of a known agonist, as well as by the specific de novo synthesis of a ligand aimed to simultaneously act on both the allosteric and orthosteric binding sites. Christopoulos’s group (51,93) characterized the earliest known partial agonist McN-A-343 as a bitopic ligand, which displays substantial differences in efficacy across receptor subtypes: as a full agonist of M1mAChR or M4mAChR but a weak partial agonist at M1mAChR. For the fragments of McN-A-343, the tetramethyl ammonium cation represents the message part occupying the orthosteric binding site, whereas the 3-chlorphenylcarbamate is the address part for the allosteric EL2 vestibule, and a rigid butynyl chain links the two parts. 3-Chlorphenylcarbamate has a substantial negative allosteric effect on the signaling efficacy of tetramethyl ammonium cation and contributes to the weak partial agonism displayed by the parent compound, McN-A-343. However, this molecule seems so small that it cannot occupy larger areas in the allosteric binding pocket and thereby gain subtype selectively for the M2mAChR since it attains similar binding affinity to the M4mAChR as well.
In a traditional view, it may be expected that bitopic ligands will gain substantially improved binding affinity with their target receptor compared to the binding affinities of the individual pharmacophores. Just as the case, Steinfeld et al. (94) generated the compound THR-160209 by connecting an orthosteric 3-benzylhydrylpyr-rolinyl building partner via a heptane chain to an allosteric 4-aminobenzylpiperidine motif, and they gained a considerably higher receptor affinity than seen with individual components (pKi (total) = 9.5 vs. pKi = 5.5 for the single compounds) and a certain preference for the M2mAChR.
The other type of dualsteric compounds may display relatively good subtype selectivity for the target receptor but gain less potency/affinity than orthosteric blockers. For example, Antony et al. (95) conjugated the allosteric modulators to iperoxo, an orthosteric muscarinic receptor agonist, and gained bitopic compounds with less binding affinity, but higher preference on M2mAChR. The differences between the two types may be due to differences in the orthosteric and allosteric pharmacophores that were combined to yield the bitopic ligands. In this case, the orthosteric moiety within THRX-160209 is an antagonist, and the allosteric moiety is a negative allosteric modulator; both pharmacophores would therefore have similar preference on receptor states. In contrast, the hybrid molecules reported by Antony et al. (95) conjugated an orthosteric agonist to a negative allosteric modulator; the individual pharmacophores are likely to prefer different states of the receptor, thus resulting in a compromised binding affinity of the bitopic ligand.
Bitopic allosteric and orthosteric ligands were designed and synthesized according to the concept applying on M2mAChR as follows. Recently, Christopoulos et al. (51,93) conjugated adenosine to VCP171, a PAM of the adenosine A1 receptor, to gain a compound (named VCP746) that binds to both the orthosteric and allosteric sites. Importantly, VCP746 was shown to protect both cardiomyoblasts and cardiomyocytes against simulated ischemia in vitro studies, but it had no effect on rat atrial heart rate in vivo.
Four combinations of orthosteric–allosteric parts are theoretically possible: active–active, active–inactive, inactive–active, and inactive–inactive. The hybrid M1mAChR agonists mentioned previously represent the constellation of “orthosteric active/allosteric inactive.” In that case, both the orthosteric and the allosteric parts should be optimized to generate a highly potent dualsteric ligand. The orthosteric agonist was considered to provide very high affinity and efficacy for the receptor activation to maintain sufficient activation for the dualsteric construct because of the functionally opposing receptor conformations for the constellation of “orthosteric active/allosteric inactive.” The allosteric binding partner should possess high affinity and subtype selectivity for the receptor. However, upon binding of the dualsteric ligand, the other binding site of the receptor may undergo conformational changes, causing negative or positive co-operations. It is important to keep “balance of power” in the constellation “orthosteric active/allosteric inactive” to adjust signaling pathway selectivity to a desired level.
Sometimes dualsteric ligands have higher affinities than the respective partners with high subtype selectivity. This can be achieved if two parts bind into their corresponding binding pockets in an ideal manner without inducing an unfavorable conformational rearrangement of the receptor protein, which can be explained by the lower total entropy cost of the ligand–receptor complex. The mostly favorable spatial proximity between the orthosteric and the allosteric binding areas contributes to a decrease in entropy (96). Conformational changes in the two binding events can either be (locally) entropic or enthalpic. This can differ case by case. The general rule is that an increase in entropy is the driving force for the docking of a polar agonist and a decrease in enthalpy in the driving force for the docking of a mostly hydrophobic antagonist (97).
According to the “Lipinski Rule Five” (98), agents with molecular mass lower than 500 Da have a better bioavailability than agents with more mass. Bitopic or dualsteric compounds are often too large (>950 Da) or too lipophilic to be efficacious effectively in the clinic. According to the analysis of allosteric modulators, rules for simply deciphering the chemical characteristics of allosteric modulators were developed by Zhang’s group (15), as follows: (i) molecular weight, MW ≤ 600; (ii) rotatable bond number, RBN ≤ 6; (iii) number of rings, 2 ≤ nR ≤ 5; (iv) SlogP, 3 ≤ SlogP ≤ 7. The “allosteric-like” rules provide a preliminary filter for the identification of allosteric modulators, guiding rational design and optimization of allosteric hits.
Valant et al. (22,23) and Lane et al. (25,26) presented synthesis methods for assisting in the discovery or validation of bitopic ligands. The orthosteric pharmacology can be confirmed by competition binding assays, while the allosteric pharmacology can be confirmed by functional assays ([35S]GTPγS and ERK1/2 phosphorylation).
The design of bitopic ligands may have multiple problems since several parts of the molecule—the orthosteric and the allosteric parts as well as the linker—must be optimized and linked in appropriate ways. No matured computational approaches have yet been developed for the discovery of bitopic ligands. However, from the concept of bitopic ligands (including different pharmacophoric units and linker), some potential computational methods should be suitable for the design of the bitopic ligands. We note especially that in computational fragment-based design, the strategy of design bitopic ligands is markedly similar to computational fragment-based design. Recently, Oguievetska et al. (99) successfully used computational fragment-based drug design to explore the hydrophobic sub-pocket of mitotic kinesin Eg5 allosteric binding site. Eg5 is a mitotic kinesin exclusively involved in the formation and function of the mitotic spindle that has attracted interest as an anticancer drug target.
Three potential allosteric sites were reported in GPCRs, as shown in Fig. 4. The structural basis of the binding pocket of D3R (29) and the docking results of D2R (26) indicated a potential allosteric binding site that extended toward the ECL2, formed by TM1, TM2, and TM7. Based on the detailed interactions between allosteric modulators and CB1 receptor (31)/M2mAChR (66), another allosteric site (allosteric site II) can be observed in the ECL2. Recently, the third allosteric site for sodium was reported by Liu et al. (53) and Xie’s group (5). The previous two allosteric sites have enough space to be occupied by allosteric modulators, which also have potential for allosteric pharmacophores for bitopic ligands. The sodium allosteric site, the allosteric pocket, only accommodates small molecules, such as amiloride, due to its limited size (53). Although this allosteric site for sodium seems conserved, the allostery of sodium or amiloride is only found in few receptors (55), indicating that this allosteric site may result in high selectivity of related ligands. All three allosteric sites can be used to construct allosteric pharmacophores for bitopic ligands with the fragment-based method, which is to apply orthosteric features and the allosteric pharmacophore separately, as shown in Fig. 5. Briefly, for orthosteric features, a library of known orthosteric compounds of corresponding targets is collected to generate an active “fragment/feature,” in which the fragment or feature may show activity, with lower than 10 μM against target protein. The allosteric fragments can be obtained using the same protocol for orthosteric features from the library of both orthosteric compounds, and from known allosteric compounds, in which the latter library is more reasonable.
Fig. 4.
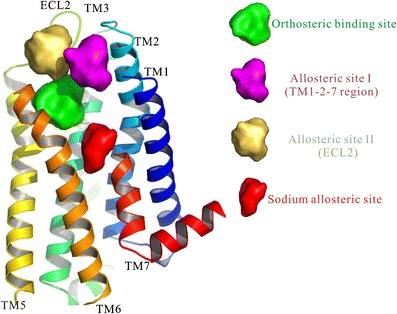
Allosteric binding site I was formed by TM1, TM2, and TM7, which is highlighted in purple. Allosteric site II can be observed in the ECL2, which is highlighted in yellow. Sodium allosteric site is highlighted in red. The orthosteric site is highlighted in green
Fig. 5.

Computational strategies for designing the bitopic ligands for GPCRs using fragment-based approaches. Design of bitopic ligands includes but is not limited to a connecting the orthosteric site and the allosteric site I, b connecting the orthosteric site and the sodium allosteric site, and c connecting the orthosteric site, allosteric site I, and the sodium allosteric site. Upon confirming the orthosteric and allosteric pharmacological properties, the linkers can be attached to each of the pharmacophores
FUTURE DIRECTIONS
Computationally identified allosteric modulators and bitopic ligands of GPCRs have advantages against orthosteric ligands. When developing allosteric modulators and bitopic ligands, chemical and pharmacological issues should be taken into consideration, such as ligand-based signaling and safety assessment.
The discovery of allosteric modulators is a milestone for designing novel GPCR ligands. The design and development of bitopic ligands is a promising and exciting strategy to discover new drugs, especially for improving selectivity of target proteins. However, it is still challenging to validate bitopic ligands by experiments. The mechanisms of action of bitopic ligands also need to be validated by bioassays, such as mutagenesis and kinetic and equilibrium experiments. More pre-clinical data is still needed to support the therapeutic application of the new bitopic compounds.
In contrast to the encouraging progress made in the discovery and functional characterization of allosteric/bitopic GPCR ligands, the detailed structural biology of their binding sites is still poorly understood. Existing experimental methods were proved extremely difficult to crystallize the allosteric binding site and cannot provide the same degree of structural data which had supported allosteric/bitopic drug design in soluble proteins such as kinases (100). The good news is that novel computational approaches can facilitate our understanding of the role of allosteric/bitopic ligands and their corresponding GPCRs in related diseases.
In order to improve accuracy to predict allosteric modulators and bitopic ligands in the future, further research developments are still needed in some computational approaches. The following improvements in the field seem likely in the future:
Robust approaches of predicting allosteric sites for GPCRs. Since the allosteric sites predicted by structural-based or topological-based approaches are selected mainly from protein kinases and enzymes, no allosteric site from GPCRs is used. As in all modeling, tuning parameters and model selection are sensitive to the completeness and representativeness of data sources for parameterization. Therefore, these models (biased application) may make an ineffective prediction for the allosteric sites in GPCRs (52).
New libraries for allosteric pharmacophores. Until now, the libraries used for virtual docking screening of allosteric modulators mainly come from the orthosteric compounds library. So far, more than 22,000 allosteric modulators have been reported, which should be collected and put together into an allosteric library.
Fragment-based allosteric analysis. Until now, no mature computational approaches are developed specifically for the discovery of allosteric modulators or bitopic ligands. Based on concept of bitopic ligands, that consist of different pharmacophoric units and linkers and the successful instance of using computational fragment-based drug design to explore the allosteric binding site of mitotic kinesin Eg5 (99), we suggest that the fragment-based allosteric analysis approach should be developed further to aid in the discovery of allosteric modulators and bitopic ligands for GPCRs.
Improvement of MD simulation. Rigorous molecular dynamics simulations can be used for the identification and validation of allosteric sites and the detailed interactions (52). However, conventional MD simulations suffer from incomplete conformation sampling for systems with the same size. Moreover, accurate MD is time-consuming, and long MD simulations are mathematically ill-conditioned. Last but not least, the traditional force-field parameters may not be accurate for the modulators or bitopic ligands.
Novel combinations of approaches. Logical set analysis of results from alternative methods can employ overlap (consensus set) or union (any result) type approaches, with varying degrees of resulting sensitivity and specificity. An objective means to setting up a rapid, tunable screening procedure could be established if various logical combinations of sets of results were studied systematically, with empirical validation, as has been done for mass-spectrometry-based proteomics peptide identification. Although the method space is huge (Table I), a comprehensive study would inform on the utility of considering more than one approach simultaneously.
Acknowledgments
The authors would like to acknowledge the funding support for the Xie laboratory at the University of Pittsburgh from the NIDA P30DA035778A1 and NIH R01DA025612. We also thank our lab members Ms. Xiaole Yang and Ms. Xiaomeng Xu for their effort on literature studies and data collection on the compounds reported and marketed. Dr. James Lyons-Weiler provided pre-submission editorial review and comment.
References
- 1.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng Z, Hou T, Li Y. Studies on the interactions between β2 adrenergic receptor and Gs protein by molecular dynamics simulations. J Chem Inf Model. 2012;52:1005–14. doi: 10.1021/ci200594d. [DOI] [PubMed] [Google Scholar]
- 3.Feng Z, Hou T, Li Y. Selectivity and activation of dopamine D3R from molecular dynamics. J Mol Model. 2012;18:5051–63. doi: 10.1007/s00894-012-1509-x. [DOI] [PubMed] [Google Scholar]
- 4.Feng Z, Hou T, Li Y. Docking and MD study of histamine H4R based on the crystal structure of H1R. J Mol Graph Model. 2013;39:1–12. doi: 10.1016/j.jmgm.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Feng Z, Alqarni MH, Yang P, Tong Q, Chowdhury A, Wang L, et al. Modeling, molecular dynamics simulation and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. J Chem Inf Model. 2014;54:2483–99. doi: 10.1021/ci5002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bridges TM, Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–41. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 7.Wenthur CJ, Gentry PR, Mathews TP, Lindsley CW. Drugs for allosteric sites on receptors. Annu Rev Pharmacol Toxicol. 2014;54:165–84. doi: 10.1146/annurev-pharmtox-010611-134525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wellendorph P, Bräuner‐Osborne H. Molecular basis for amino acid sensing by family CG‐protein‐coupled receptors. Brit J Pharmacol. 2009;156:869–84. doi: 10.1111/j.1476-5381.2008.00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flor PJ, Acher FC. Orthosteric versus allosteric GPCR activation: the great challenge of group-III mGluRs. Biochem Pharmacol. 2012;84:414–24. doi: 10.1016/j.bcp.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–5. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–74. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 13.Lane JR, Abdul-Ridha A, Canals M. Regulation of G protein-coupled receptors by allosteric ligands. ACS Chem Neurosci. 2013;4:527–34. doi: 10.1021/cn400005t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, et al. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem. 2012;55:1445–64. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Z, Mou L, Shen Q, Lu S, Li C, Liu X, et al. ASD v2.0: updated content and novel features focusing on allosteric regulation. Nucleic Acids Res. 2014;42:D510–6. doi: 10.1093/nar/gkt1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Möhler H, Fritschy J, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 17.Epping-Jordan M, Le Poul E, Rocher J-P. Allosteric modulation: a novel approach to drug discovery. Innovations Pharm Technol. 2007;24:24–6. [Google Scholar]
- 18.Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol. 2011;81:691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 19.Dalton J, Gómez-Santacana X, Llebaria A, Giraldo J. A computational analysis of negative and positive allosteric modulator binding and function in metabotropic glutamate receptor 5 (In) activation. J Chem Inf Model. 2014;54:1476–87. doi: 10.1021/ci500127c. [DOI] [PubMed] [Google Scholar]
- 20.Kamal M, Jockers R. Bitopic ligands: all-in-one orthosteric and allosteric. F1000 Biol Rep. 2009;1:77. doi: 10.3410/B1-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davie BJ, Christopoulos A, Scammells PJ. Development of M1 mAChR allosteric and bitopic ligands: prospective therapeutics for the treatment of cognitive deficits. ACS Chem Neurosci. 2013;4:1026–48. doi: 10.1021/cn400086m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valant C, Sexton PM, Christopoulos A. Orthosteric/allosteric bitopic ligands. Mol Interv. 2009;9:125. doi: 10.1124/mi.9.3.6. [DOI] [PubMed] [Google Scholar]
- 23.Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2012;52:153–78. doi: 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- 24.Jo E, Bhhatarai B, Repetto E, Guerrero M, Riley S, Brown SJ, et al. Novel selective allosteric and bitopic ligands for the S1P3 receptor. ACS Chem Biol. 2012;7:1975–83. doi: 10.1021/cb300392z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lane JR, Sexton PM, Christopoulos A. Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci. 2013;34:59–66. doi: 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, et al. A new mechanism of allostery in a G protein–coupled receptor dimer. Nat Chem Biol. 2014;10:745–52. doi: 10.1038/nchembio.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maggio R., Scarselli M., Capannolo M., Millan M. J. Novel dimensions of D3 receptor function: focus on heterodimerisation, transactivation and allosteric modulation. Eur Neuropsychopharm. 2014. [DOI] [PubMed]
- 28.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–82. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lane JR, Chubukov P, Liu W, Canals M, Cherezov V, Abagyan R, et al. Structure-based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol Pharmacol. 2013;84:794–807. doi: 10.1124/mol.113.088054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–6. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shore DM, Baillie GL, Hurst DH, Navas F, Seltzman HH, Marcu JP, et al. Allosteric modulation of a cannabinoid G protein-coupled receptor binding site elucation and relationship to G protein signaling. J Biol Chem. 2014;289:5828–45. doi: 10.1074/jbc.M113.478495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldman T, Kabaleeswaran V, Jang SB, Antczak C, Djaballah H, Wu H, et al. A class of allosteric caspase inhibitors identified by high-throughput screening. Mol Cell. 2012;47:585–95. doi: 10.1016/j.molcel.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jahnke W, Rondeau J-M, Cotesta S, Marzinzik A, Pellé X, Geiser M, et al. Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat Chem Biol. 2010;6:660–6. doi: 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- 35.Plosker GL. Cinacalcet Pharm Econ. 2011;29:807. doi: 10.2165/11207220-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 36.Maraviroc: Reactivation of hepatitis B virus infection in an elderly patient: case report. Reactions Weekly 2014; 1508: 24–24.
- 37.Zarbock A, Allegretti M, Ley K. Therapeutic inhibition of CXCR2 by Reparixin attenuates acute lung injury in mice. Brit J Pharmacol. 2008;155:357–64. doi: 10.1038/bjp.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Addex presents ADX10059 GERD data at conference. Chem Bus Newsbase. 2007: 1.
- 39.Addex announces ADX10059 phase IIa acute anxiety data. Chem Bus Newsbase. 2008: 1.
- 40.Berg D, Godau J, Trenkwalder C, Eggert K, Csoti I, Storch A, et al. AFQ056 treatment of levodopa-induced dyskinesias: results of 2 randomized controlled trials. Mov Disord. 2011;26:1243–50. doi: 10.1002/mds.23616. [DOI] [PubMed] [Google Scholar]
- 41.Girard F, Keywood C, Poli SP, Mutel V. Anti-parkinsonian effects of ADX48621 a mGluR5 negative allosteric modulator, in the rat haloperidol induced catalepsy model. Mov Disord. 2010;25:S282. doi: 10.1002/mds.22851. [DOI] [Google Scholar]
- 42.Sourial M, Cheng C, Doering LC. Progress toward therapeutic potential for AFQ056 in fragile X syndrome. J Exp Pharmacol. 2013;2013:45–54. doi: 10.2147/JEP.S27044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.In; Wiley Periodicals, Inc: 2009; Vol. 11, p 7.
- 44.Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I, et al. Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H] JNJ-40068782. J Pharmacol Exp Ther. 2013;346:514–27. doi: 10.1124/jpet.113.204990. [DOI] [PubMed] [Google Scholar]
- 45.Hopkins CR. Is there a path forward for mGlu2 positive allosteric modulators for the treatment of schizophrenia? ACS Chem Neurosci. 2013;4:211–3. doi: 10.1021/cn400023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macdonald GJ, Lindsley CW. A unique industrial–academic collaboration towards the next generation of schizophrenia therapeutics. Curr Top Med Chem. 2014;14:304–12. doi: 10.2174/1568026613666131127154443. [DOI] [PubMed] [Google Scholar]
- 47.Lindsley CW, Hopkins CR. Metabotropic glutamate receptor 4 (mGlu4)-positive allosteric modulators for the treatment of Parkinson’s disease: historical perspective and review of the patent literature. Expert Opin Ther Pat. 2012;22:461–81. doi: 10.1517/13543776.2012.679437. [DOI] [PubMed] [Google Scholar]
- 48.Taylor SG, Riley G, Hunter AJ, Stemp G, Routledge C, Hagan JJ, et al. SB-269652 is a selective D3 receptor antagonist in vitro and in vivo. Eur Neuropsychopharm. 1999;9:266. doi: 10.1016/S0924-977X(99)80282-X. [DOI] [Google Scholar]
- 49.Silvano E, Millan MJ, la Cour CM, Han Y, Duan L, Griffin SA, et al. The tetrahydroisoquinoline derivative SB269, 652 is an allosteric antagonist at dopamine D3 and D2 receptors. Mol Pharmacol. 2010;78:925–34. doi: 10.1124/mol.110.065755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tahtaoui C, Parrot I, Klotz P, Guillier F, Galzi J-L, Hibert M, et al. Fluorescent pirenzepine derivatives as potential bitopic ligands of the human M1 muscarinic receptor. J Med Chem. 2004;47:4300–15. doi: 10.1021/jm040800a. [DOI] [PubMed] [Google Scholar]
- 51.Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, et al. A novel mechanism of G protein-coupled receptor functional selectivity muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J Biol Chem. 2008;283:29312–21. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu S, Huang W, Zhang J. Recent computational advances in the identification of allosteric sites in proteins. Drug Discov Today. 2014;19:1595–600. doi: 10.1016/j.drudis.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 53.Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–6. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fenalti G, Giguere PM, Katritch V, Huang X-P, Thompson AA, Cherezov V, et al. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191–6. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tesileanu T., Colwell L. J., Leibler S. Protein sectors: statistical coupling analysis versus conservation. Quant. Biol. 2014: 1–31. [DOI] [PMC free article] [PubMed]
- 56.Gasper PM, Fuglestad B, Komives EA, Markwick PR, McCammon JA. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc Natl Acad Sci U S A. 2012;109:21216–22. doi: 10.1073/pnas.1218414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Wart AT, Durrant J, Votapka L, Amaro RE. Weighted Implementation of Suboptimal Paths (WISP): an optimized algorithm and tool for dynamical network analysis. J Chem Theory Comput. 2014;10:511–7. doi: 10.1021/ct4008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McLaughlin RN, Jr, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–42. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reynolds KA, McLaughlin RN, Ranganathan R. Hotspots for allosteric regulation on protein surfaces. Cell. 2011;147:1564–75. doi: 10.1016/j.cell.2011.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinforma. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang W, Lu S, Huang Z, Liu X, Mou L, Luo Y, et al. Allosite: a method for predicting allosteric sites. Bioinformatics. 2013;29:2357–9. doi: 10.1093/bioinformatics/btt399. [DOI] [PubMed] [Google Scholar]
- 62.Baskaran SK, Goswami N, Selvaraj S, Muthusamy VS, Lakshmi BS. Molecular dynamics approach to probe the allosteric inhibition of PTP1B by chlorogenic and cichoric acid. J Chem Inf Model. 2012;52:2004–12. doi: 10.1021/ci200581g. [DOI] [PubMed] [Google Scholar]
- 63.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–9. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 64.Matosin N, Fernandez-Enright F, Frank E, Deng C, Wong J, Huang X-F, et al. Metabotropic glutamate receptor mGluR2/3 and mGluR5 binding in the anterior cingulate cortex in psychotic and nonpsychotic depression, bipolar disorder and schizophrenia: implications for novel mGluR-based therapeutics. J Psychiatr Neurosci. 2014;39:407. doi: 10.1503/jpn.130242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan Y, Pei J, Lai L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr Pharm Des. 2013;19:2326–33. doi: 10.2174/1381612811319120019. [DOI] [PubMed] [Google Scholar]
- 66.Ivetac A, McCammon JA. Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach. Chem Biol Drug Des. 2010;76:201–17. doi: 10.1111/j.1747-0285.2010.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ngan CH, Bohnuud T, Mottarella SE, Beglov D, Villar EA, Hall DR, et al. FTMAP: extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 2012;40:W271–5. doi: 10.1093/nar/gks441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qi Y, Wang Q, Tang B, Lai L. Identifying allosteric binding sites in proteins with a two-state G̅o model for novel allosteric effector discovery. J Chem Theory Comput. 2012;8:2962–71. doi: 10.1021/ct300395h. [DOI] [PubMed] [Google Scholar]
- 69.Bahar I, Lezon TR, Bakan A, Shrivastava IH. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem Rev. 2009;110:1463–97. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Panjkovich A, Daura X. PARS: a web server for the prediction of protein allosteric and regulatory sites. Bioinformatics. 2014;30:1314–5. doi: 10.1093/bioinformatics/btu002. [DOI] [PubMed] [Google Scholar]
- 71.Goncearenco A, Mitternacht S, Yong T, Eisenhaber B, Eisenhaber F, Berezovsky IN. SPACER: server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 2013;41:W266–72. doi: 10.1093/nar/gkt460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitternacht S, Berezovsky IN. A geometry-based generic predictor for catalytic and allosteric sites. Protein Eng Des Sel. 2011;24:405–9. doi: 10.1093/protein/gzq115. [DOI] [PubMed] [Google Scholar]
- 73.Mitternacht S, Berezovsky IN. Binding leverage as a molecular basis for allosteric regulation. PLoS Comput Biol. 2011;7:l.e1002148. doi: 10.1371/journal.pcbi.1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hocker HJ, Rambahal N, Gorfe AA. LIBSA—a method for the determination of ligand-binding preference to allosteric sites on receptor ensembles. J Chem Inf Model. 2014;54:530. doi: 10.1021/ci400474u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Castelli MP, Casu A, Casti P, Lobina C, Carai MA, Colombo G, et al. Characterization of COR627 and COR628, two novel positive allosteric modulators of the GABAB receptor. J Pharmacol Exp Ther. 2012;340:529–38. doi: 10.1124/jpet.111.186460. [DOI] [PubMed] [Google Scholar]
- 76.Urwyler S, Mosbacher J, Lingenhoehl K, Heid J, Hofstetter K, Froestl W, et al. Positive allosteric modulation of native and recombinant γ-aminobutyric acidB receptors by 2, 6-Di-tert-butyl-4-(3-hydroxy-2, 2-dimethyl-propyl)-phenol (CGP7930) and its aldehyde analog CGP13501. Mol Pharmacol. 2001;60:963–71. [PubMed] [Google Scholar]
- 77.Guery S, Floersheim P, Kaupmann K, Froestl W. Syntheses and optimization of new GS39783 analogues as positive allosteric modulators of GABAB receptors. Bioorg Med Chem Lett. 2007;17:6206–11. doi: 10.1016/j.bmcl.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malherbe P, Masciadri R, Norcross R, Knoflach F, Kratzeisen C, Zenner MT, et al. Characterization of (R, S)-5, 7-di-tert-butyl-3-hydroxy-3-trifluoromethyl-3H-benzofuran-2-one as a positive allosteric modulator of GABAB receptors. Brit J Pharmacol. 2008;154:797–811. doi: 10.1038/bjp.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bowery N. Historical perspective and emergence of the GABAB receptor. Adv Pharmacol. 2010;58:1–18. doi: 10.1016/S1054-3589(10)58001-3. [DOI] [PubMed] [Google Scholar]
- 80.Kubas H, Meyer U, Krueger B, Hechenberger M, Vanejevs M, Zemribo R, et al. Discovery, synthesis, and structure–activity relationships of 2-aminoquinazoline derivatives as a novel class of metabotropic glutamate receptor 5 negative allosteric modulators. Bioorg Med Chem Lett. 2013;23:4493–500. doi: 10.1016/j.bmcl.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 81.Renner S, Noeske T, Parsons CG, Schneider P, Weil T, Schneider G. New allosteric modulators of metabotropic glutamate receptor 5 (mGluR5) found by ligand‐based virtual screening. ChemBioChem. 2005;6:620–5. doi: 10.1002/cbic.200400332. [DOI] [PubMed] [Google Scholar]
- 82.Noeske T, Jirgensons A, Starchenkovs I, Renner S, Jaunzeme I, Trifanova D, et al. Virtual screening for selective allosteric mGluR1 antagonists and structure–activity relationship investigations for coumarine derivatives. ChemMedChem. 2007;2:1763–73. doi: 10.1002/cmdc.200700151. [DOI] [PubMed] [Google Scholar]
- 83.Hamon V., Bourgeas R., Ducrot P., Theret I., Xuereb L., Basse M. J., Brunel J. M., Combes S., Morelli X., Roche P. 2P2IHUNTER: a tool for filtering orthosteric protein–protein interaction modulators via a dedicated support vector machine. J R Soc Interface. 2013; 11. [DOI] [PMC free article] [PubMed]
- 84.Wang CI, Lewis RJ. Emerging opportunities for allosteric modulation of G-protein coupled receptors. Biochem Pharmacol. 2013;85:153–62. doi: 10.1016/j.bcp.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 85.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–5. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Katritch V, Rueda M, Abagyan R. Ligand-guided receptor optimization. Meth Mol Biol. 2012;857:189–205. doi: 10.1007/978-1-61779-588-6_8. [DOI] [PubMed] [Google Scholar]
- 87.Abagyan RA, Orry A, Raush E, Budagyan L, Totrov M. ICM manual. La Jolla: MolSoft LLC; 2012. [Google Scholar]
- 88.Radestock S, Weil T, Renner S. Homology model-based virtual screening for GPCR ligands using docking and target-biased scoring. J Chem Inf Model. 2008;48:1104–17. doi: 10.1021/ci8000265. [DOI] [PubMed] [Google Scholar]
- 89.Feng Z., Ma S., Hu G., Xie X.-Q. Allosteric binding site and activation mechanism of class C G-protein coupled receptors: metabotropic glutamate receptor family. AAPS J. 2015: 1–17. [DOI] [PMC free article] [PubMed]
- 90.Schwyzer R. ACTH: a short introductory review*. Ann NY Acad Sci. 1977;297:3–26. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- 91.Portoghese PS. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol Sci. 1989;10:230–5. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- 92.Halazy S. G-protein coupled receptors bivalent ligands and drug design. Expert Opin Ther Pat. 1999;9:431–46. doi: 10.1517/13543776.9.4.431. [DOI] [Google Scholar]
- 93.Christopoulos A, Mitchelson F. Pharmacological analysis of the mode of interaction of McN-A-343 at atrial muscarinic M2 receptors. Eur J Pharmacol. 1997;339:153–6. doi: 10.1016/S0014-2999(97)01379-4. [DOI] [PubMed] [Google Scholar]
- 94.Steinfeld T, Mammen M, Smith JAM, Wilson RD, Jasper JR. A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol Pharmacol. 2007;72:291–302. doi: 10.1124/mol.106.033746. [DOI] [PubMed] [Google Scholar]
- 95.Antony J, Kellershohn K, Mohr-Andrä M, Kebig A, Prilla S, Muth M, et al. Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 2009;23:442–50. doi: 10.1096/fj.08-114751. [DOI] [PubMed] [Google Scholar]
- 96.Mammen M, Choi S-K, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Edit. 1998;37:2754–94. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 97.Maksay G. Allostery in pharmacology: thermodynamics, evolution and design. Prog Biophys Mol Biol. 2011;106:463–73. doi: 10.1016/j.pbiomolbio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 98.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 99.Oguievetskaia K, Martin-Chanas L, Vorotyntsev A, Doppelt-Azeroual O, Brotel X, Adcock SA, et al. Computational fragment-based drug design to explore the hydrophobic sub-pocket of the mitotic kinesin Eg5 allosteric binding site. J Comput-Aid Mol Des. 2009;23:571–82. doi: 10.1007/s10822-009-9286-z. [DOI] [PubMed] [Google Scholar]
- 100.Lewis JA, Lebois EP, Lindsley CW. Allosteric modulation of kinases and GPCRs: design principles and structural diversity. Curr Opin Chem Biol. 2008;12:269–80. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]