

Abstract
The derivation of somatic motoneurons (MNs) from ES cells (ESCs) after exposure to sonic hedgehog (SHH) and retinoic acid (RA) is one of the best defined, directed differentiation strategies to specify fate in pluripotent lineages. In mouse ESCs, MN yield is particularly high after RA + SHH treatment, whereas human ESC (hESC) protocols have been generally less efficient. In an effort to optimize yield, we observe that functional MNs can be derived from hESCs at high efficiencies if treated with patterning molecules at very early differentiation steps before neural induction. Remarkably, under these conditions, equal numbers of human MNs were obtained in the presence or absence of SHH exposure. Using pharmacological and genetic strategies, we demonstrate that early RA treatment directs MN differentiation independently of extrinsic SHH activation by suppressing the induction of GLI3. We further demonstrate that neural induction triggers a switch from a poised to an active chromatin state at GLI3. Early RA treatment prevents this switch by direct binding of the RA receptor at the GLI3 promoter. Furthermore, GLI3 knock-out hESCs can bypass the requirement for early RA patterning to yield MNs efficiently. Our data demonstrate that RA-mediated suppression of GLI3 is sufficient to generate MNs in an SHH-independent manner and that temporal changes in exposure to patterning factors such as RA affect chromatin state and competency of hESC-derived lineages to adopt specific neuronal fates. Finally, our work presents a streamlined platform for the highly efficient derivation of human MNs from ESCs and induced pluripotent stem cells.
SIGNIFICANCE STATEMENT Our study presents a rapid and efficient protocol to generate human motoneurons from embryonic and induced pluripotent stem cells. Surprisingly, and in contrast to previous work, motoneurons are generated in the presence of retinoic acid but in the absence of factors that activate sonic hedgehog signaling. We show that early exposure to retinoic acid modulates the chromatin state of cells to be permissive for motoneuron generation and directly suppresses the induction of GLI3, a negative regulator of SHH signaling. Therefore, our data point to a novel mechanism by which retinoic acid exposure can bypass the requirement for extrinsic SHH treatment during motoneuron induction.
Keywords: directed differentiation, Gli3, motoneurons, pluripotent stem cells, retinoic acid, SHH signaling
Introduction
Somatic motoneurons (MNs) are characterized by their unique ability to transmit output from the CNS to peripheral muscle targets. Loss of MN function is associated with several devastating neurological disorders such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy. Pluripotent stem cells (PSCs) have developed into a powerful tool for studying basic mechanisms of neuronal fate specification and for applications in human disease modeling and regenerative medicine. The derivation of somatic MNs from mouse ESCs was one of the first successful protocols for directing neuronal cell fate in vitro using developmental patterning cues (Renoncourt et al., 1998; Wichterle et al., 2002). The combined exposure to sonic hedgehog (SHH) and retinoic acid (RA) triggers specification of brachial level MNs by providing appropriate dorsoventral and anteroposterior cues (Wichterle et al., 2002). Similar results have been obtained using SHH/RA-based MN differentiation protocols in human PSCs (hPSCs) (Li et al., 2005; Lee et al., 2007; Hu and Zhang, 2009; Placantonakis et al., 2009), but the efficiency of MN yield is typically lower than in mouse PSCs. Although progress has been made in improving MN yield from hPSCs (Amoroso et al., 2013; Qu et al., 2014), the ability to derive human MNs efficiently and with great reproducibility across hPSC lines remains an important challenge (Davis-Dusenbery et al., 2014). Furthermore, there is very limited mechanistic understanding of the interplay of patterning factors such as SHH and RA with genetic and epigenetic factors that determine competency and MN yield during hPSC differentiation.
We have previously described the use of small-molecule inhibitors of BMP and TGF-β signaling [the dual-SMAD inhibition (dSMADi) protocol] to trigger neural induction at high efficiencies from human ESCs (hESCs) and induced PSCs (iPSCs) (Chambers et al., 2009). Here, we systematically optimized MN derivation under dSMADi conditions. Similar to the derivation of other neuronal subtypes, such as floor plate and midbrain dopamine neurons (Fasano et al., 2010; Kriks et al., 2011), we observed that early exposure to patterning factors is essential to obtaining HB9+ MNs at high yield. Surprisingly, using this early patterning paradigm, we found that the MN yield was identical in the absence or presence of extrinsic SHH activation. Even more remarkably, high-yield MN derivation was achieved in the presence of SHH antagonists such as CUR61414 and cyclopamine that inhibit signaling at the level of the smoothened (SMO) receptor, suggesting that RA may act on SHH signaling downstream of SMO. We report that early RA exposure prevents the induction of GLI3 (a downstream negative regulator of SHH signaling) during neural differentiation. Inducible knock-down or CRISPR/Cas9-mediated knock-out of GLI3 extends the developmental window during which hPSC-derived precursors are competent to differentiate into MNs at high efficiencies in response to RA. Furthermore, early patterning with RA prevents a switch to an active chromatin state at the GLI3 promoter during neural induction based on ChIP in combination with sequencing (ChIP-seq) analysis for histone marks and RA receptor (RAR) binding. Our data present a highly efficient and rapid protocol to generate MNs from hESCs and iPSCs and reveal a novel mechanism of RA action during MN specification that bypasses the requirement for extrinsic SHH exposure.
Materials and Methods
PSC culture
Undifferentiated hESCs and human iPSCs of either sex (see details on each line below) were used and were maintained on mitotically inactivated mouse embryonic fibroblasts (MEFs) in hESC medium containing 20% knock-out serum replacement (KSR) and supplemented with 6 ng/ml FGF2 (R&D Systems) as described previously (Chambers et al., 2009; Kriks et al., 2011). hESC lines used for this study included H9 (WA-09; XX), the previously generated HB9::GFP bacterial artificial chromosome transgenic hESC reporter line (Placantonakis et al., 2009), the newly generated H9-based shRNAmir GLI3-RFP (clone 5.6) and H9-based CRISPR (GLI3.1, GLI3.2, and GLI3.3) hESC lines, and the iPSC lines 11a, 18b, and 27b (XY, XX, and XX, respectively; Boulting et al., 2011).
MN differentiation
Neural induction according to the previously described dSMADi strategy was performed using the specific inhibitors LDN193189 (LDN; Stemgent) and SB431542 (SB; Tocris Bioscience) (Chambers et al., 2009; Fasano et al., 2010; Chambers et al., 2012). Briefly, cells were dissociated to single cells and plated at high density on matrigel in MEF-conditioned medium or hESC medium (Chambers et al., 2009) supplemented with an additional 10 ng/ml FGF2 and 10 μm Y27632 (ROCK inhibitor; Tocris Bioscience). Once cells attained 90–100% confluency, the medium was switched to KSR medium (“day 0”) containing LDN (500 nm) and SB (10 μm). The medium was transitioned incrementally to N2 medium starting on day 4, as described previously (Chambers et al., 2009). LDN and SB were added until day 10 of differentiation, at which point the medium was switched to 100% N2 medium (Chambers et al., 2009) supplemented with BDNF (20 ng/ml; R&D Systems), ascorbic acid (AA, 0.2 mm; Sigma-Aldrich), and glial cell line-derived neurotrophic factor (GDNF, 20 ng/ml; R&D Systems). To optimize MN fate specification, C25II SHH (125 ng/ml), purmorphamine (Pur; 1–2 μm; Calbiochem), and RA (1 μm; R&D Systems) were added in various temporal combinations as described in the Results. The hedgehog antagonists CUR61414 (5 μm; Santa Cruz Biotechnology) and cyclopamine (10 μm; Tocris Bioscience) were used. For optimal MN induction, RA alone or RA + Pur (1 μm) were added at day 1 and maintained until day 14–16 of differentiation followed by passage onto matrigel or polyornithine precoated plates in media supplemented with Y27637 for 24 h post replate. At 2–4 d after passaging, DAPT (10 nm; Tocris Bioscience) was added to the medium or cells were treated with mitomycin C (1 μg/ml for 1–2 h) to minimize the presence of proliferating precursor cells. Cells were maintained without further passaging in N2 medium containing BDNF, AA, and GDNF.
shRNAmir GLI3-RFP hESC line derivation and experimental conditions
Viral preparation and transduction.
GLI3 microRNA-adapted shRNA (shRNAmir) oligonucleotides were designed as described previously (Dow et al., 2012) and synthesized (4 nmol of ultramer; IDT). Several oligonucleotides were designed against GLI3. The oligonucleotide sequence used for all the experiments presented in the current study was as follows: 5′-ACAGCAGAATACTATCATCAGA-3′. shRNAmir oligonucleotides were cloned into the pTRIPZ lentiviral doxycycline (DOX)-inducible vector. For viral transduction undifferentiated, dissociated H9 ESCs were exposed to lentiviral vectors, followed by plating at clonal densities to derive subclones with stable expression (e.g., clone 5.6).
Cell culture.
To induce shRNAmir GLI3-RFP expression, DOX (2 μg/ml) was added to the culture medium and replenished with every medium change. To determine the impact of GLI3 suppression on MN patterning, cells were replated as single cells in medium containing DOX. DOX was maintained for the first 6 d of neural induction, at which point the medium was switched. For “late patterning” condition, the medium was switched to include 1 μm RA.
Engineering of the GLI3 knock-out hESC lines
Gene targeting.
The GLI3 locus was targeted using the Cas9 system (Mali et al., 2013). To induce frameshift mutations, three guide RNAs (gRNAs) were designed to target a constitutive exon affecting all known isoforms of GLI3. After nucleofection with the gRNAs and GFP-fused Cas9, GFP+ cells were isolated by FACS and plated at low density on irradiated MEFs. Individual colonies were isolated 2 weeks later and harvested for genomic DNA. Frameshift deletions were confirmed by PCR amplification of the targeted GLI3 locus and subsequent sequencing. Protein was isolated from positive clones and immunoblotted for GLI3 to further confirm the absence of protein, as described previously (Wang et al., 2000).
Cell culture.
Targeted clones (GLI3.1 and GLI3.2) and the nontargeted clone (GLI3.3) were expanded and differentiated as knock-out and control lines, respectively. For the “early patterning” condition, neural induction medium included 1 μm RA from days 1–16. For the “late patterning” condition, neural induction medium included 1 μm RA from days 6–21.
Flow cytometry
Cells were dissociated with accutase for 20–30 min at 37°C, followed by resuspension in medium containing Y27637 (10 μm) and removal of cell clusters using a 40 μm filter. Cell sorting was performed on FACSAria (BD Biosciences) and purified cells were replated in medium containing Y27637 (10 μm) for 24 h after sorting. Data analysis was performed with FlowJo software (TreeStar).
Immunocytochemistry
Fixation and staining.
Cultures were fixed with 4% paraformaldehyde for 15 min and washed with PBS. Blocking was performed in PBS containing 1% BSA supplemented with 0.3% Triton for permeabilization for 30 min to 1 h. Cells were incubated in primary antibodies diluted in blocking buffer (without Triton) overnight at 4°C, followed by several washes with PBS, and then incubation in Alexa Fluor-labeled secondary antibodies (Invitrogen) diluted 1:400 in blocking buffer for 30 min to 1 h. Please refer to the antibody list in Table 1 for details on sources and concentrations.
Table 1.
Primary antibodies
| Antibody | Species, isotype | Company | Dilution |
|---|---|---|---|
| GFP | Chicken | Abcam | 1:1000 |
| HB9 | Mouse, IgG1 | DSHB | 1:100 |
| NKX6.1 | Mouse, IgG1 | DSHB | 1:100 |
| ISL1/2 | Mouse, IgG2b | DSHB | 1:100 |
| LIM3 | Mouse, IgG1 | DSHB | 1:100 |
| CHAT | Goat | Chemicon | 1:100 |
| TUJ1 | Mouse or rabbit | Covance | 1:500 |
| MAP2 | Mouse | Sigma-Aldrich | 1:200 |
| PAX6 | Rabbit | Covance | 1:75 |
| SMI32 | Mouse | Millipore | 1:1000 |
| OLIG2 | Rabbit | Millipore | 1:200 |
| FOXA2 | Goat | Santa Cruz Biotechnology | 1:100 |
| GLI3 | Rabbit | Wang et al., 2000 | 1:100 |
DSHB, Developmental Studies Hybridoma Bank.
Image quantification.
For marker quantification, images were acquired using the Operetta (PerkinElmer) with a 20× objective and analyzed with Harmony High Content Analysis Software version 3.0.
Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer's protocol (Invitrogen). cDNA was reverse transcribed from 1 μg of RNA using the QuantiTect Reverse Transcription kit (QIAGEN). HPRT was used as a reference gene for normalization. For quantitative real-time PCR (qRT-PCR), primers were acquired from Applied Biosystems (excluding RAR-β2, which was designed individually) and reactions were run according to manufacturer's protocol on the Mastercycler RealPlex2 (Eppendorf).
Global transcriptome profiling.
RNA was isolated using TRIzol with additional purification using the RNEasy column (QIAGEN) and submitted to the Memorial Sloan Kettering Cancer Center (MSKCC) Integrated Genomics Operation Facility for processing. Two biological replicates were submitted per condition. Paired-end (50 × 50 bp) sequencing was performed on an Illumina HiSeq 2000. For RNA-seq datasets, all paired end reads were mapped to hg19 using Tophat version 1.3.3 with default parameters. Mapped reads were then quantified using Cufflinks and Cuffdiff or exported to the Partek Genomics Suite. Significant differences were determined using a fold change of >2 and a q value <0.05.
ChIP sequencing
Cell culture.
Samples were fixed in 1% formaldehyde after 24 h of neural induction before initiating patterning (day 1) or after 8 d of neural induction in the absence or presence of patterning factors. Patterning factors were 1 μm RA or 1 μm Pur initiated at day 1 and maintained for the experiment duration.
ChIP sequencing and analysis.
ChIP was performed using the SimpleChip Plus Enzymatic Chromatin IP kit with magnetic beads (#9005; Cell Signaling Technologies) following the manufacturer's protocol. Chromatin fragments were immunoprecipitated overnight at 4°C using the specific antibodies against H3K4me3 (Abcam 8580; 5 μg per reaction), H3K27me3 (Millipore 07-449; 5 μg per reaction), RAR-α (Santa Cruz Biotechnology sc-551X; 5 μg per reaction), RAR-β (Santa Cruz Biotechnology sc-552X; 5 μg per reaction), and IgG control (Abcam 46540). Samples and their respective 2% input controls were submitted to the MSKCC Integrated Genomics Operation Facility for processing. Paired-end (50 × 50 bp) sequencing was performed on an Illumina HiSeq 2000. For ChIP-seq data, all paired reads were mapped to the human reference genome (hg19) using bowtie version 0.12.7 with default settings allowing for one mismatch. Peak calling was performed using HOMER using the findPeaks algorithm (Heinz et al., 2010). Each sample was compared with their respective 2% input control treatment. Aligned reads were visualized using the Integrative Genomics Viewer (IGV).
Electrophysiology
Whole-cell current-clamp recordings were performed at room temperature (23–24°C) using a Multiclamp 700B amplifier (Molecular Devices) as described previously (Ying and Goldstein, 2005; Ying et al., 2007). Recorded neurons were selected using a Nikon Eclipse FN-1 microscope equipped with a 4× objective and 40× water-immersion objective. Data were acquired using pClamp 10.2 at 10 kHz, and digitized using a Digidata 1440A A/D interface (Molecular Devices). During recordings, cells were continuously perfused with freshly prepared extracellular solution, which contained the following (in mm): 126 NaCl, 26 NaHCO3, 3.6 KCl, 1.2 NaH2PO4, 5 MgCl2, 0.5 CaCl2, and 10 glucose, and the solution was saturated with 95% O2–5% CO2. The intracellular solution contained the following (in mm): 135 K-gluconate, 5 NaCl, 10 HEPES, 0.5 EGTA, 3 Mg-ATP, 0.2 Na-GTP, and 10 Na2-phosphocreatine, pH adjusted to 7.3 with KOH, with an osmolarity of ∼290 mOsm. Biocytin (0.5%) was added to the intracellular solution for post hoc analysis. Input resistance was determined by the current response elicited by applying a 75 ms, 5 mV hyperpolarizing voltage step at an initial membrane potential of −70 mV. Action potential (AP) firing was evoked using depolarizing DC current step injections (5–50 pA; 5 pA/step, 1000 ms duration). Liquid junction potentials were calculated and corrected online. Data were analyzed using Clampfit (Molecular Devices) and SigmaPlot 11 (Systat Software) and are presented as mean ± SEM.
Calcium imaging
RA-alone- and RA + Pur-derived MN cultures plated on PO/LAM/FN-coated 40 mm glass coverslips were subjected to calcium imaging between days 55 and 70, as described previously (Barreto-Chang and Dolmetsch, 2009). Briefly, cells were incubated with 5 μm Fura-2 (Life Technologies) for 30 min at 37°C before imaging. Coverslips were mounted in a FCS2 imaging chamber (Bioptechs) and cells were perfused with normal Tyrode's saline solution that contained the following (in mm): 125 NaCl, 30 glucose, 25 HEPES, 5 KCl, 2 CaCl2, and 1 MgCl2 with the addition of 0.1% BSA w/v, pH 7.4. For activation, cells were perfused with Tyrode's solution containing glutamate (100 μm) in the presence or absence of the glutamate receptor antagonists AP5 (50 μm) and/or DNQX (10 μm). Images were acquired every 5 s at 340 and 380 nm wavelengths using an Axiovert 200M Inverted Microscope with a 40 × 1.3 numerical aperture oil-immersion objective (Zeiss). Ratiometric analysis was performed using MetaMorph Software (Molecular Devices).
C2C12 muscle coculture
C2C12 myoblasts (ATCC) were cultured as described previously (Son et al., 2011). Briefly, C2C12 myoblasts were cultured with 50-d-old RA-derived MN cultures for 8 d and spontaneous contractions were measured. Application of 2 μm vecuronium was used to inhibit acetylcholine-dependent contractions.
Statistics
Unless otherwise noted, results are reported as the mean ± SEM of at least three independent experiments. One-way ANOVA with Dunnett's test was used to analyze multiple conditions against a control condition. Unpaired t tests were used to compare two conditions. Comparisons were determined statistically significant if p ≤ 0.05 with degree of significance denoted by asterisks (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). Data were analyzed and presented using Prism version 6.0c (GraphPad).
Results
High-yield MN differentiation from hESCs requires early exposure to patterning factors
Neural induction is a complex process involving temporal epigenetic, transcriptional, and molecular changes that result in divergent cellular states exhibiting different responses to patterning cues. We sought to understand how these stage-dependent competencies toward specific signaling pathways contribute to MN specification from hPSCs. To this end, we first defined the optimal window of competency for MN derivation under dSMADi conditions. The specific inhibitors used for dual-SMAD inhibition in this study were LDN193189 and SB431542 (LSB). The dSMADi protocol is particularly suitable for such studies because it converts hPSCs into a nearly homogenous population of neuroepithelial cells in a synchronized manner (Chambers et al., 2009). Furthermore, it has been shown previously that dual SMAD inhibition is suitable for MN induction and can enhance the robustness of MN derivation across cell lines (Boulting et al., 2011). Using the HB9::GFP bacterial artificial chromosome transgenic hESC line that specifically expresses GFP in somatic MNs in the spinal cord and hindbrain (Placantonakis et al., 2009), we varied the timing of exposure to the canonical patterning factors RA and the SMO agonist (Pur) known to activate SHH signaling. RA + Pur exposure was initiated at day 1, 2, 4, or 6 of differentiation and maintained up to day 21 (with equal patterning duration for all conditions), when cells were assessed for the percentage of GFP+ cells by FACS.
We observed striking, time-dependent differences in the yield of HB9::GFP+ cells. RA + Pur exposure starting at the earliest time point (24 h after triggering differentiation) achieved the highest yield, with up to 30% of total cells expressing HB9 (Fig. 1A). The overall efficiency of MN induction is likely even higher given the presence of ISL1+/HB9− MNs (Amoroso et al., 2013). The ability to specify MNs rapidly decreased over time, with the lowest induction efficiency observed when patterning was initiated at day 6 of differentiation (when PAX6 is already expressed). Interestingly, the yield obtained under those suboptimal, day 6 patterning conditions was comparable to previously published results using neural-rosette-based MN protocols (Lee et al., 2007; Placantonakis et al., 2009). Furthermore, this patterning window is even earlier than recently published strategies using LSB treatment in combination with embryoid-body-based neural differentiation (Amoroso et al., 2013) or compound C for neural induction (Qu et al., 2014). Although most published hPSC-based MN induction protocols initiate patterning at the early PAX6+ neural progenitor stage, our results point to an earlier window of competency for achieving optimal HB9+ MN yields using dSMADi.
Figure 1.
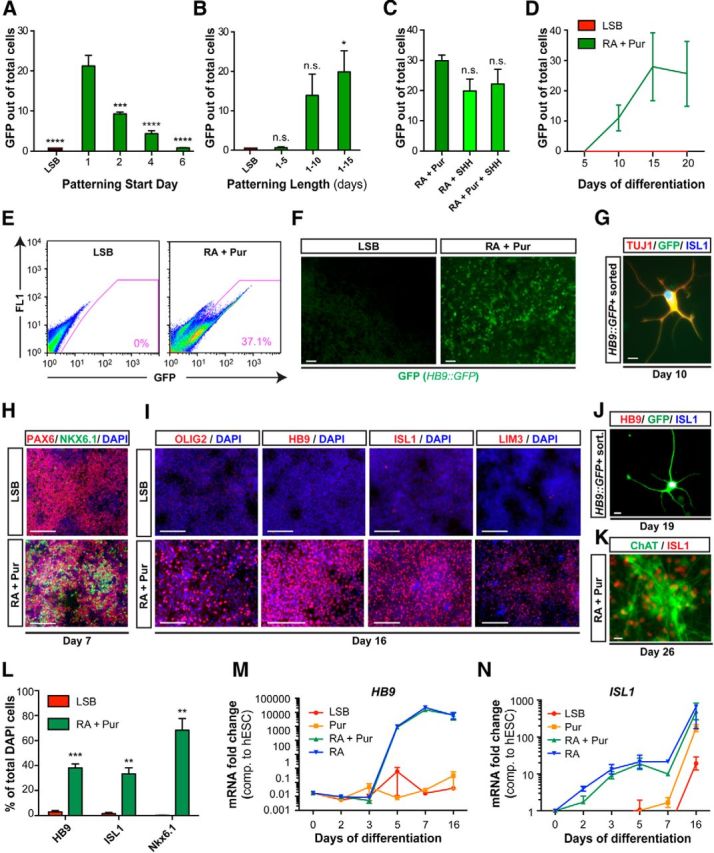
Efficient derivation of MNs from hESCs upon early exposure to patterning cues. Exposure to patterning factors at a very early neural stage (A) during dSMADi for at least 2 weeks (B) efficiently specifies MNs as assessed by percentage of GFP+ cells using an HB9::GFP BAC hESC line. Data are shown as mean ± SEM; n = 3 independent experiments; ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05 versus RA initiated at day 1 (A) and LSB (B), Dunnett test. C, SHH exposure in place of or in addition to Pur does not improve yields. Data are shown as mean ± SEM; n = 2 independent experiments versus RA + Pur, Dunnett test. D, Acquisition of GFP occurs rapidly, with cultures expressing up to 40% GFP+ after 2 weeks of patterning. E, Representative FACS plot for GFP at day 16 for LSB and RA + Pur. F, Live GFP expression in LSB and RA + Pur cultures at day 16. Scale bar, 10 μm. G, Isolated GFP+ cells at day 10 already coexpress ISL1 (blue) and TUJ1 (red) by immunocytochemistry. Scale bar, 10 μm. H, Immunocytochemistry at day 7 for PAX6 (red) and NKX6.1 (green) demonstrates presence of MN progenitors within 1 week of differentiation. Scale bars, 100 μm. I, Immunocytochemistry at day 16 for OLIG2, HB9, ISL1, and LIM3 verifies MN identity. Scale bars, 100 μm. J, K, After 3 weeks of differentiation, MNs exhibit more mature features including neuronal morphology as shown by immunocytochemistry for HB9 (red), ISL1 (blue), and GFP (green) of GFP+ isolated cells at day 19 and by expression of ChAT (green) and ISL1 (red) at day 26. Scale bars, 10 μm. L, Quantification of markers presented in I. M, N, Evaluation of patterning factors individually reveals striking ability of RA to induce MN markers HB9 and ISL1 efficiently in the presence and absence of SHH activation (Pur) as assessed by qRT-PCR.
Signaling pathways influence cell fate specification not only by the strength of their activation but by also the duration of their signal (Ericson et al., 1996; Dessaud et al., 2007). Because we observed that optimal efficiency requires patterning at very early stages of neural induction, we next wanted to define the length of patterning cue exposure required for efficient MN specification. RA + Pur was introduced at day 1 and maintained for various time periods (5, 10, and 15 d). Cultures were assessed for percentage of GFP+ cells at day 16. We observed that 15 d of patterning resulted in optimal MN yield, a timeframe similar to that reported in rosette-based protocols, with no significant improvement upon continued exposure (Fig. 1B). These data suggest that there are stage-specific differences in competency toward MN fate in response to RA + SHH signaling, but that the optimal length of exposure to patterning factors is independent of starting stage. Furthermore, neither substituting Pur with the recombinant protein SHH C25II nor combining SHH and Pur treatment improved efficiency (Fig. 1C). Therefore, using a systematic approach to studying the timing and duration of the patterning signals during neural induction, we demonstrate that exposure at very early stages of differentiation, before neural commitment, is critical to deriving human MNs at high yields.
The 2 week requirement of exposure to RA + Pur suggests a lag between competency and commitment. To determine how quickly commitment occurs under these optimized temporal conditions, we assessed the sequence of MN marker acquisition. We first monitored the percentage of GFP+ cells by FACS over time. Significant induction of GFP occurred by 10 d of differentiation, with up to 40% of cultures expressing GFP by day 14–16 (Fig. 1D–F). Sorting for GFP at day 10 isolated cells that coexpress the neuronal marker TUJ1 and the MN marker Islet1 (Fig. 1G). Because we observed such rapid induction of the postmitotic MN marker HB9, we investigated onset of expression of the MN progenitor marker NKX6.1, as well as other independent markers of MN fate (Fig. 1H–L). Striking coexpression of NKX6.1 and PAX6 was observed as early as 1 week after differentiation (Fig. 1H), with 68.3 ± 9.5% of the cultures acquiring NKX6.1 within 16 d (Fig. 1L). The canonical MN markers OLIG2, HB9, ISL1, and LIM3 were expressed by week 2, whereas coexpression of ISL1 with ChAT (the enzyme for acetylcholine) was observed by 3–4 weeks of differentiation (Fig. 1I–K).
RA in the absence of extrinsic SHH activators yield MNs at high efficiencies from both hESCs and iPSCs
After identification of the neural stage most competent for MN induction, we next sought to determine the contribution of each signaling pathway to this rapid specification. Derivation of other tissue types such as floor plate and midbrain dopamine neurons that rely on induction by SHH require early patterning; therefore, we speculated that our narrow window of competency might relate to this dependence on sonic signaling (Fasano et al., 2010; Kriks et al., 2011). To address whether early SHH exposure is the critical factor for high-yield MN derivation, we manipulated the timing of patterning cues individually and assessed induction of canonical MN markers. We first introduced RA alone, Pur alone, or RA + Pur on day 1 and evaluated the temporal transcription of HB9 and ISL1 over the first 2 weeks of differentiation. Although Pur alone did not induce HB9 expression, treatment with RA alone triggered upregulation of both ISL1 and HB9 transcripts as early as day 5 (Fig. 1M,N). RA-only exposure drove transcription of both markers with nearly identical kinetics and at comparable levels of induction compared with RA + Pur. In addition to inducing MN-related transcripts, we observed that both the total number of GFP+ cells and the kinetics of HB9 acquisition as assessed by FACS was indistinguishable in cultures treated with RA alone versus those treated with RA + Pur (Fig. 2A). This was surprising because SHH is an essential morphogen during MN development and mice lacking SHH fail to generate ventral neural cell types, including MNs (Litingtung and Chiang, 2000; Bai et al., 2004). Our data reveal that early RA treatment triggers MN differentiation efficiently without the need for adding extrinsic activators of SHH signaling, suggesting a dual role for RA in providing both caudal and ventral cues to hPSCs.
Figure 2.
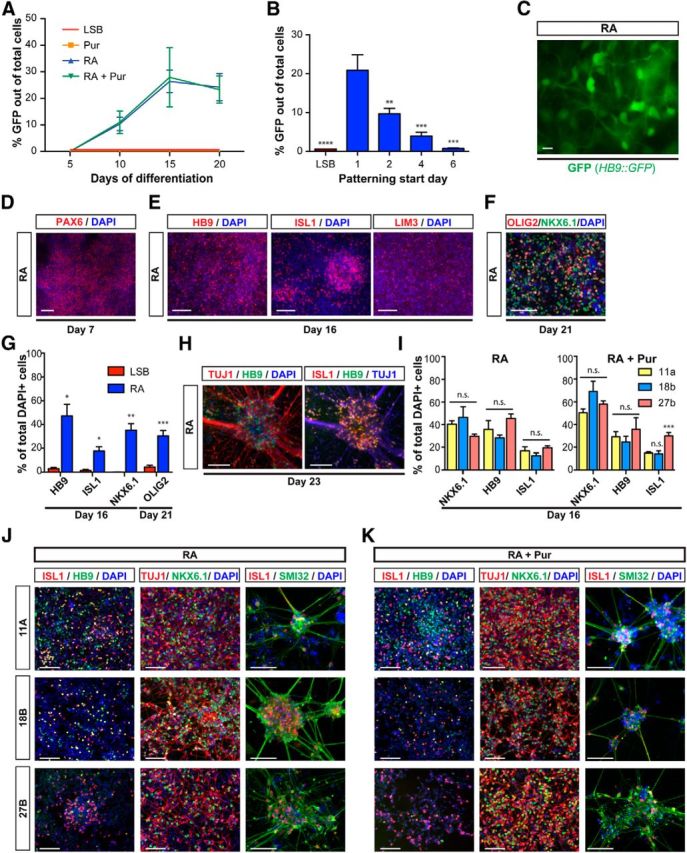
RA induces MNs from hESCs and iPSCs in the absence of extrinsic SHH activation. A, Early exposure to RA efficiently derives MNs from hPSCs with comparable yields and timing in the presence and absence of Pur. B, Patterning with RA achieves highest MN yields when initiated at a very early neural stage (day 1). Data are shown as mean ± SEM; n = 3 independent experiments; ***p < 0.001; **p < 0.01; *p < 0.05 versus RA initiated at day 1, Dunnett test. C, Live GFP expression in RA-patterned culture at day 16. Scale bar, 10 μm. Immunocytochemistry at day 7 for PAX6 (D), at day 16 for HB9, ISL1, and LIM3 (E), and at day 21 for NKX6.1 (green) and OLIG2 (red; F) confirms MN identity of RA-derived cultures. G, Quantification of markers presented in E and F. Immunocytochemistry at day 23 for HB9, ISL1, and TUJ1 (H) shows MNs at a more mature stage. I–K, Immunocytochemistry at day 16 for ISL1 (red) and HB9 (green; left), NKX6.1 (green) and TUJ1 (red; middle), and at day 23 for SMI32 (green) and ISL1 (red; right) for MNs derived from control (11A and 18B) and ALS patient-derived (27B) iPSCs via patterning with RA (J) or RA + Pur (K). I, Quantification of markers in J and K demonstrating similar, efficient propensity to generate MNs across multiple lines regardless of disease status. Scale bars, 100 μm unless labeled otherwise.
We next evaluated how the timing of RA exposure affects MN yield. If RA alone is sufficient to drive MN induction, then its window of patterning should remain constant regardless of exposure to an SHH agonist. Measuring HB9::GFP+ cells at week 3 of differentiation, we obtained the highest percentage of MNs upon very early RA-alone treatment (starting on day 1), with yields progressively declining after delayed RA treatments (Fig. 2B). Therefore, the early, narrow patterning window for high-yield MN induction depends primarily on RA exposure.
The derivation of HB9+ cells by RA was associated with the expression of appropriate neural and neuronal markers (PAX6 and TUJ1), as well as various additional MN markers (OLIG2, HB9, ISL1, NKX6.1, LIM3), confirming their identity (Fig. 2D–H). Importantly, RA-alone treatment was sufficient to trigger MN induction efficiently across all hPSC lines tested, including iPSC lines derived from an ALS patient (27b) and healthy controls (11a, 18b) that had been shown previously to exhibit highly variable MN yield (Boulting et al., 2011). Quantification of the MN markers HB9, ISL1, and NKX6.1 after 2 weeks of differentiation revealed no significant differences between lines that previously exhibited lower (11a) or higher (18b) MN potential (Fig. 2I–K). MN generation was not observed during standard neural induction (LSB) without patterning. These data highlight the robustness of our protocol and its applicability for disease-modeling studies. Therefore, we present here a stage-dependent, robust strategy to direct the differentiation of MNs from hPSCs using RA alone in the absence of SHH.
RA-induced MNs exhibit appropriate functional properties
To determine whether RA alone can pattern hPSCs into mature, functionally active MNs, we analyzed the biophysical properties of MNs derived upon early RA treatment in the presence and absence of Pur. Whole-cell patch-clamp recordings (Fig. 3A) were performed in parallel on RA-alone- and RA + Pur-patterned cultures at 38–45 d of differentiation. There were no significant differences in resting membrane potential (−50.1 ± 1.9 mV, n = 15, and −58.4 ± 2.8 mV, n = 9, respectively). However, a difference in input resistance [1233 ± 133 (n = 15) and 870 ± 105 (n = 9), p = 0.02, for RA alone and RA + Pur respectively; Fig. 3B] suggested a slight difference in maturity. Spontaneous AP firing was observed in both groups with AP overshoot in excess of 0 mV (Fig. 3C).
Figure 3.
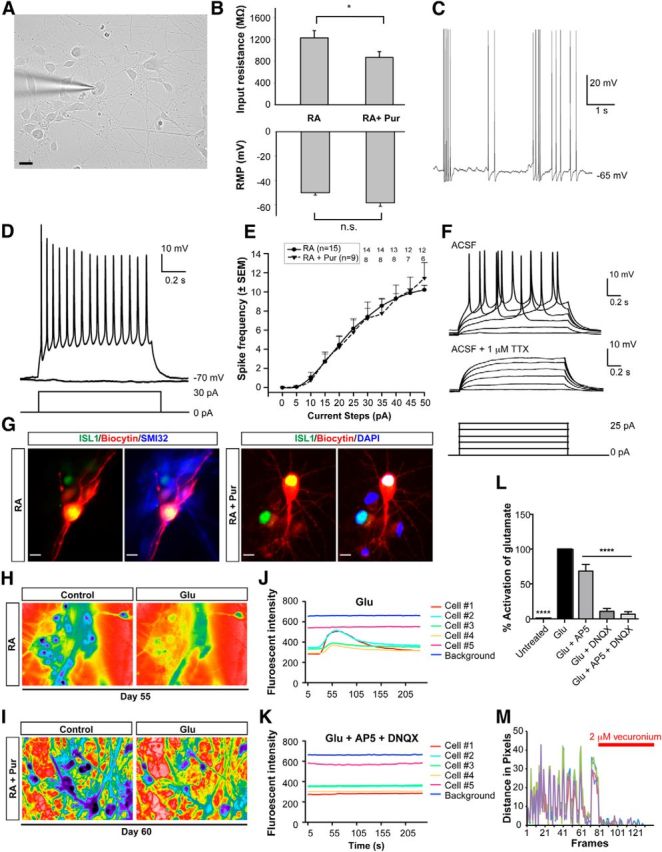
Functional characterization of MNs patterned with RA alone versus RA + Pur. A, Differential interference contrast photomicrograph of a representative patch-clamped MN at 38 d of differentiation. Scale bar, 20 μm. B, Graphs showing the comparison of input resistance and resting membrane potential (RMP) of RA-alone- versus RA + Pur-induced MNs. *p < 0.05. C, Spontaneous APs (at the indicated membrane potential) were readily observed in an RA + Pur-induced MN. D, In a different RA + Pur-derived neuron than in C, a representative voltage trace demonstrating sustained, tonic AP firing in response to a 30 pA depolarizing current step (current protocol shown below). E, Graph showing relation between firing frequency and injected current pulse. Data are shown as mean ± SEM of all the data points from RA (n = 15) and RA + Pur (n = 9) until 25 pA, after which the number of data points are indicated for each current step. F, AP firing was blocked by the Na+ channel blocker tetrodotoxin (TTX) in an RA-only-derived neuron. G, Immunocytochemistry for ISL1 and SMI32 (left) or DAPI (right) in biocytin-filled cells confirms MN identity of recorded cells corresponding to D and F. Scale bars, 10 μm. H–L, Both RA- and RA + Pur-derived MNs exhibit an excitatory calcium response after glutamate exposure. Representative ratiometric images of identical MNs in the absence and presence of glutamate (100 μm) patterned with RA (H) or RA + Pur (I). Calcium traces in response to glutamate activation (J) and inhibition of this response by application of glutamate receptor antagonists AP5 (50 μm) and DNQX (10 μm; K) in RA-derived MNs. L, Quantification of activation in response to glutamate exposure in the presence and absence of AP5 and/or DNQX (RA n = 16, RA + Pur n = 16). There was no significant difference in activation levels between MNs derived with RA versus those derived with RA + Pur. Data are shown as mean ± SEM; ****p < 0.0001 versus GLU, Dunnett test. M, Quantification of spontaneous contractions observed upon coculture of RA-derived MNs and C2C12 myoblasts that are suppressed by application of the acetylcholine receptor antagonist vecuronium (2 μm).
The progression from firing single to trains of APs reflects the gradual maturation of MNs as shown by in vitro studies of primary rat, mouse, and hESC-derived MNs (Miles et al., 2004; Soundararajan et al., 2006; Takazawa et al., 2012). MNs patterned with RA both in the presence and absence of Pur exhibited the repetitive firing behavior characteristic of mature MNs. From an initial membrane potential of −70 ± 2 mV, tonic AP firing was evoked by injection of depolarizing currents. All of the cells that were recorded exhibited APs and sustained firing (Fig. 3D). In both groups of neurons, the AP frequency increased with the amplitude of the current pulse (Fig. 3E) and there was no statistically significant difference between the groups. APs were blocked by tetrodotoxin (Fig. 3F, n = 2), consistent with their generation resulting from the activation of voltage-dependent Na+ channels. Recorded neurons were filled with biocytin and later stained with ISL1 to confirm MN identity (Fig. 3G). These data demonstrate that RA specifies MNs with appropriate biophysical properties independently of SHH agonist exposure.
Spinal MNs receive their afferent input from glutamatergic upper MNs that reside in the motor cortex. If our cells represent bona fide MNs, then they should be able to respond to glutamatergic stimulation. To test this directly, we infused glutamate into 55- to 60-d-old RA-alone and RA + Pur-derived MN cultures while measuring calcium response to monitor neuronal activity. Glutamate exposure activated the vast majority of neurons (14/16 and 13/16) for both RA-alone- and RA + Pur-derived neurons, respectively (Fig. 3H,I). Application of the glutamate receptor specific antagonists for NMDA (AP5) and kainate and AMPA (DNQX) inhibited the calcium response to glutamate, verifying the functionality of these receptors (Fig. 3J–L). These data demonstrate that MNs derived both in the presence and absence of exogenous SHH readily respond to glutamatergic stimuli appropriate for their physiological role within the motor circuit.
Upon stimulation by cortical MNs, spinal MNs relay this activity to the periphery, where they release acetylcholine into the neuromuscular junction to contract muscle. Coculturing ESC-derived MNs with C2C12 myoblasts models neuromuscular junction formation in vitro and presents a platform for studying MN functionality (Harper et al., 2004; Miles et al., 2004; Son et al., 2011; Umbach et al., 2012). Using this assay, we observed myotube formation upon 1 week of coculturing RA-patterned MNs with C2C12 mouse myoblasts, followed by the appearance of spontaneous contractions. Muscle contractions were inhibited by treatment with the acetylcholine receptor antagonist vecuronium (2 μm), verifying that the contractions were a direct response to acetylcholine release (Fig. 3M). RA-alone-derived MNs, therefore, display the gold standard of functional MN maturity—the ability to contract skeletal muscle. Because the presence or absence of exogenous SHH neither affected the propensity to generate MNs nor the ability of these neurons to mature and display characteristic functional properties, we show here that, when initiated early during neural induction, RA acts as the primary and sufficient stimulus to trigger MN induction under dSMADi conditions.
RNA sequencing demonstrates highly similar transcriptional profiles in HB9+ cells patterned with RA alone versus RA + Pur
Both molecular and functional parameters suggest that the MN populations generated in the presence or absence of extrinsic SHH activation are highly comparable. However, these analyses do not take into account the possibility that the two conditions yield distinct MN subtypes. Diverse transcriptional programs define the different subtypes and topographical organization of MNs within the spinal cord and hindbrain (Dasen et al., 2003; Dasen et al., 2005; Peljto et al., 2010; Mazzoni et al., 2013a). To address whether exogenous SHH activation influences the identity of our MNs, we performed RNA sequencing in purified HB9+ MNs derived in the absence or presence of SHH agonists and in parallel unsorted LSB cultures that represent anterior neuroectodermal precursors (all at day 21 of differentiation; Fig. 4A). Patterning factors were removed 3 d before analysis. Our data confirmed the induction of transcripts characteristic of MN fate (HB9, ISL1, NKX6.1) in both RA-alone- and RA + Pur-derived GFP+ but not in LSB populations (Fig. 4B). Expression of the MN-specific NMDA subunit NR3B and the cholinergic-neuron-specific ChAT transporter (SLC5A7) genes were also enriched in both RA-alone- and RA + Pur-derived HB9+ populations compared with LSB (Fig. 4B).
Figure 4.
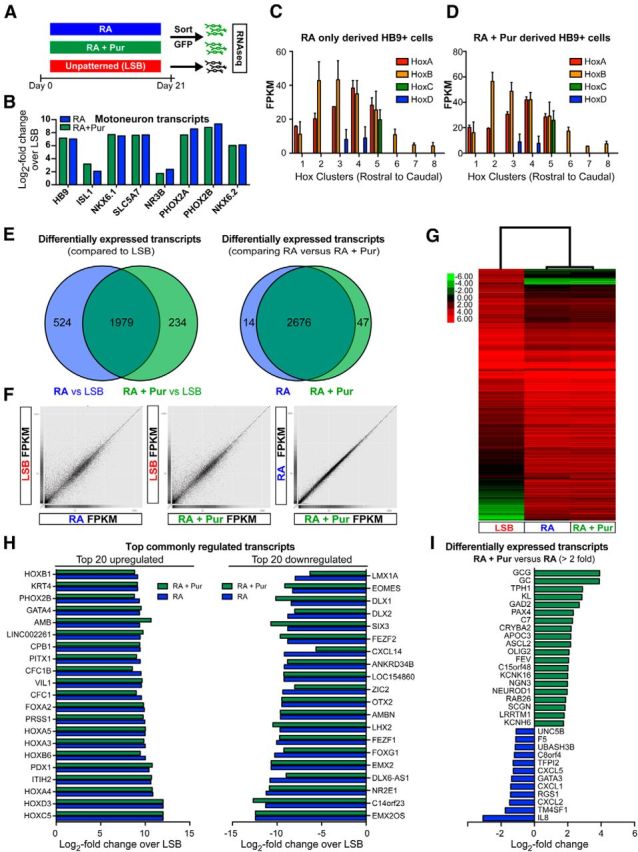
Transcriptome analysis of HB9::GFP-purified MNs derived by RA alone versus RA + Pur treatment. A, Schematic illustration depicting experimental paradigm for RNA sequencing. B, Selective expression of MN transcripts in isolated GFP+ cells derived with RA or RA + Pur compared with LSB. Hox gene expression demonstrates nearly identical anteroposterior identity of MNs derived in the absence (C) and presence (D) of extrinsic SHH. E, Comparison of genes differentially expressed between LSB and RA-alone- or RA + Pur-derived GFP+ populations shows significant overlap of MN populations. Of those differentially expressed genes compared with LSB, only 14 and 47 exhibit significant expression between RA-alone- and RA + Pur-derived MNs, demonstrating striking similarity between the populations. Scatterplots comparing FPKMs of individual genes (F) and hierarchical clustering of differentially expressed genes (G) further corroborate remarkable similarity between RA-alone- and RA + Pur-patterned MN populations. H, The top 20 commonly upregulated (left) and downregulated (right) genes expressed by RA- and RA + Pur-derived MNs compared with LSB. Genes are ranked by RA. I, Genes exhibiting significantly different (>2-fold) expression between RA + Pur and RA MN populations.
We next compared Hox gene expression profiles in the GFP+ populations to determine whether exposure to SHH agonists affects regional identity of the resulting MNs. Whereas LSB cultures were enriched for forebrain markers FOXG1 and OTX2, both HB9 populations expressed nearly identical patterns of Hox genes, with particularly high levels of clusters 2–5 reflecting a caudal hindbrain or cervical spinal cord fate (Fig. 4C,D). We further analyzed genes differentially expressed between MN and LSB conditions for both RA alone and RA + Pur. Interestingly, there was a highly significant overlap (p < 0.000001), with 1979 transcripts shared by both groups compared with LSB and merely 524 and 234 transcripts showing differential expression versus LSB selectively in RA alone or in RA + Pur conditions, respectively (Fig. 4E). Furthermore, the difference in the fold changes for these genes were minimal. In fact, only 14 and 47 genes exhibited unique expression patterns when comparing RA-alone- versus RA + Pur-derived MNs directly (Fig. 4E). Correlation matrices and hierarchical clustering of differentially expressed genes further confirmed the striking similarity between HB9 populations derived with or without extrinsic SHH exposure (Fig. 4F,G). Gene ontology analysis for the top commonly enriched transcripts in MN versus LSB conditions reflected changes in anteroposterior pattern formation, neurogenesis, neural fate specification, and hindbrain development, whereas downregulated terms included dorsoventral pattern formation, forebrain development, and neuron fate commitment (Fig. 4H). We found no statistically significant biological categories for genes uniquely expressed by RA-alone- or RA + Pur-patterned cells (Fig. 4I). In conclusion, our gene expression data show that early RA treatment triggers nearly identical transcriptional programs to specify MN identity regardless of exposure to SHH agonists.
RA drives high MN yield in presence of SHH antagonists
SHH signaling plays a crucial role in defining dorsoventral position within the spinal cord, as evidenced by the absence of ventral progenitor domains, including the MN progenitor domain, in the spinal cords of mice lacking SHH or its receptor SMO (Litingtung and Chiang, 2000; Wijgerde et al., 2002; Bai et al., 2004). Previously published in vitro strategies for deriving MNs from mouse ESCs and hPSCs recapitulate these findings because they require SHH exposure to attain ventral cell types including MNs at high yield (Wichterle et al., 2002; Singh Roy et al., 2005; Placantonakis et al., 2009). Based on our current data, RA produces MNs largely indistinguishable from those derived in the presence of SHH agonists, raising the possibility that RA triggers endogenous activation of SHH signaling. To address whether RA treatment can trigger SHH signaling, we first assessed the levels of endogenous SHH transcript. We observed a striking temporal upregulation of SHH mRNA after early RA exposure (Fig. 5A) that occurs in a dose-dependent manner similar to the dose-dependent upregulation observed for the known RA-inducible gene RAR-β2 (Fig. 5B).
Figure 5.

Early RA exposure yields MNs in the presence of SMO antagonist. A, Early exposure to RA both alone or in combination with Pur progressively upregulates SHH transcript levels during neural induction. B, RA induces SHH transcripts in a dose-dependent manner that correlates with RA activity as measured by RA-inducible RAR-β2 mRNA expression. Data are shown as mean ± SEM; n = 3 independent experiments; ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05 versus 0 μm RA, Dunnett test. C, qRT-PCR for SHH mRNA at day 21 demonstrates that very early RA exposure is required for optimal SHH upregulation. Patterning duration (2 weeks) held constant between all conditions. Data are shown as mean ± SEM; n = 3 independent experiments; ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05 versus RA or RA + Pur initiated at day 1, respectively, Dunnett test. D, E, Early RA exposure in the absence or presence of Pur upregulated transcripts of floor plate markers FOXA2 (D) and F-SPONDIN and NETRIN-1 (E). F, Transcripts of the floor plate markers FOXA2 and NETRIN-1 require exposure to RA or RA + Pur at very early time points during neural induction as measured by qRT-PCR. G, Immunocytochemistry at day 16 for HB9 (green) and ISL1 (red; top) and NKX6.1 (red; bottom) reveals that RA induces postmitotic and progenitor MN markers in the presence of 5 μm CUR61414. H, Quantification of markers present in G. Data are shown as mean ± SEM; n = 3 independent experiments; ****p < 0.0001 versus RA, Dunnett test. Scale bars, 10 μm. I, Immunocytochemistry at day 11 for FOXA2 demonstrates that the SMO antagonist CUR61414 (5 μm) inhibits SHH-mediated, but not RA-mediated, induction of FOXA2.
We next addressed whether the competency of RA to induce SHH expression depends on the stage of cell differentiation. We initiated exposure to RA alone or RA + Pur at day 1, 2, 4, or 6 of differentiation for a fixed period of 2 weeks, followed by assessment of SHH transcript (day 21). We observed that the ability of RA to upregulate SHH was limited to early stages of differentiation (Fig. 5C) that parallel the window of competency for MN specification.
One potential explanation for the ability of RA to trigger MN differentiation is that induction of endogenous SHH obviates the need for extrinsic SHH exposure. The notochord and floor plate are the known sources of endogenous SHH during development. We observed induction of the floor plate markers FOXA2, F-SPONDIN, and NETRIN-1 (Fig. 5D–F), but not the notochord marker CNOT1 (data not shown), upon early RA exposure. To address whether floor plate-derived SHH is essential for RA-mediated MN derivation, we blocked hedgehog signaling using the SMO antagonist CUR61414, which should suppress activity emanating from all known hedgehog ligands (Sonic, Indian, and Desert hedgehog). We hypothesized that early hedgehog inhibition might block the formation of the SHH-secreting floor plate-like cells, whereas late treatment may prevent the secreted, endogenous SHH from specifying MN progenitors. We assessed the impact of both early (days 1–6) and late (days 6–16) treatment with CUR61414 (5 μm) during RA-mediated MN induction (days 1–16). Surprisingly, neither treatment condition significantly affected the induction of the progenitor or postmitotic MN markers NKX6.1 and HB9 (Fig. 5G,H). To ascertain that CUR61414 treatment is capable of inhibiting SMO-dependent responses of SHH signaling, we confirmed suppression of FOXA2 induction after early patterning with SHH (Fig. 5I). In addition, treatment with another SMO antagonist, cyclopamine, throughout RA treatment (days 1–16) did not prevent induction of the MN transcripts HB9 and ISL1 (data not shown). Therefore, our data demonstrate that induction of endogenous SHH does not account for the ability of RA to induce MNs in the absence of extrinsic activators of SHH signaling.
Early RA exposure modulates SHH signaling by suppressing GLI3 induction
SHH is thought to release the repression of SMO by patched, allowing SMO to relay signaling via the GLI proteins to induce hedgehog-dependent transcription (for review, see Dessaud et al., 2008). Because GLI transcription factors transduce the SHH signal downstream of the SMO receptor, we sought to determine whether RA could affect SHH signaling at this level. To this end, we compared expression of GLI1, GLI2, and GLI3 transcripts during the first 2 weeks of differentiation after patterning with RA alone, Pur, RA + Pur, or in the absence of patterning factors (LSB). RA modulated GLI1 transcription in a manner similar to Pur treatment by partially preventing the decrease in GLI1 levels observed during neural induction with LSB, although these data did not reach statistical significance (Fig. 6A). In contrast, RA exposure slightly decreased GLI2 levels compared with either LSB or Pur treatment (Fig. 6A).
Figure 6.

Early RA treatment suppresses the induction of GLI3 during neural induction. A, Patterning with RA does not upregulate transcripts of the canonical active SHH transducers GLI1 and GLI2, but prevents the progressive upregulation of GLI3 transcripts that occurs during LSB as measured by qRT-PCR. Data are shown as mean ± SEM; n = 3 independent experiments; *p < 0.05 versus LSB at day 15, Dunnett test. B, qRT-PCR of GLI3 mRNA at day 8 reveals that RA is capable of preventing GLI3 upregulation even in the presence of 5 μm CUR61414 (days 1–8). Data are shown as mean ± SEM; n = 3 independent experiments; *p < 0.05 versus RA, Dunnett test. C, qRT-PCR of GLI1, GLI2, and GLI3 transcript levels after a 4 d exposure to patterning cues initiated at different neural stages (day 1, day 6, or rosette) reveals reduced ability of RA to influence GLI expression at later neural stages. Data are shown as mean ± SEM; n = 3 independent experiments; ***p < 0.001; **p < 0.01; *p < 0.05 versus LSB for each time point, Dunnett test.
We next addressed whether RA could modulate GLI3, which commonly represses SHH signaling during development (Ruiz i Altaba, 1998; Litingtung and Chiang, 2000; Wijgerde et al., 2002; Bai et al., 2004). During LSB differentiation, we observed a progressive upregulation of GLI3, with levels reaching a 154.6 ± 63.7-fold increase in expression from day 0–15 of differentiation (p = 0.003). Remarkably, treatment with RA inhibited this induction as effectively as treatment with Pur, resulting in GLI3 levels only moderately elevated above those observed during pluripotency (7.26 ± 3.34-fold over day 0 for RA and 14.23 ± 6.42-fold for Pur; Fig. 6A). Treatment with SMO antagonists (CUR61414 or cyclopamine) during RA exposure (Fig. 6B; data not shown for cyclopamine) failed to block the RA-mediated suppression of GLI3 transcription, indicating that RA acts downstream of SMO within the SHH signaling pathway. This inhibition of GLI3 expression could explain how RA patterning in the absence of SHH exposure induces ventralization of neural precursors, enabling MN specification.
It has been shown previously that ectopic GLI3 expression can convert ventral domains toward more dorsal fates in vivo, disrupting MN differentiation (Persson et al., 2002). We wondered whether the upregulation of GLI3 during LSB-mediated neural induction could similarly restrict human MN fate and contribute to the narrow window of patterning observed in our studies. To address this, we first determined whether RA retains the competency to return GLI3 to pluripotency levels when introduced at later neural stages. We initiated RA alone, RA + Pur, or Pur at day 1, day 6, or at neuroepithelial precursor (rosette) stage of differentiation for a 4 d duration and quantified GLI expression. As expected, RA exposure initiated at day 1 maintained GLI3 to levels not significantly elevated above those at the pluripotency stage (4.2 ± 0.2-fold over day 0), but significantly reduced compared with neural induction using LSB only (16.5 ± 4.0-fold over day 0; Fig. 6C). When treatment was initiated at the early Pax6 stage (day 6), RA exposure did not yield a statistically significant suppression of GLI3 compared with LSB (31.7 ± 10.4-fold vs 119.3 ± 62.0-fold over day 0) and GLI3 levels were clearly elevated above those at the pluripotency stage. When starting treatment at the even later rosette stage, we observed no effect of RA on GLI3. In contrast to the suppression of GLI3 and GLI2, GLI1 levels showed a trend toward increased expression upon early RA exposure (Fig. 6C).
Suppressing GLI3 extends window of competency for MN derivation
SHH promotes ventral specification during spinal cord development by counteracting the repressive activity of GLI3 and its default induction of dorsal fates. Inactivating GLI3 in mice lacking SHH or SMO rescues the development of ventral progenitor domains in the telencephalon (Rallu et al., 2002) and spinal cord (Litingtung and Chiang, 2000; Wijgerde et al., 2002; Bai et al., 2004), suggesting that SHH acts in a derepressive manner. We wondered whether the narrow window of patterning could reflect the time period during which GLI3 levels are below a threshold permissible to MN specification. To address this hypothesis, we transduced hESCs to establish stable lines carrying a DOX-inducible RFP tagged GLI3 shRNAmir and verified that treatment with DOX for the first week of differentiation suppressed GLI3 upregulation in RFP+ isolated cells (Fig. 7A). We next tested whether suppression of GLI3 extends the window of patterning and enables RA to achieve high MN efficiency when initiated at later stages. Cells were exposed to RA for 2 weeks starting either at the early stage (day 1, “MN patterning”) or the late stage (day 6, “late patterning”) of differentiation in cells maintained in the presence or absence of DOX (days 0–6; Fig. 7B). MN yield was quantified by the percentage of cells expressing HB9 and ISL1 at day 21 (Fig. 7C). We observed robust induction of MNs using a late RA-patterning paradigm after DOX treatment (Fig. 7C,D). However, late RA exposure without DOX pretreatment or DOX treatment alone did not efficiently induce MNs. Although knock-down of GLI3 followed by RA treatment resulted in percentages of HB9+ cells that were not significantly different from those achieved upon early RA treatment, there was only a partial rescue for inducing ISL1+ cells. DOX exposure alone did not induce MN markers, but, in combination with early RA treatment, it slightly improved yields above our normal MN patterning condition.
Figure 7.
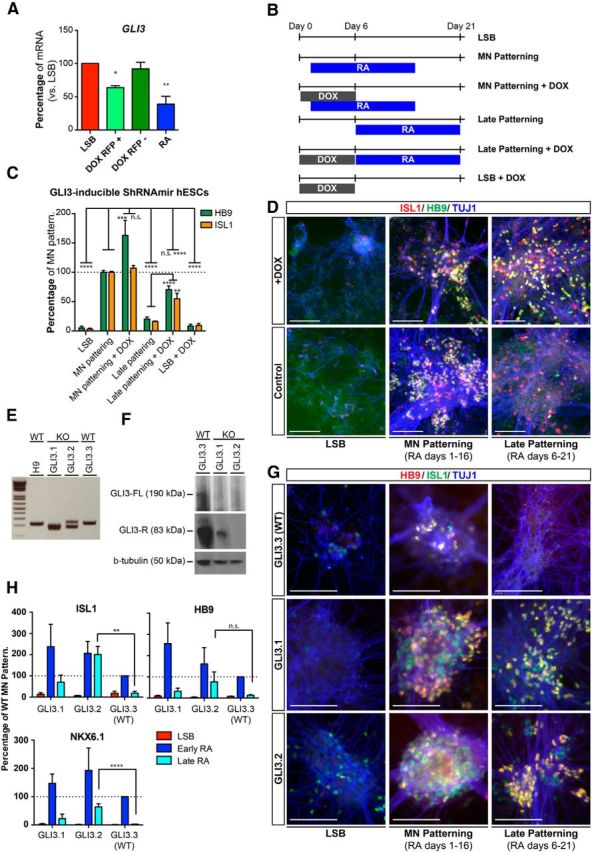
Genetic suppression of GLI3 extends the window of competency toward MN fate. A, qRT-PCR confirms suppression of GLI3 mRNA during neural induction in DOX-treated RFP+ cells but not RFP− cells or untreated cells using our DOX-inducible shRNAmir GLI3-RFP hESC line. Data are shown as mean ± SEM; n = 3 independent experiments; **p < 0.01; *p < 0.05 versus LSB, Dunnett test. B, Diagram depicting patterning paradigm for experiment. C, Quantification of immunocytochemistry at day 21 for HB9 and ISL1 shows that GLI3 suppression via DOX treatment significantly rescues ability of RA to derive MNs from hPSCs at a later neural stage. Data are shown as mean ± SEM; n = 3 independent experiments; ****p < 0.0001; ***p < 0.001; **p < 0.01 versus RA, Dunnett test; late patterning compared with late patterning + DOX using unpaired t tests, two-tailed. D, Immunocytochemistry at day 23 for ISL1 (red), HB9 (green), and TUJ1 (blue) demonstrates the ability to specify MNs with similar efficiency to early patterning when later patterning stages are combined with DOX-induced GLI3 suppression. Scale bars, 100 μm. E, PCR amplification of GLI3 from genomic DNA of control (H9 and GLI3.3) and knock-out lines (GLI3.1 and GLI3.2) confirms a migrational shift of the PCR products from the knock-out lines consistent with genomic deletion. F, Western blot for GLI3 verifies loss of GLI3 protein in CRISPR GLI3 knock-out hESC lines (GLI3.1 and GLI3.2) but not the untargeted control line (GLI3.3). G, Immunocytochemistry at day 29 for HB9 (red), ISL1 (green), and TUJ1 (blue) shows knock-out but not control lines have the ability to specify MNs at later patterning stages with similar efficiency to early patterning of control line. H, Quantification of immunocytochemistry at day 21 for ISL1, HB9, and NKX6.1 shows that GLI3 knock-out rescues the ability of RA to derive MNs from hPSCs at a later neural stage. Data are shown as mean ± SEM; n = 3 independent experiments; ****p < 0.0001; **p < 0.01 versus late RA in GLI3.3 line, Dunnett test.
To further validate that GLI3 suppression extends the window of competency, we generated stable GLI3 knock-out lines using CRISPR technology. Amplification of the GLI3 locus by PCR and subsequent sequencing revealed that lines GLI3.1 and GLI3.2 had targeted frameshift genomic deletions in exon 3 of GLI3, but line GLI3.3 did not (Fig. 7E and data not shown). Analysis of GLI3 protein expression by Western blot verified reduced GLI3 levels in GLI3.1 and lack of GLI3 in GLI3.2 (Fig. 7F). Therefore, we used GLI3.3 as a control line (WT) because it underwent the same targeting procedure but lacked alterations at GLI3. Repeating the same experimental paradigm performed for the shRNAmir lines, we quantified expression of the MN markers ISL1, HB9, and NKX6.1 upon early and late RA exposure in the CRIPSR lines. We observed a robust rescue of MN induction for ISL1 and NKX6.1 upon late RA exposure in the GLI3.2 line, whereas the GLI3.1 line showed only a trend toward improved MN marker expression. Rescue of HB9 was not significant in GLI3.1 or GLI3.2 lines at day 21 of differentiation, but continued to increase by day 29 (Fig. 7G). This indicates that residual GLI3 expression in GLI3.1 may hinder the rescue (Fig. 7G,H). Our data indicate that GLI3 restricts the window of patterning during MN specification and that manipulating its expression either by early patterning or via functional interference enables the efficient RA-dependent derivation of MNs from hPSCs. Therefore, we uncovered a novel mechanism for RA to affect SHH signaling and thereby bypass the need for extrinsic SHH treatment during MN specification.
RA prevents transition to active chromatin state at GLI3 during neural induction
The mechanisms of GLI3 regulation at the protein level and its function in developmental biology have been studied extensively (Litingtung and Chiang, 2000; Persson et al., 2002; Wijgerde et al., 2002; Ahn and Joyner, 2004; Bai et al., 2004). However, there is a lack of understanding regarding mechanisms controlling its transcriptional regulation. Recently, studies have begun to address such mechanisms, including the discovery of enhancer elements that regulate its developmental spatiotemporal expression (Abbasi et al., 2007; Abbasi et al., 2010; Coy et al., 2011; Hasenpusch-Theil et al., 2012). We showed that the induction of GLI3 by dSMADi is repressed with early exposure to RA and that repression of GLI3 extended the patterning window, allowing for MN induction by the addition of RA at later stages of differentiation. We next tested whether RA influences the chromatin state of GLI3. Previously published ENCODE data (Xie et al., 2013) suggest that, during neural induction using dSMADi, the chromatin landscape surrounding GLI3 transitions from a bivalent state (marked by the deposition of H3K4me3 and H3K27me3 together) in ESCs toward becoming solely H3K4me3 in neural precursor cells, suggesting its transcriptional activation. Interestingly, at the ESC stage, GLI3 has H3K27me3 deposited within the second intron. We hypothesized that RA may silence GLI3 signaling by promoting either the spreading of the intronic H3K27me3 to a greater repressive domain or resolving the bivalency toward the loss of H3K4me3. To this end, we performed ChIP-seq for H3K4me3 and H3K27me3 during MN differentiation. We compared four differentiation conditions: (1) the early stage of hESC differentiation when cells exhibit optimal competency toward patterning (day 1); (2) during RA-mediated MN induction shown to suppress GLI3 induction (day 8); (3) during activation of SHH signaling using Pur only (day 8); and (4) the standard neural induction protocol without any additional patterning factors (LSB; day 8) (Fig. 8A).
Figure 8.
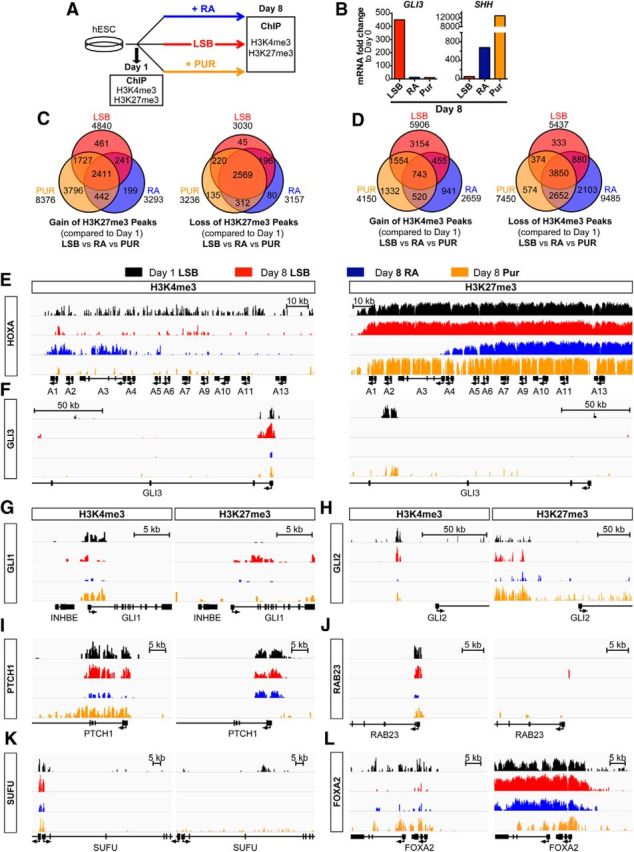
Early RA exposure prevents chromatin switch of GLI3 to active state during neural induction. A, Schematic illustration of experimental conditions for ChIP-seq of the histone modification marks H3K4me3 and H3K27me3. B, qRT-PCR for GLI3 and SHH on day 8 RNA extracted from parallel samples confirms appropriate GLI3 suppression and SHH induction upon RA or Pur exposure. C, Genes associated with gain (left) or loss (right) of ChIP-seq H3K27me3 peak enrichments for day 8 LSB, RA, and Pur compared with day 1. D, Genes associated with gain (left) or loss (right) of ChIP-seq H3K4me3 peak enrichments for day 8 LSB, RA, and Pur compared with day 1. E, H3K4me3 and H3K27me3 binding at the HoxA cluster exhibits characteristic gain of active and loss of repressive marks, respectively, at RA-mediated anteriorly expressed Hox genes upon RA exposure but not other treatment conditions. F, Enrichment for H3K4me3 in the promoter of GLI3 occurs during neural induction. Treatment with RA or Pur prevents this enrichment. H3K4me3 and H3K27me3 binding patterns demonstrate dynamic temporal changes during standard LSB neural induction at SHH-related genes, including: GLI1 (G), GLI2 (H), PTCH1 (I), RAB23 (J), SUFU (K), and FOXA2 (L). Early RA or Pur exposure attenuates these epigenetic events. All graphs have a y-axis (represents reads per million per base pair, RPM/bp) height of 8 on the IGV log scale except for the H3K27me3 graphs for HOXA, PTCH1, and FOXA2, which have a height of 30 on the IGV log scale.
Gene expression analysis was performed in parallel samples to confirm upregulation of SHH and suppression of GLI3 transcripts for RA and Pur compared with LSB (Fig. 8B). Comparison of genes that acquired H3K27me3 peaks (Fig. 8C) demonstrates acquisition of repressive marks in genes related to Wnt signaling and nonneural fates shared across patterning conditions (top gene ontology categories include digestive system development and TBOX—involved in limb and heart development). There was also a particularly high degree of overlap in genes that lost H3K27me3 peaks among all three patterning conditions. Gene ontology revealed loss of repressive marks predominantly related to neural induction, with top categories including: developmental proteins, neuron differentiation, embryonic morphogenesis, neuron development, and axonogenesis. These epigenetic changes are consistent with progressive neural fate acquisition.
Similarly, comparison of genes that acquired or lost active H3K4me3 peaks (Fig. 8D) among patterning conditions demonstrates a shared profile of acquiring peaks in genes involved in neural tube development and losing peaks in genes related to nonneural fates (i.e., top gene ontology categories included neural crest cell, uretric bud, and mesenchymal cell development). Interestingly, in contrast to LSB and Pur, RA primarily showed a loss rather than gain of unique H3K4me3 peaks, indicating that RA may act by restricting competency toward non-RA-mediated cell fates. Gene ontology revealed that RA exposure induced a loss of active marks at genes predominantly involved in cell division, with top categories including: cell cycle, centrosome, and microtubule organizing center. These results suggest that early RA exposure may limit competency while driving cells to differentiate. Furthermore, the ChIP-seq data showed expected RA-specific decreases in H3K27me3-repressive marks and increases in H3K4me3 active marks at the 3′ HOXA (Fig. 8E) and HOXB clusters (data not shown), confirming previous reports during mouse ESC differentiation (Mazzoni et al., 2013b).
To identify genes that acquire an active chromatin state during dSMADi-based neural induction, we determined the differential H3K4me3 peaks between LSB (day 8) and day 1. Unbiased analysis of the top 20 differential H3K4me3 peaks revealed dynamic changes occurring at genes associated with anterior neural progenitor fate, including PAX6, OTX1, OTX2, and SOX11. Remarkably, also among these top 20 unbiased peaks was a peak at the promoter of GLI3 that exhibited significant temporal dynamics (Fig. 8F). Initially in a bivalent or weakly active state at day 1, neural induction triggered a switch at the GLI3 locus to a strong active chromatin state at this site. Strikingly, early exposure to RA or Pur prevented this switch. The significance of this suppression is highlighted by the fact that this GLI3 peak also ranked among the top 20 unbiased differential peaks comparing LSB versus RA or LSB versus Pur.
We observed additional significant changes in chromatin state occurring at sites mapping to other members of the SHH pathway (Fig. 8G–L). Functional clustering of genes associated with changes in H3K4me3 peaks identified the hedgehog signaling pathway (Biocarta and KEGG pathways) as a significant category when comparing LSB with day 1 and with RA (p values 2.65E-07 and 1.92E-06, respectively). Significant changes accompanying neural induction primarily involved the acquisition of active marks or, occasionally, loss of repressive marks at genes that play inhibitory roles within the sonic signaling cascade, including SUFU and FKBP8 (Fig. 8K and data not shown). These modifications indicate an active chromatin state at negative regulators of hedgehog signaling, which may act to limit competency toward SHH signaling at the more advanced, Pax6+ neural precursor cell stage.
We next investigated whether exposure to early patterning cues alters those changes in epigenetic state that favor attenuated sonic signaling at later neural stages. Similar to its effect on GLI3, early patterning with RA prevented these switches to active chromatin state at these genes that typically counteract SHH signaling (PTCH1, RAB23, FKBP8, SUFU, and HHIP; Fig. 8I–K and data not shown). Comparison with Pur treatment revealed that RA exposure influenced the chromatin state as efficiently, if not more so, than activation of the canonical SMO-mediated SHH pathway at these genes. Intriguingly, despite both RA and Pur exposure readily inducing FOXA2 transcript levels during differentiation, their treatments exhibit divergent effects on the epigenetic state of FOXA2 (Fig. 8L). Unlike RA, Pur exposure enriches for H3K4me3 and reduces H3K27me3 marks around FOXA2. This is of particular interest because we have demonstrated earlier that Pur and RA induce FOXA2 expression via different mechanisms that are dependent or independent of SMO activation, respectively. Their different effects on the chromatin state of FOXA2 could reflect different mechanisms of action, thus providing an example of how epigenetic state may be not merely a readout of transcriptional activity, but can actually point to mechanism-specific actions. However, we also cannot rule out that the finding reflects a dosage effect because FOXA2 mRNA was induced to a greater extent in Pur versus RA in this experiment. Interestingly, the changes in chromatin state at PTCH1, a negative feedback regulator of SHH signaling, further indicate significant differences between Pur- and RA-treated cultures. Whereas Pur promotes an active chromatin state, reflected by the loss of H3K27me3 and an increase in H3K4me3, RA appears to promote the opposite effect, as indicated by the decrease in H3K4me3 and maintenance of the repressive H3K27me3 mark. Furthermore, in the case of the SHH-activating gene GLI1, which acquired H3K27me3 marks upon neural induction, both RA and Pur exposure prevented acquisition of this repressive mark (Fig. 8G).
These data suggest that epigenetic changes occur during dSMADi-based neural induction both at positive and negative mediators of SHH signaling and that RA prevents those changes in a pattern consistent with promoting chromatin states permissible to SHH signaling. Therefore, these effects on epigenetic state, particularly at the GLI3 promoter, could modulate competency toward MNs, and potentially other SHH-dependent fates, by restricting the window of patterning to very early stages of neural induction.
RAR binds to the GLI3 promoter upon early RA exposure
We next addressed whether RA triggers the changes in chromatin state observed at the GLI3 promoter and other SHH-signaling genes via a direct or indirect mechanism. RA mediates signaling through RARs, which can regulate transcription. We performed ChIP-seq using antibodies against the two RAR isoforms (α and β) primarily expressed in our MNs. We compared the binding pattern of RAR isoforms at day 8 during RA-mediated MN induction and standard neural induction (LSB; Fig. 9A). qRT-PCR for the RAR-β promoter was performed to confirm expected enrichment for RAR-α and RAR-β upon RA treatment at a known RAR target (Fig. 9B).
Figure 9.
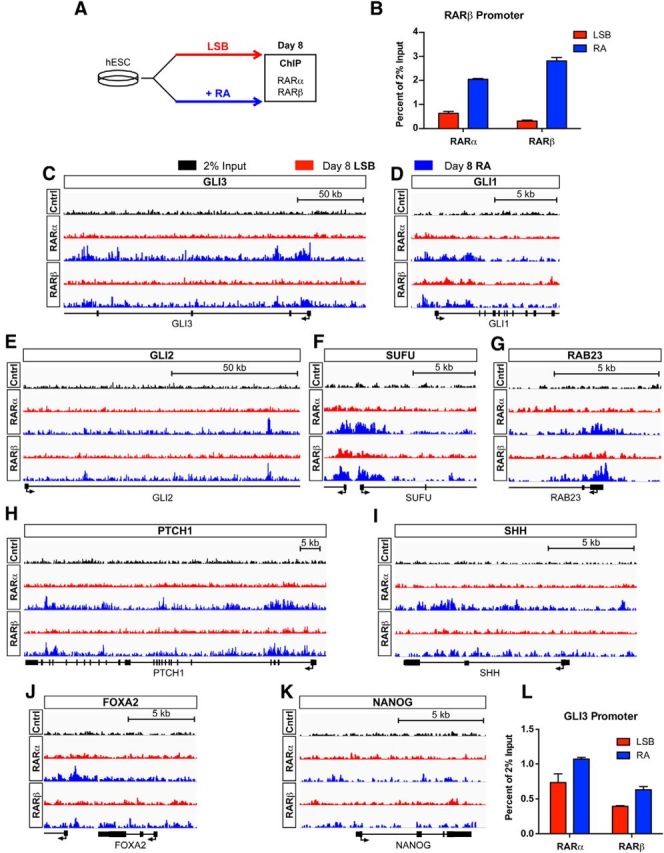
Early RA treatment induces binding of RAR at the GLI3 promoter and other SHH signaling genes during neural induction. A, Schematic illustration of experimental conditions for ChIP-seq of the RAR isoforms α and β. B, qRT-PCR for RAR-β promoter on day 8 ChIP confirms binding enrichment for RAR-α and RAR-β upon RA exposure compared with LSB. C, Enrichment for RAR-α and RAR-β in the promoter of GLI3 upon RA treatment. RA exposure alters RAR-α and RAR-β binding patterns compared with LSB at SHH-related genes, including: GLI1 (D), GLI2 (E), SUFU (F), RAB23 (G), PTCH1 (H), and SHH (I). RA treatment did not enrich for RAR-α or RAR-β binding at FOXA2 (J) or NANOG (K). All ChIP-seq graphs have a y-axis (represents reads per million per base pair, RPM/bp) height of 15 on the IGV linear scale. L, qRT-PCR for GLI3 promoter on day 8 ChIP confirms binding enrichment for RAR-α and RAR-β upon RA exposure.
ChIP-seq revealed a striking enrichment of RAR-α and RAR-β binding at the GLI3 promoter in RA compared with LSB (Fig. 9C). We also found differential binding at other SHH signaling genes for which we previously observed altered chromatin marks upon RA treatment, including GLI1, GLI2, SUFU, RAB23, and PTCH1 (Fig. 9D–H). Slight enrichment for RAR binding upon RA treatment was also observed for SHH (primarily for RAR-α), as well as FKBP8 and HHIP (Fig. 9I and data not shown). In contrast, RA treatment did not enhance RAR-α or RAR-β binding at either the FOXA2 or NANOG loci (Fig. 9J,K). Lack of RAR enrichment at FOXA2 upon RA exposure is particularly interesting because, unlike Pur, RA treatment did not change the epigenetic state of FOXA2. These data further support alternate mechanisms of action for RA and Pur in FOXA2 induction. One potential mechanism for RA-mediated FOXA2 induction could be via FOXA1 because it has been shown to bind to the TSS of FOXA2 (Wang et al., 2013). We observed enrichment of RAR (particularly RAR-α) binding at the FOXA1 locus upon RA exposure, supporting such a mechanism. The binding of RAR to the SHH-related genes for which RA induced epigenetic changes indicates a previously undescribed, direct role for RA in modulating SHH signaling.
To corroborate our novel finding that early RA exposure induces binding of RARs to the GLI3 promoter, we performed confirmatory qRT-PCR for the GLI3 promoter on parallel ChIP samples. Enrichment of both RAR-α and RAR-β upon RA treatment verifies RA-mediated binding of RAR at GLI3 (Fig. 9L). We demonstrate here a novel, direct mechanism of action by which RA acts on genes involved in SHH signaling.
Discussion
The reproducible derivation of disease-relevant cell types from iPSCs remains a major challenge for applications in human disease modeling. We describe here a novel, robust strategy for deriving functional MNs from hPSCs using RA only as a single patterning factor. Although recent advances have been made in improving MN yield from hPSCs (Amoroso et al., 2013; Qu et al., 2014), the current study further streamlines and optimizes MN derivation. Our key finding, however, is on the molecular interplay of patterning factors and precursor cell competency. Unlike previous MN induction protocols, in which MN patterning follows an initial neural induction step (Li et al., 2005; Lee et al., 2007; Placantonakis et al., 2009; Amoroso et al., 2013), we report the highest competency of hPSC derivatives toward MN lineage before the onset of neural marker expression. The identification of such an early and narrow window of patterning enabled the derivation of MNs under monolayer culture conditions, with postmitotic HB9+ MNs peaking by week 2 and the acquisition of neurochemical properties such as ChAT expression starting by week 3 of differentiation.
Despite the very early window of competency, a delay exists between initiation of MN patterning (day 1) and expression of MN markers (day 7–8). We reported previously that early exposure to SHH is required for efficient floor plate derivation with a similar delay between initiation of SHH treatment (day 1) and upregulation floor plate markers (day 7–8; Fasano et al., 2010). However, unlike floor plate induction, which depends on SHH signaling, we show that extrinsic SHH exposure is dispensable for MN generation because early exposure to RA alone elicited a nearly identical upregulation of MN transcripts compared with patterning with both RA + Pur.
Previous studies have used various SHH agonists for the derivation of MNs from mouse or hPSCs (Wichterle et al., 2002; Li et al., 2008; Amoroso et al., 2013) and some studies have shown MN derivation even in the absence of extrinsic RA exposure (Wichterle et al., 2002; Li et al., 2005; Li et al., 2008; Peljto et al., 2010; Patani et al., 2011; Amoroso et al., 2013). However, the ability to efficiently derive MNs from hPSCs without extrinsic SHH signaling activators has not been reported previously. Our data demonstrate that early patterning with RA alone triggers both neural (PAX6, TUJ1, and SMI32) and MN (HB9, ISL1, OLIG2, NKX6.1, and LIM3) markers at levels comparable to RA + Pur. High expression levels of NKX6.1, a canonical SHH-induced class II protein (Briscoe et al., 2000), reflects the ability of RA to elicit endogenous SHH signaling. These molecular features of RA-only-induced MNs translate into appropriate functional properties, as shown by our electrophysiological data and coculture studies with C2C12 myoblasts demonstrating acetylcholine-dependent spontaneous muscle contractions. Although these molecular and functional characterizations demonstrate a high degree of similarity between MNs generated in the presence and absence of extrinsic SHH activation, we cannot rule out the possibility that RA-only MNs could be subtly compromised in some way. Importantly, the early RA patterning strategy readily generates MNs across multiple iPSC lines, including lines previously reported to display poor MN yield upon EB-based differentiation (Boulting et al., 2011). Future studies will need to address whether this SHH-independent strategy extends to mouse PSCs or reflects a unique species-specific property. The RA treatment paradigm is therefore not only sufficient to trigger MN induction during dSMADi, but presents a robust platform to generate disease-specific MNs.
SHH and RA initiate distinct transcriptional cascades that, in combination, are known to specify MNs during development. RA plays a role in defining anteroposterior identity, whereas SHH induces ventral cell types during the dorsoventral fate specification of neural precursor cells. However, our RNA-sequencing data reveal that MN identity and anteroposterior and dorsoventral marker expression of RA-alone- versus RA + Pur-treated purified HB9::GFP+ cells appear nearly identical. These striking transcriptional similarities further support the argument that exposure to RA at early differentiation stages can trigger an endogenous SHH-related transcriptional program comparable to that induced after extrinsic SHH agonist exposure.
An obvious potential source of endogenous SHH is the presence of RA-induced hPSC-derived floor plate cells. Indeed, we observed the efficient induction of floor plate markers including SHH expression in response to early RA exposure. However, blocking SHH signaling with the SMO antagonist CUR61414 did not significantly affect the yield of RA-induced HB9+ MNs. Therefore, our data indicate that, whereas RA can trigger floor-plate induction from hPSCs, floor plate-derived SHH is not essential for MN derivation.
Because GLI proteins transduce SHH signaling downstream of SMO, it was essential to address whether RA affects GLI expression directly to elicit endogenous SHH signaling. Our results indicate that early exposure to RA prevents the progressive increase in GLI3 expression during dSMADi. In contrast, RA treatment at later differentiation stages (PAX6+ or neural rosette stage) is inadequate for triggering SHH-related MN programs, likely due in part to insufficient suppression of GLI3 below threshold levels (Fig. 10). The ability of RA to repress GLI3 upregulation was not affected by treatment with SMO antagonists, indicating direct modulation of GLI3 by RA that was independent of endogenous SHH signaling. Previous studies in SHH−/− mice have shown that inactivating GLI3 can rescue SHH-dependent defects in ventral spinal cord domain development (Litingtung and Chiang, 2000; Bai et al., 2004; Lei et al., 2004). Therefore, the RA-mediated repression of GLI3 may represent the key mechanism for enabling high MN yields in the absence of SHH. Interestingly, other studies in mice showed that GLI3 loss and ectopic RA signaling during development result in a similar polydactyly phenotype that is generally associated with a gain-of-function in SHH signaling (Bynum, 1991; Hui and Joyner, 1993; Fawcett et al., 1995). Furthermore, during limb bud formation, RA acts as a permissive signal allowing for subsequent SHH exposure to induce the ZPA axis (Ogura et al., 1996), representing additional in vivo evidence for a link between RA and SHH signaling. Hill et al. (2009) report an SHH-independent regulation of GLI3 activity during limb bud formation that they hypothesize is caused by a novel mechanism of GLI3 transcriptional control. Our data suggest RA as a potential candidate for this SHH-independent GLI3 transcriptional control. Future studies should address whether our results in hPSCs on the regulation of GLI3 could provide a potential mechanism for SHH-related RA phenotypes in vivo.
Figure 10.

Schematic illustration depicting mechanism by which patterning with RA enables efficient MN derivation via GLI3 suppression. A, Upregulation of GLI3 transcript levels during neural induction restrict competency toward the MN lineage. RA suppresses GLI3 to levels comparable to those expressed at pluripotency when initiated early during neural induction, enabling high MN yields from hPSCs in the absence of extrinsic SHH activation. B, Early RA treatment modulates GLI3 expression by inducing RAR binding at the GLI3 promoter and preventing the chromatin switch from a bivalent to active state. C, RA exposure initiated at later neural stages is not capable of returning GLI3 levels back to baseline, and these elevated levels act like a “brake” on MN specification. Removal of this “brake” via GLI3 knock-out extends the window of competency for MN specification.
Ectopic GLI3 expression hinders MN formation (Ruiz i Altaba, 1998; Persson et al., 2002). Our results argue for a stage-dependent model in which elevated GLI3 levels, such as those observed at later differentiation stages during dSMADi, may reach a threshold that results in suppressing neural precursor cell competency toward the MN lineage (Fig. 10). Our RA exposure and GLI3 loss-of-function studies at early differentiation stages confirm that GLI3 levels can be modulated to extend competency of neural precursors toward an MN fate. We demonstrate that dynamic epigenetic changes are associated with the early, narrow window of competency and that extrinsic patterning factors such as RA can interfere with those changes. The striking switch of GLI3 to an active chromatin state during dSMADi extended to many other negative regulators of SHH signaling and was paralleled by the acquisition of repressive chromatin states at genes encoding active transducers of SHH signaling. The changes in chromatin state correlated with reduced induction of SHH-related targets, including FOXA2 and SHH, observed upon exposure to patterning cues at later differentiation stages. We show that early RA exposure induced RAR binding at GLI3 and other SHH-related genes for which RA altered the epigenetic state, supporting a direct role for RA in their regulation. The ability of RA to direct RAR binding and prevent the chromatin switch at the GLI3 locus and at the loci of other members of the SHH signaling cascade argues that RA could exert its role in part through such an epigenetic mechanism. RA has been shown previously to play a direct role in remodeling chromatin of the Hox clusters (Mazzoni et al., 2013b); therefore, it is plausible that RA plays an active role in controlling the output of developmental signals with which it interacts closely. RA may repress H3K4me3 accumulation at GLI3 via active recruitment of histone decaetylase or DNA methylation complexes to the promoter. Our results point to a novel mechanism through which RA can affect SHH signaling directly and circumvent the need for exogenous SHH exposure during MN specification.
Together, our findings support a model in which SHH transduction evolves from a more passive process to one requiring direct activation during neural differentiation—equivalent to a gas pedal and brake scenario. At very early stages of differentiation, low GLI3 levels enhance sensitivity to SHH signaling, as reflected by the ability of RA to influence GLI1 only at early differentiation stages. As differentiation progresses, epigenetic changes accompanied by increasing GLI3 levels apply a brake on SHH signaling, necessitating exposure to agonists to achieve activation. During development, such evolving responses to a given signal may generate waves of divergent cell populations to achieve complex cellular diversity from a finite number of morphogens. Similar changes may have important consequences for the specification of other SHH-dependent cell types requiring early patterning, such as floor plate (Fasano et al., 2010) or midbrain dopamine neurons (Kriks et al., 2011). Therefore, we present an interplay between chromatin state and developmental signaling pathways during the neural differentiation of hPSCs that may have general implications for modulating cellular competency.
Footnotes
This work was supported by the National Cancer Institute–National Institutes of Health (Grant P30CA008748), NYSTEM (Contract C024413 to V.T. and L.S.), the Starr Foundation (V.T., L.S.), Project ALS (L.S.), and the Department of Anesthesiology, Weill Cornell Medical College (P.A.G.). J.T. is supported by a Starr stem cell scholarship. J.A.S. is supported by a Deutsche Forschungsgemeinschaft fellowship. We thank the Memorial Sloan Kettering Cancer Center (MSKCC) molecular cytology facility for help with calcium imaging, the MSKCC integrated genomics operation facility for RNA and ChIP-sequencing studies, the MSKCC stem cell research facility for general support, the MSKCC flow cytometry facility for FACS based analysis and cell purification, K. Eggan and H. Wichterle for providing ALS and control-donor-derived iPSC lines, and B. Wang for providing the GLI3 antibody.
The authors declare no competing financial interests.
References
- Abbasi AA, Paparidis Z, Malik S, Goode DK, Callaway H, Elgar G, Grzeschik KH. Human GLI3 intragenic conserved non-coding sequences are tissue-specific enhancers. PLoS One. 2007;2:e366. doi: 10.1371/journal.pone.0000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbasi AA, Paparidis Z, Malik S, Bangs F, Schmidt A, Koch S, Lopez-Rios J, Grzeschik KH. Human intronic enhancers control distinct sub-domains of Gli3 expression during mouse CNS and limb development. BMC Dev Biol. 2010;10:44. doi: 10.1186/1471-213X-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Joyner AL. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell. 2004;118:505–516. doi: 10.1016/j.cell.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Amoroso MW, Croft GF, Williams DJ, O'Keeffe S, Carrasco MA, Davis AR, Roybon L, Oakley DH, Maniatis T, Henderson CE, Wichterle H. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci. 2013;33:574–586. doi: 10.1523/JNEUROSCI.0906-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/S1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Barreto-Chang OL, Dolmetsch RE. Calcium imaging of cortical neurons using Fura-2 AM. J Vis Exp. 2009;pii:1067. doi: 10.3791/1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, Rodolfa CT, Dimos JT, Mikkilineni S, MacDermott AB, Woolf CJ, Henderson CE, Wichterle H, Eggan K. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Pierani A, Jessell TM, Ericson J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell. 2000;101:435–445. doi: 10.1016/S0092-8674(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Bynum SV. Morphogenesis of retinoic acid-induced postaxial polydactyly in mice. Teratology. 1991;43:1–9. doi: 10.1002/tera.1420430102. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, Shi SH, Studer L. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coy S, Caamaño JH, Carvajal J, Cleary ML, Borycki AG. A novel Gli3 enhancer controls the Gli3 spatiotemporal expression pattern through a TALE homeodomain protein binding site. Mol Cell Biol. 2011;31:1432–1443. doi: 10.1128/MCB.00451-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasen JS, Liu JP, Jessell TM. Motor neuron columnar fate imposed by sequential phases of Hox-c activity. Nature. 2003;425:926–933. doi: 10.1038/nature02051. [DOI] [PubMed] [Google Scholar]
- Dasen JS, Tice BC, Brenner-Morton S, Jessell TM. A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell. 2005;123:477–491. doi: 10.1016/j.cell.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Davis-Dusenbery BN, Williams LA, Klim JR, Eggan K. How to make spinal motor neurons. Development. 2014;141:491–501. doi: 10.1242/dev.097410. [DOI] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, Briscoe J. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450:717–720. doi: 10.1038/nature06347. [DOI] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–2503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, Park Y, Dickins RA, Hannon GJ, Lowe SW. A pipeline for the generation of shRNA transgenic mice. Nat Protoc. 2012;7:374–393. doi: 10.1038/nprot.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87:661–673. doi: 10.1016/S0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- Fasano CA, Chambers SM, Lee G, Tomishima MJ, Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. 2010;6:336–347. doi: 10.1016/j.stem.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fawcett D, Pasceri P, Fraser R, Colbert M, Rossant J, Giguère V. Postaxial polydactyly in forelimbs of CRABP-II mutant mice. Development. 1995;121:671–679. doi: 10.1242/dev.121.3.671. [DOI] [PubMed] [Google Scholar]
- Harper JM, Krishnan C, Darman JS, Deshpande DM, Peck S, Shats I, Backovic S, Rothstein JD, Kerr DA. Axonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron-injured adult rats. Proc Natl Acad Sci U S A. 2004;101:7123–7128. doi: 10.1073/pnas.0401103101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenpusch-Theil K, Magnani D, Amaniti EM, Han L, Armstrong D, Theil T. Transcriptional analysis of Gli3 mutants identifies Wnt target genes in the developing hippocampus. Cereb Cortex. 2012;22:2878–2893. doi: 10.1093/cercor/bhr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill P, Götz K, Rüther U. A SHH-independent regulation of Gli3 is a significant determinant of anteroposterior patterning of the limb bud. Dev Biol. 2009;328:506–516. doi: 10.1016/j.ydbio.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Hu BY, Zhang SC. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat Protoc. 2009;4:1295–1304. doi: 10.1038/nprot.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet. 1993;3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Shamy GA, Elkabetz Y, Schofield CM, Harrsion NL, Panagiotakos G, Socci ND, Tabar V, Studer L. Directed differentiation and transplantation of human embryonic stem cell-derived motoneurons. Stem Cells. 2007;25:1931–1939. doi: 10.1634/stemcells.2007-0097. [DOI] [PubMed] [Google Scholar]
- Lei Q, Zelman AK, Kuang E, Li S, Matise MP. Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development. 2004;131:3593–3604. doi: 10.1242/dev.01230. [DOI] [PubMed] [Google Scholar]
- Li XJ, Du ZW, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA, Zhang SC. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- Li XJ, Hu BY, Jones SA, Zhang YS, Lavaute T, Du ZW, Zhang SC. Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells. 2008;26:886–893. doi: 10.1634/stemcells.2007-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Chiang C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat Neurosci. 2000;3:979–985. doi: 10.1038/79916. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, Wichterle H. Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat Neurosci. 2013a;16:1219–1227. doi: 10.1038/nn.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Peljto M, Patel T, Thornton SR, McCuine S, Reeder C, Boyer LA, Young RA, Gifford DK, Wichterle H. Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat Neurosci. 2013b;16:1191–1198. doi: 10.1038/nn.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles GB, Yohn DC, Wichterle H, Jessell TM, Rafuse VF, Brownstone RM. Functional properties of motoneurons derived from mouse embryonic stem cells. J Neurosci. 2004;24:7848–7858. doi: 10.1523/JNEUROSCI.1972-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, Alvarez IS, Vogel A, Rodríguez C, Evans RM, Izpisúa Belmonte JC. Evidence that Shh cooperates with a retinoic acid inducible co-factor to establish ZPA-like activity. Development. 1996;122:537–542. doi: 10.1242/dev.122.2.537. [DOI] [PubMed] [Google Scholar]
- Patani R, Hollins AJ, Wishart TM, Puddifoot CA, Alvarez S, de Lera AR, Wyllie DJ, Compston DA, Pedersen RA, Gillingwater TH, Hardingham GE, Allen ND, Chandran S. Retinoid-independent motor neurogenesis from human embryonic stem cells reveals a medial columnar ground state. Nat Commun. 2011;2:214. doi: 10.1038/ncomms1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peljto M, Dasen JS, Mazzoni EO, Jessell TM, Wichterle H. Functional diversity of ESC-derived motor neuron subtypes revealed through intraspinal transplantation. Cell Stem Cell. 2010;7:355–366. doi: 10.1016/j.stem.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M, Stamataki D, te Welscher P, Andersson E, Böse J, Rüther U, Ericson J, Briscoe J. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002;16:2865–2878. doi: 10.1101/gad.243402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placantonakis DG, Tomishima MJ, Lafaille F, Desbordes SC, Jia F, Socci ND, Viale A, Lee H, Harrison N, Tabar V, Studer L. BAC transgenesis in human embryonic stem cells as a novel tool to define the human neural lineage. Stem Cells. 2009;27:521–532. doi: 10.1634/stemcells.2008-0884. [DOI] [PubMed] [Google Scholar]
- Qu Q, Li D, Louis KR, Li X, Yang H, Sun Q, Crandall SR, Tsang S, Zhou J, Cox CL, Cheng J, Wang F. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat Commun. 2014;5:3449. doi: 10.1038/ncomms4449. [DOI] [PubMed] [Google Scholar]
- Rallu M, Machold R, Gaiano N, Corbin JG, McMahon AP, Fishell G. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development. 2002;129:4963–4974. doi: 10.1242/dev.129.21.4963. [DOI] [PubMed] [Google Scholar]
- Renoncourt Y, Carroll P, Filippi P, Arce V, Alonso S. Neurons derived in vitro from ES cells express homeoproteins characteristic of motoneurons and interneurons. Mech Dev. 1998;79:185–197. doi: 10.1016/S0925-4773(98)00189-0. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development. 1998;125:2203–2212. doi: 10.1242/dev.125.12.2203. [DOI] [PubMed] [Google Scholar]
- Singh Roy N, Nakano T, Xuing L, Kang J, Nedergaard M, Goldman SA. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp Neurol. 2005;196:224–234. doi: 10.1016/j.expneurol.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Son EY, Ichida JK, Wainger BJ, Toma JS, Rafuse VF, Woolf CJ, Eggan K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell. 2011;9:205–218. doi: 10.1016/j.stem.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan P, Miles GB, Rubin LL, Brownstone RM, Rafuse VF. Motoneurons derived from embryonic stem cells express transcription factors and develop phenotypes characteristic of medial motor column neurons. J Neurosci. 2006;26:3256–3268. doi: 10.1523/JNEUROSCI.5537-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazawa T, Croft GF, Amoroso MW, Studer L, Wichterle H, Macdermott AB. Maturation of spinal motor neurons derived from human embryonic stem cells. PLoS One. 2012;7:e40154. doi: 10.1371/journal.pone.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JA, Adams KL, Gundersen CB, Novitch BG. Functional neuromuscular junctions formed by embryonic stem cell-derived motor neurons. PLoS One. 2012;7:e36049. doi: 10.1371/journal.pone.0036049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/S0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wang H, Meyer CA, Fei T, Wang G, Zhang F, Liu XS. A systematic approach identifies FOXA1 as a key factor in the loss of epithelial traits during the epithelial-to-mesenchymal transition in lung cancer. BMC Genomics. 2013;14:680. doi: 10.1186/1471-2164-14-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/S0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, Yang H, Wang T, Lee AY, Swanson SA, Zhang J, Zhu Y, Kim A, Nery JR, Urich MA, Kuan S, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Goldstein PA. Propofol-block of SK channels in reticular thalamic neurons enhances GABAergic inhibition in relay neurons. J Neurophysiol. 2005;93:1935–1948. doi: 10.1152/jn.01058.2004. [DOI] [PubMed] [Google Scholar]
- Ying SW, Jia F, Abbas SY, Hofmann F, Ludwig A, Goldstein PA. Dendritic HCN2 channels constrain glutamate-driven excitability in reticular thalamic neurons. J Neurosci. 2007;27:8719–8732. doi: 10.1523/JNEUROSCI.1630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]