

Abstract
Protein aggregates containing ubiquitin (Ub) are commonly observed in neurodegenerative disorders, implicating the involvement of the ubiquitin proteasome system (UPS) in their pathogenesis. Here, we aimed to generate a mouse model for monitoring UPS function using a green fluorescent protein (GFP)-based substrate that carries a “noncleavable” N-terminal ubiquitin moiety (UbG76V). We engineered transgenic mice expressing a fusion protein, consisting of the following: (1) UbG76V, GFP, and a synaptic vesicle protein synaptobrevin-2 (UbG76V-GFP-Syb2); (2) GFP-Syb2; or (3) UbG76V-GFP-Syntaxin1, all under the control of a neuron-specific Thy-1 promoter. As expected, UbG76V-GFP-Syb2, GFP-Syb2, and UbG76V-GFP-Sytaxin1 were highly expressed in neurons, such as motoneurons and motor nerve terminals of the neuromuscular junction (NMJ). Surprisingly, UbG76V-GFP-Syb2 mice developed progressive adult-onset degeneration of motor nerve terminals, whereas GFP-Syb2 and UbG76V-GFP-Syntaxin1 mice were normal. The degeneration of nerve terminals in UbG76V-GFP-Syb2 mice was preceded by a progressive impairment of synaptic transmission at the NMJs. Biochemical analyses demonstrated that UbG76V-GFP-Syb2 interacted with SNAP-25 and Syntaxin1, the SNARE partners of synaptobrevin. Ultrastructural analyses revealed a marked reduction in synaptic vesicle density, accompanying an accumulation of tubulovesicular structures at presynaptic nerve terminals. These morphological defects were largely restricted to motor nerve terminals, as the ultrastructure of motoneuron somata appeared to be normal at the stages when synaptic nerve terminals degenerated. Furthermore, synaptic vesicle endocytosis and membrane trafficking were impaired in UbG76V-GFP-Syb2 mice. These findings indicate that UbG76V-GFP-Syb2 may compete with endogenous synaptobrevin, acting as a gain-of-function mutation that impedes SNARE function, resulting in the depletion of synaptic vesicles and degeneration of the nerve terminals.
SIGNIFICANCE STATEMENT Degeneration of motor nerve terminals occurs in amyotrophic lateral sclerosis (ALS) patients as well as in mouse models of ALS, leading to progressive paralysis. What causes a motor nerve terminal to degenerate remains unknown. Here we report on transgenic mice expressing a ubiquitinated synaptic vesicle protein (UbG76V-GFP-Syb2) that develop progressive degeneration of motor nerve terminals. These mice may serve as a model for further elucidating the underlying cellular and molecular mechanisms of presynaptic nerve terminal degeneration.
Keywords: neurodegeneration, neuromuscular junction, synapse, synaptic degeneration, synaptic transmission, transgenic mice
Introduction
Synaptobrevin2 (Syb2)/vesicle-associated membrane protein 2 (VAMP2) is a SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) protein that is highly expressed in chemical synapses (Baumert et al., 1989; Elferink et al., 1989; Archer et al., 1990; Takamori et al., 2006). Syb2 plays crucial roles in chemical synaptic transmission through the formation of SNARE complexes between vesicle SNAREs (e.g., Syb2) and target membrane SNAREs (e.g., SNAP-25 and Syntaxin-1). The SNARE complexes, together with the Sec1/Munc-18-like (SM) protein Munc18–1, undergo a cycle of association during fusion and dissociation after fusion (for review, see Jahn and Scheller, 2006; Südhof and Rothman, 2009; Jahn and Fasshauer, 2012; Rizo and Südhof, 2012), and a disruption of this cycle may result in neurodegeneration (Chandra et al., 2005; Sharma et al., 2011, 2012a).
How alteration of SNARE proteins may lead to neurodegeneration remains unclear. Genetic deletion of SNARE proteins or the SM protein in mice invariably leads to embryonic lethality, precluding further investigation of their roles in the adult nervous system (Verhage et al., 2000; Schoch et al., 2001; Washbourne et al., 2002). Here we report a new line of transgenic (Tg) mice in which neuron-specific expression of a biochemically modified Syb2 (UbG76V-GFP-Syb2) causes a progressive, adult-onset neurodegeneration.
Our initial motivation for this study was to generate a mouse model to monitor the ubiquitin proteasome system (UPS) in synapses, as impairment of the UPS is associated with various neurodegenerative diseases (for review, see Ciechanover and Brundin, 2003; Dennissen et al., 2012; Dantuma and Bott, 2014). Our strategy was based upon a well characterized proteasome substrate carrying a ubiquitin fusion degradation signal and a N-terminally linked ubiquitin that has been mutated at the C terminus (UbG76V; Bachmair and Varshavsky, 1989; Johnson et al., 1995) and fused to green fluorescent protein (GFP). This substrate (UbG76V-GFP) is short lived in cells and is rapidly degraded by the proteasome (Lindsten et al., 2001). However, functional impairment of the UPS would result in accumulation of the substrate and increases in GFP fluorescence that can be detected under a microscope. A previous study (Lindsten et al., 2003) has reported transgenic mice expressing UbG76V-GFP under the control of a β-actin promoter. However, the widespread expression of the transgene makes it impossible to distinguish individual synapses. To circumvent this problem, we fused UbG76V-GFP in-frame to a synaptic vesicle (SV) protein Syb2 (to target the transgenic protein to synapses) under the control of a neuron-specific Thy1 promoter (Vidal et al., 1990; Caroni, 1997; Feng et al., 2000; Probst et al., 2000; Han et al., 2005).
UbG76V-GFP-Syb2 transgenic mice appear grossly normal during the first 5–6 months, but, unexpectedly, they become progressively paralyzed after 7 months of age. These unexpected results prompted us to investigate how neuron-specific expression of UbG76V-GFP-Syb2 may lead to synaptic degeneration. Examination of the neuromuscular synapses reveals age-dependent functional impairments and synaptic degeneration, accompanied by a marked reduction in synaptic vesicle density. Our data suggest that UbG76V-GFP-Syb2 acts as a gain-of-function mutation that disrupts endogenous SNARE function, resulting in the depletion of synaptic vesicles and the degeneration of nerve terminals.
Materials and Methods
Generation of transgenic mice
Construction of transgenic vectors.
Plasmids were constructed using standard subcloning procedures. Briefly, pThy1/enhanced cyan fluorescent protein (ECFP)-Syb2 was generated through the following steps. First, a 700 bp (XhoI-BsrGI) ECFP coding sequence was subcloned into a Thy1 mini-gene vector (a gift from Dr. M. Goedert, MRC Laboratory, Cambridge, UK) that contains the Thy1 gene without exon 3 and its flanking introns so that neuron-specific expression is achieved (Vidal et al., 1990; Caroni, 1997; Probst et al., 2000). This generated an intermediate vector that we named pThy1/ECFP-C1. Second, an XhoI site was introduced by mutagenesis to the N terminus of bovine Syb2 in pCMV18-1a to generate pCMV18-1a-wp using the following oligonucleotides: 5′-CCG CTG CCA AGT CCT CGA GTC CGC TGG CCC CCG C and 5′-GCG GGG GCC AGC GGA CTC GAG GAC TTG GCA GCG G. This generated a vector named pThy1-ECFP-C1 to generate pCMV18-1a-wp. Finally, a 1.5 kb XhoI-XhoI fragment from pCMV18-1a-wp was inserted into the XhoI site of pThy1-ECFP-C1 to generate the final transgenic vector pThy1/ECFP-Syb2. Plasmid pThy1/UbG76V-enhanced GFP (EGFP)-Syb2 was generated by replacing ECFP in pThy1/ECFP-Syb2 with a 0.9 kb EcoRI/BsrGI fragment from pUbG76V-GFP (a gift from Dr. Nico Dantuma, Karolinska Institute, Stockholm, Sweden). Using similar subcloning strategies, we generated plasmid pThy1/UbG76V-ECFP-Syb2 by replacing EGFP from pThy1/UbG76V-EGFP-Syb2 with ECFP, and plasmid pThy1/UbG76V-EGFP-Syntaxin1 (Syx1) by replacing Syb2 from pThy1/EGFP-Syb2 with Syntaxin1. All constructs were sequence confirmed.
Generation of transgenic mice.
The transgenes were gel purified and injected into fertilized oocytes from BL6SJL/F1 hybrid mice using procedures previously described (Hogan et al., 1994). Founders were identified by Southern blot using GFP cDNA as a probe and reconfirmed by PCR using specific primers. Transgenic lines were established by backcross first to BL6SJL/F1 hybrid mice, and then to C57BL/6 mice for >15 generations.
The genotypes of Thy1/ECFP-Syb2 transgenic mice were determined by PCR using the following primers, wh76 (TGG TGA ACC GCA TCG AGC TG, from ECFP) and wh79 (CGT TCA CCC TCA TGA TGT CC, from Syb2). Thy1/UbG76V-EGFP-Syb2 and UbG76V-ECFP-Syb2 transgenic mice were genotyped using the following primers: forward, TCC GTC TCA GAG GTG TGG TG; and reverse, GTT GTG GCT GTT GTA GTT GTA). Thy1/UbG76V-EGFP-Syntaxin1 transgenic mice were genotyped using the following primers: forward, TCC GCC CTG AGC AAA GAC; and reverse, CGA CGA GCG GTT CAG AC.
Multiple lines of transgenic mice were generated for each transgene, including Thy1/UbG76V-EGFP-Syb2 (two lines), Thy1/UbG76V-ECFP-Syb2 (three lines), Thy1/ECFP-Syb2 (two lines), and Thy1/UbG76V-EGFP-Syntaxin1 (three lines). Both Thy1/UbG76V-EGFP-Syb2 and Thy1/UbG76V-ECFP-Syb2 mice developed adult-onset neurodegeneration; whereas, Thy1/ECFP-Syb2 and Thy1/UbG76V-EGFP-Syntaxin1 mice were normal and viable, indicating that the development of the neurodegenerative phenotype was unrelated to spectral variations of GFP (e.g., EGFP vs ECFP). To simplify the nomenclature, we hereafter refer to both EGFP and ECFP collectively as GFP. Therefore, Thy1/UbG76V-EGFP-Syb2 and Thy1/UbG76V-ECFP-Syb2 transgenic mice are referred to as UbG76V-GFP-Syb2 transgenic mice, Thy1/ECFP-Syb2 transgenic mice as GFP-Syb2 transgenic mice, and Thy1/UbG76V-EGFP-Syntaxin1 mice as UbG76V-GFP-Syntaxin1. UbG76V-GFP transgenic mice were provided by Dr. Nico Dantuma (Karolinska Institute, Stockholm, Sweden). The analyses were performed using hemizygous mice from each line, unless otherwise specified. Both male and female mice were used for this study.
Primary antibodies
The following antibodies were used for Western blotting: SNAP-25 [mouse monoclonal, 1:5000; CL71.1, Synaptic Systems (http://www.sysy.com)]; Synaptobrevin 2 (mouse monoclonal, 1:2000; CL 69.1, Synaptic Systems); Syntaxin 1 (rabbit polyclonal, 1:1000; U6251); Munc18 (rabbit polyclonal, 1:1000; K329); Complexin I (rabbit polyclonal, 1:1000; L668); Complexin II (rabbit polyclonal, 1:1000; P942), NSF (rabbit polyclonal, 1:250; J372); CSPα (rabbit polyclonal, 1:1000; Synaptic Systems); αSNAP (mouse monoclonal, 1:1000; CL77.2, Synaptic Systems); Synaptotagamin (mouse monoclonal, 1:10,000; CL41.1, Synaptic Systems); Synapsin (rabbit polyclonal, 1:1000; E028); Synaptojanin (rabbit, 1:1000; T694); Synaptogyrin (rabbit, 1:1000; P925); SV2a (mouse monoclonal, 1:1000; 17G10, Synaptic Systems); SV2b (mouse monoclonal, 1:1000; 246EG, Synaptic Systems); Velis (rabbit polyclonal, 1:1000; Butz et al., 1998); Rab3a (mouse monoclonal, 1:1000; CL42.1, Synaptic Systems); Rab5a (mouse monoclonal, 1:1000; CL 621.3, Synaptic Systems); Rabphilin (rabbit polyclonal, 1:1000; Fykse et al., 1995); Ubiquitin (mouse monoclonal, 1:1000; FK2, ENZO Life Sciences); valosin-containing protein (VCP, rabbit polyclonal, 1:1000; provided by Dr. Cezary Wójcik, Indiana University, Bloomington, IN); and GFP (rabbit polyclonal, 1:1000, affinity purified; provided by Dr. William Wickner, Dartmouth University, Hanover, NH).
For immunofluorescence staining, the following antibodies were used: GM130 (mouse monoclonal, 1:1000; 95131, BD Transduction Laboratories); Syb2 (rabbit polyclonal, 1:1000; P939; Liu et al., 2011); Syt2 (rabbit polyclonal, 1:1000; I735; Pang et al., 2006); SV2 (rabbit polyclonal, 1:1000; P916); glial fibrillary acidic protein (GFAP; rabbit polyclonal, 1:1000; Millipore Bioscience Research Reagents); synaptophysin (rabbit polyclononal, 1:1000; DAKO); NF150 (rabbit polyclonal, 1:1000; Millipore Bioscience Research Reagents); and UChL1 (rabbit polyclonal, 1:1000; Enzo Life Sciences).
Biochemical Analyses
Detection and quantification of protein expression.
Whole brain/spinal cords from aged-matched littermate mice were homogenized in 50 mm Tris buffer, pH 7.2, 100 mm NaCl, 1 mm EDTA (Sigma-Aldrich), 1 mm phenylmethanesulfonyl fluoride (Sigma-Aldrich), and protease inhibitor cocktail (Roche). Equal concentrations (25 μg of total protein) from each sample were solubilized in SDS loading buffer and boiled for 10 min. For qualitative immunoblotting, total proteins from each sample were separated by SDS-PAGE followed by peroxidase-conjugated secondary antibodies, and visualized with enhanced chemiluminescence. For quantitative immunoblotting, I125-labeled secondary antibodies (Bio-Rad) were applied, followed by analyses with a phosphor-imager (Bio-Rad). Signals were normalized to VCP or endogenous Syb2 on the same blots.
Coimmunoprecipitation.
Spinal cord homogenates were prepared in 50 mm HEPES-NaOH, pH 7.2, 100 mm NaCl, 4 mm EGTA, 1 mm DTT, and 2 mm MgCl2, and extracted with 1% Triton X-100 for 2 h at 4°C. Following centrifugation at 11,000 × g at 4°C for 30 min, the soluble fraction was precleared with protein A agarose for 30 min at 37°C. SNARE proteins were immunoprecipitated using polyclonal antibodies to GFP, Syntaxin1 (U6251), or synaptobrevin2 (P939) for 1 h at 37°C; and coimmunoprecipitated synaptic proteins were visualized with enhanced chemiluminescence.
SNARE core complex assay.
Analyses were performed in vitro to assess the formation of SNARE complexes resistant to ionic detergent such as SDS, but sensitive to boiling (Hayashi et al., 1994). HEK293 cells were cotransfected using Fugene 6 reagent (Roche) with plasmids pCMV/SNAP-25A and pCMV/Syntaxin1A (residues 1–243; Dulubova et al., 1999; Matos et al., 2003; Khvotchev and Südhof, 2004), together with one of the following expression plasmids: pCAGGS/UbG76V-GFP, pCAGGS/UbG76V-GFP-Syb2, or pCAGGS/UbG76V-GFP-Syb2 mutant, which carries five point mutations in its SNARE motif. The expression plasmids pCAGGS/UbG76V-GFP and pCAGGS/UbG76V-GFP-Syb2 were constructed by subcloning UbG76V-GFP or UbG76V-GFP-Syb2 into a pCAGGS vector (Bolliger et al., 2011), respectively. UbG76V-GFP-Syb2 mutant vector was generated by site-directed mutagenesis (Kunkel, 1985) using UbG76V-GFP-Syb2 as a backbone. The UbG76V-GFP-Syb2 mutant carries five point mutations in its SNARE motif. The following primer pair was used for inverse PCR to generate UbG76V-GFP-Syb2 mutant: forward, ACC TCA CTA GTA ACA GGA GAG ACC AGC AGA CCC AGG CCC AGG ACG ATG AGG ACG TGG ACA TCG ATA GGG TGA ACG TAG ACA AGG ACT TGG AGC GG; and reverse, GCT GCA CTA GTT TCA AAC TGG GAG GCC CCC GCC TGG AGG GCG TCG GCA CGG TCG TCC AGC TCC GAC AGC TTC TGG TCC CGC TCC AAG TCC TTG TC. All PCR products were sequence confirmed.
Two days after transfection, cells were extracted in lysis buffer (150 mm NaCl, 25 mm HEPES, pH 7.4, 1% Triton X-100, 1 mm EDTA, and protease inhibitors). Cell lysate (10 μg) was then mixed with SDS-PAGE loading buffer and was either boiled or not boiled. Samples were then analyzed by immunoblotting for Syntaxin1.
Electrophysiology
Intracellular recordings were performed in lumbrical muscles from mice (from transgenic mice and their nontransgenic littermate control mice) at 1, 3, 5, 8, 10, and 12 months of age. Lumbrical muscles with plantar nerves attached were acutely isolated in oxygenated (95% O2, 5% CO2) Ringer's solution (136.8 mm NaCl, 5 mm KCl, 12 mm NaHCO3, 1 mm NaH2PO4, 1 mm MgCl2, 2 mm CaCl2, and 11 mm d-glucose, pH 7.3; Liley, 1956). End-plate regions identified under a water-immersion objective (BX51WI, Olympus) were impaled with glass micropipettes (resistance, 20–40 MΩ) filled with 2 m potassium citrate and 10 mm potassium chloride. Evoked end-plate potentials (EPPs) were elicited by suprathreshold stimulation (2 V, 0.1 ms) of the nerve via a suction electrode connected to an extracellular stimulator (SD9, Grass-Telefactor). To prevent muscle contraction, μ-conotoxin GIIIB (2 μm; Peptides International) was added to the bath solution 30 min before recording. Miniature EPPs (mEPPs) and EPPs were acquired using an intracellular amplifier (AxoClamp-2B) and were digitized with a Digidata 1332 (Molecular Devices). Data were analyzed with pClamp version 9.0 (Molecular Devices) and the Mini Analysis Program (Synaptosoft). Quantal content (i.e., the number of acetylcholine quanta released in response to a single nerve impulse) was estimated using the following direct method: dividing the mean amplitude of EPPs by the mean amplitude of mEPPs of the same cell (Boyd and Martin, 1956; Hubbard et al., 1969; Wood and Slater, 2001).
Light microscopy
Immunostaining of spinal cord sections.
Frozen sections of lumbar spinal cord segments were briefly rinsed three times with PBS and then preincubated in antibody dilution buffer (500 mm NaCl, 0.01 m phosphate buffer, 3% BSA, and 0.01% thimerosal) for 30 min at room temperature. Sections were then incubated with primary antibodies overnight at room temperature, followed by secondary antibodies (Texas Red-conjugated goat anti-rabbit or anti-mouse IgG) for 1 h at room temperature. To label the nucleus, spinal cord sections were incubated with TO-PRO-3 (1:5000 in PBS; Life Technologies), a carbocyanine monomer nucleic acid stain, for 30 min at room temperature. Then sections were washed with PBS and mounted in Vectashield antifade mounting medium (H-1000, Vector Laboratories). Images were acquired using an Olympus BX51 Upright Fluorescence Microscope equipped with a Hamamatsu ORCA-285 camera, or using a Zeiss LSM 510 Meta Confocal Microscope.
Analysis of the neuromuscular junction morphology and denervation.
Morphological analysis of the neuromuscular junction (NMJ) was performed in triangularis sterni muscle, lumbrical muscle, and teased thigh muscles in UbG76V-GFP-Syb2, GFP-Syb2, UbG76V-GFP-Syntaxin1, and age-matched nontransgenic control mice, using previously described procedures (Chen et al., 2010). To examine muscle for denervation, the fourth lumbrical muscle from UbG76V-GFP-Syb2 and their nontransgenic littermate control mice were fixed in 2% paraformaldehyde (PFA) in 0.1 m phosphate buffer, pH 7.3, overnight at 4°C. Three pairs of mice were analyzed at each age group (1, 3, 5, and 8 months). After extensively washing with PBS, muscles were teased into small bundles under a dissection microscope. Muscle bundles were then incubated with Texas-Red-conjugated α-bungarotoxin (α-bgt; to label end plates; 2 nm; Invitrogen) for 30 min, followed by anti-Syt2 (I735) antibodies (to label presynaptic motor nerve terminals; Pang et al., 2006). After three washes (45 min each), muscles were incubated with Alexa Fluor 647-conjugated goat anti-rabbit IgG (1:600) overnight at 4°C. Muscle fibers were examined under an Olympus BX51 Upright Fluorescence Microscope. Muscle fibers were scored, according to Fischer et al. (2004), as “innervated” (complete overlap of end plates and nerve terminals), “partially denervated” (partial overlap of end plates and nerve terminals), or “totally denervated” (end plate with no nerve terminal).
Motor neuron counting.
Motor neuron counts were performed according to previously described procedures (Clarke and Oppenheim, 1995). UbG76V-GFP-Syb2 and their nontransgenic littermate control mice (three pairs of each, 6 and 8 months of age) were analyzed. Mice were anesthetized by isoflurane and perfused first with ice-cold PBS and then with 4% PFA. Spinal cords were dissected and post-fixed in 4% PFA overnight at 4°C. The lumbar segments (L3–L5) were transversally sectioned at a thickness of 10 μm and stained with cresyl violet acetate (C5042, Sigma-Aldrich). Motor neurons were examined under a microscope; they were counted only if they exhibited all of the following criteria: large soma, a clear nucleus with intact nuclear membrane, and at least one large clump of nucleolar material.
Electron microscopy
Conventional electron microscopy and morphometric analysis.
UbG76V-GFP-Syb2 transgenic and nontransgenic littermate control mice at various ages (3, 5, and 8 months of age) were fixed by cardiac perfusion with a mixture of 1% glutaraldehyde and 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The lumbar segments of the spinal cord and lumbrical muscles were dissected and further fixed overnight in the same fixative at 4°C. After extensive rinsing with 0.1 m phosphate buffer, the tissues were trimmed to small pieces and post-fixed with 1% osmium tetroxide in 0.1 m phosphate buffer for 3 h on ice. Then the tissues were dehydrated with graded series of ethanol, infiltrated with propylene oxide, and embedded in Epon 812 (Polysciences). For light microscopy, Epon blocks were cut at 1 μm thickness and stained with Toluidine Blue. Ultrathin sections were cut at 70 nm and mounted on Formvar film-lined grids. The grids were stained with uranyl acetate and lead citrate, and were observed with a Tecnai Electron Microscope operated at 120 kV. Electron micrographs were acquired by a SIS Morada 11 megapixel side-mount CCD camera (Soft Imaging Solutions, Olympus).
Motor nerve terminal profiles of NMJs from lumbrical muscles acquired by electron microscopy (EM) were analyzed using National Institutes of Health ImageJ software. The following measurements were made in a given profile: nerve terminal area, perimeter length, synaptic contact length, total synaptic vesicle number, and docked synaptic vesicle number. In addition, the postsynaptic membrane length, excluding junctional folds, was also measured. The morphological criteria were adapted from previous observations in wild-type mice (Kelly and Zacks, 1969). Briefly, the synaptic contact was defined as the length of the presynaptic plasma membrane that was apposed to the postsynaptic specialization of muscle membrane at a distance of 50–80 nm. Docked synaptic vesicles were defined as vesicles that attached to the presynaptic plasma membrane directly facing postsynaptic specialization. Synaptic vesicles in each nerve terminal profile were counted for total synaptic vesicle number. Three parameters, contact ratio, presynaptic/postsynaptic ratio, and synaptic vesicle density were calculated from the measurements in each nerve terminal profile. The contact ratio was calculated from synaptic contact length/perimeter length and was expressed as a percentage. The presynaptic/postsynaptic ratio was calculated from synaptic contact length/postsynaptic membrane length and was expressed as a percentage. The synaptic vesicle density was calculated from total synaptic vesicle number/nerve terminal area and was expressed the number of vesicles per square micrometer.
Analysis of synaptosome.
Synaptosomes were prepared from whole brains of 8-month-old Tg and non-Tg mice using a procedure described previously (De Camilli et al., 1983), with minor modifications. Mouse brains were quickly isolated and homogenized with ice-cold 0.32 m sucrose in 5 mm phosphate buffer, pH 7.4. The homogenates were centrifuged at 1000 × g for 10 min at 4°C. The supernatants were collected and centrifuged again at 9200 × g for 15 min at 4°C. The resulting pellets were resuspended with oxygenated normal mouse Ringer's solution; this preparation is referred to as the “synaptosomes,” hereafter. Synaptosomes were then incubated with 8 mg/ml horseradish peroxidase (HRP; Sigma-Aldrich) in Ringer's solution containing 60 mm KCl (high-K stimulated) or normal Ringer's solution (nonstimulated) at 37°C for 15 min. At the end of the incubation time, cold normal Ringer's solution was added; the synaptosomes were immediately centrifuged, and the supernatant was discarded. The pellets were then fixed with 2% glutaraldehyde in 0.01 m phosphate buffer, pH 7.4, containing 0.3 m sucrose on ice for 30 min. After extensive washing with 0.1 m phosphate buffer, washed pellets were incubated with 0.5 mg/ml diaminobenzidine (DAB; Sigma-Aldrich) in 0.175 m Tris-HCl, pH 7.6, for 10–15 min at room temperature. The incubation solution was then switched to the DAB solution containing 0.01% hydrogen peroxide and were incubated for another 15 min at room temperature. The reaction was terminated by adding cold 0.1 m phosphate buffer and washing the pellets three times. Pellets were then embedded in agarose as described previously (De Camilli et al., 1983), and synaptosome-containing agarose blocks were post-fixed with osmium tetroxide and processed as the conventional EM preparations described above.
Statistical analysis
Data are presented as the mean ± SEM. Statistical significance was assessed by two-tailed Student's t test.
Measurement of proteasome activity
Proteasome activity was measured using synthetic peptide substrates linked to a fluorometric reporter aminomethylcoumarin (AMC; Calbiochem) according to a procedure described previously (Puttaparthi et al., 2003). Briefly, lumbar spinal cords from three pairs of UbG76V-GFP-Syb2 transgenic and nontransgenic littermate control mice (8 months of age) were freshly dissected, washed in PBS, and gently homogenized in ice-cold Buffer H (20 mm Tris, 20 mm NaCl, 1 mm EDTA, 5 mm β-mercaptoethanol, pH 7.6). The spinal cord homogenates were then centrifuged (10,000 K, 4°C) and the supernatants were used for quantifying protein concentration and for measuring proteasome enzymatic activity. The spinal cord lysates were then incubated with specific substrate-Suc-LLVYAMC for chymotrypsin-like (ChT-L) activity; Z-LLEAMC for peptidyl-glutamyl-peptide hydrolase (PGPH); and Z-VVR-AMC for trypsin-like activity. The enzymatic reaction of each specific substrate was quantified in a FL600 Plate Reader (Bio-Tek) with 360/460 nm excitation/emission wavelengths. Enzymatic activity was normalized according to protein concentration and was presented as a percentage of activity relative to control.
Neuronal cell culture
Dissociated hippocampal cultures were prepared as previously described (Kavalali et al., 1999; Schoch et al., 2001). Briefly, whole hippocampi were dissected from postnatal day (P0) to P3 Sprague Dawley rats. Tissue was trypsinized (10 mg/ml trypsin) for 10 min at 37°C, mechanically dissociated by pipetting, and plated on Matrigel (BD Biosciences)-coated coverslips. Cytosine arabinoside (4 μm AraC, Sigma) was added at day 1 in vitro (DIV) and reduced to 2 μm at 4 DIV. All experiments were performed on cultures at 14–21 DIV.
Lentiviral preparation
Lentiviral expression constructs encoding either GFP-Syb2 or UbG76V-GFP-Syb2, tagged at their C termini with mOrange, a pH-sensitive fluorescent DsRed variant, were generated. Recombinant viruses were prepared by transfection of HEK293 T cells using Fugene 6 (Roche Applied Science) with the plasmids of interest together with plasmids encoding viral packaging and coating proteins (pRsv-Rev, pPRE-MALG, and pVSVG; Ramirez et al., 2012). The virus was harvested from the HEK293 T-conditioned medium 3 days post-transfection and was added to the neuronal culture medium. Dissociated hippocampal cultures were infected with virus at 4 DIV. Using these techniques, we consistently obtained infection frequencies approaching 100%.
Live cell imaging
Live cell imaging experiments were performed on a Zeiss LSM510 Confocal Microscope using previously described procedures (Ramirez et al., 2012). Experiments were performed in a modified Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4. The Tyrode's solution also contained the glutamate receptor blockers AP-5 (50 μm) and CNQX (10 μm) to prevent excitotoxicity. An argon laser was used to excite pHluorin at 488 nm, and an HeNe laser was used to excite mOrange at 543 nm. The use of specific filters (500–550 bandpass filter for the green channel and a 543 long-pass filter for the red channel) and sequential scanning of the red and green channels enables reliable detection of each fluorophore in the time frame of the experiment with negligible bleedthrough. Solutions were perfused by gravity into the chamber. Neurons were subjected to 20 Hz stimulation for 20 s followed by NH4Cl treatment (50 mm in Tyrode's solution with equivalent reduction in NaCl concentration) after the return to baseline. Images were recorded approximately every 5 s. Between 65 and 100 synaptic puncta of constant diameter were selected from each coverslip from images at the end of the experiment (after NH4Cl treatment) to obtain the time courses of fluorescence changes per bouton. Peak fluorescence values were calculated by normalizing the peak (at 20 Hz stimulation) to the peak after NH4Cl treatment.
Results
Generation of transgenic mice expressing GFP-Syb2 or UbG76V-GFP-Syb2 in neurons
Using a neuron-specific Thy-1 promoter as a driver (Vidal et al., 1990; Caroni, 1997; Feng et al., 2000; Probst et al., 2000; Han et al., 2005), we generated transgenic mice expressing a fusion protein consisting of either (1) “nonremovable” N-terminal ubiquitin moiety (UbG76V; Johnson et al., 1995; Dantuma et al., 2000), GFP, and Syb2 (UbG76V-GFP-Syb2); or (2) GFP and Syb2 (GFP-Syb2; Fig. 1A). Western blot analysis of total brain and spinal cord homogenates from GFP-Syb2 or UbG76V-GFP-Syb2 transgenic mice (3 months of age) detected a single band predicted by the size of the fusion protein, either GFP-Syb2 or UbG76V-GFP-Syb2, as recognized by anti-GFP antibodies (Fig. 1B). To determine the expression level of these transgenic proteins, we performed quantitative Western blot analyses using 125I-labeled antibodies. We quantified the level of UbG76V-GFP-Syb2 relative to endogenous Syb2 in UbG76V-GFP-Syb2 mice and observed that the expression level of transgenic UbG76V-GFP-Syb2 was ∼10% of the endogenous Syb2 level (Fig. 1C,D). The UbG76V-GFP-Syb2 protein was maintained at similar levels at 3, 6, and 8 months of age (Fig. 1G,H). In GFP-Syb2 mice, the expression level of GFP-Syb2 was 95% of the endogenous Syb2 level (Fig. 1E,F).
Figure 1.

Generation of transgenic mice expressing a fusion protein, either UbG76V-GFP-Syb2 or GFP-Syb2. A, Schematic diagrams of constructs for generating transgenic mice expressing either UbG76V-GFP-Syb2 or GFP-Syb2, directed by a neuron-specific Thy1 promoter. The asterisk (ub*) denotes a point mutation (G76V) at the last residue of the C terminus of the Ub, which prevents the removal of Ub from its substrate. B, Western blot analysis of total brain and spinal cord homogenates prepared from 3-month-old nontransgenic control, GFP-Syb2, and UbG76V-GFP-Syb2 mice, probed with anti-GFP antibodies and then reprobed with antibodies against VCP. Anti-GFP antibodies recognize a single band predicted by the size of the fusion protein, either GFP-Syb2 or UbG76V-GFP-Syb2. C, D, Western blot analysis of spinal cord homogenates prepared from nontransgenic (control), UbG76V-GFP-Syb2 transgenic (line 1), and UbG76V-GFP-Syb2 transgenic (line 2) mice (three mice in each group, 3 months of age) and probed with antibodies against Syb2, which recognizes both endogenous Syb2 and UbG76V-GFP-Syb2. The blot was reprobed with anti-syntaxin1 antibodies. The endogenous Syb2 was abundant in both nontransgenic and transgenic mice, whereas UbG76V-GFP-Syb2 was detected only in the transgenic mice at a level markedly lower than those of the endogenous Syb2. This is quantified by the bar graph on the right comparing the levels of UbG76V-GFP-Syb2 expression relative to the endogenous Syb2 (D). E, F, Western blot analysis of spinal cord homogenates from nontransgenic (control), GFP-Syb2 transgenic, and UbG76V-GFP-Syb2 transgenic (line 1) mice (three mice in each group, 3 months of age), probed with antibodies against Syb2. Relative expression levels (compared with endogenous Syb2) are shown in the bar graph (F). G, H, Western blot analysis of spinal cord homogenates from nontransgenic control and UbG76V-GFP-Syb2 transgenic mice (3, 6, and 8 months of age, three pairs of mice in each age group). Expression levels of UbG76V-GFP-Syb2 remained at a steady-state level at 3, 6, and 8 months of age (H).
Consistent with a previous report that VAMP2/Syb2-CFP fusion protein is targeted to both preterminal nerves and nerve terminals (Sampo et al., 2003), we observed GFP fluorescence along the nerve trunk and intensely concentrated at nerve terminals at the NMJs in UbG76V-GFP-Syb2 mice, as early as P0 (Fig. 2A). Examination under high-magnification objectives further revealed that GFP fluorescence appeared as distinct puncta within the nerve terminal (Fig. 2A, inset). Such a localization pattern was detected at all ages in UbG76V-GFP-Syb2 mice. Furthermore, the GFP fluorescence signal within the nerve terminals was colocalized with immunofluorescence revealed by antibodies to synaptic proteins such as Syntaxin1, Syb2, SV2, and Syt2 (Fig. 2B,C). Similarly, the GFP fluorescence signal was also detected along the nerve and at the NMJs in GFP-Syb2 mice (Fig. 2C).
Figure 2.

Localization of UbG76V-GFP-Syb2 or GFP-Syb2 in motor axons and nerve terminals. A, Whole-mount immunofluorescence staining of triangularis sterni muscles in UbG76V-GFP-Syb2 and nontransgenic control mice at P0, viewed under GFP fluorescence (green), anti-Syntaxin1 staining (blue), and Texas-Red-conjugated α-bungarotoxin for AChRs (red). GFP fluorescence is detected along the nerve trunk (arrow) and nerve terminals (arrowheads) in UbG76V-GFP-Syb2 mice. Insets at the top right corner of each panel show high-power views of individual nerve terminals; GFP puncta were readily detectable at the nerve terminal. B, Images of single nerve terminals obtained from teased thigh muscle fibers in UbG76V-GFP-Syb2 mice (5 months of age). Top green panels show GFP fluorescence; middle red panels show nerve terminals labeled by antibodies against Syntaxin1, Syb2, or SV2. Merged images show the localization of GFP fluorescence at the nerve terminals. C, Confocal images of whole-mount triangularis sterni muscle from nontransgenic control, UbG76V-GFP-Syb2, and GFP-Syb2 mice (5 months of age), viewed under GFP fluorescence (green), anti-synaptotagmin-2 (cyan), and Alexa Fluor 647-conjugated α-bungarotoxin for AChR (red). GFP fluorescence is localized along preterminal axons (arrows) and nerve terminals (arrowheads) in both UbG76V-GFP-Syb2 and GFP-Syb2 transgenic mice. Scale bars: A, 100 μm; A, insets, 20 μm; B, 20 μm; C, 50 μm.
In addition, GFP-positive neurons were detected throughout the brain and spinal cord in both UbG76V-GFP-Syb2 and GFP-Syb2 transgenic mice. For example, in the ventral horn of lumbar spinal cord, GFP fluorescence was detected in the neuropil surrounding motor neuron somata in both GFP-Syb2 (Fig. 3A) and UbG76V-GFP-Syb2 mice (Fig. 3B–D). Furthermore, motor neuron somata in UbG76V-GFP-Syb2 mice were heavily labeled. The GFP signal appeared as numerous puncta in the perinuclear region of the cytoplasm and was detected at all ages (examples at 1, 5, and 8 months of age are shown in Fig. 3B–D). These GFP-positive puncta were closely associated (but did not overlap) with the cis-Golgi that were labeled by anti-GM130 antibodies (Fig. 3M). The presence of UbG76V-GFP-Syb2 within motor neuron somata did not appear to affect the localization of endogenous Syb2, since endogenous Syb2 was localized as puncta in neuropil surrounding the motor neuron soma, in a pattern similar to that in non-Tg control mice (Fig. 3N–S).
Figure 3.

Expression and localization of UbG76V-GFP-Syb2 and GFP-Syb2 in motor neurons. A–D, Cross sections of the ventral horn of lumbar spinal cords from GFP-Syb2 (A, 5-month-old) and UbG76V-GFP-Syb2 mice (B, 1-month-old; C, 5-month-old; D, 8-month-old), counterstained with TO-PRO-3 to label nuclei (A, arrow). Merged images are shown to illustrate the distribution of GFP fluorescence (green) and nuclei (red; an example is shown by an arrow in A). In both GFP-Syb2 and UbG76V-GFP-Syb2 mice, GFP-positive labeling is detected in the neuropil (*) surrounding the somata. In addition, GFP-positive puncta (arrowheads in B–D) are detected within the somata of motor neurons in UbG76V-GFP-Syb2 mice. A similar labeling pattern is observed in UbG76V-GFP-Syb2 mice at 1, 5, and 8 months of age. E–M, Confocal images of spinal motor neurons from non-Tg control (top), GFP-Syb2 (middle), and UbG76V-GFP-Syb2 mice (bottom), immunolabeled with anti-GM130 antibodies (red). Merged image shows that GFP-positive puncta are closely associated with cis-Golgi (labeled by anti-GM130 antibodies). N–S, High-magnification images of motor neurons in the ventral horn of lumbar spinal cord, immunolabeled with anti-Syb2 antibodies (red) that recognize endogenous Syb2 but not the fusion protein UbG76V-GFP-Syb2. In both UbG76V-GFP-Syb2 and non-Tg control mice, Syb2-positive puncta (arrowheads in P and S) are detected outside of the motor neuron somata. Dashed lines in A–D, G, J, M, P, and S demarcate the boundary of the motor neuron somata. Scale bars: A–D, 50 μm; E–M, 20 μm; N–S, 20 μm.
Neuron-specific expression of UbG76V-GFP-Syb2, but not GFP-Syb2, leads to adult-onset paralysis and progressive degeneration of motor nerve terminals
Surprisingly, UbG76V-GFP-Syb2 mice developed adult-onset paralysis. Initially, for the first 5–6 months, UbG76V-GFP-Syb2 mice were normal and indistinguishable from their littermate nontransgenic control mice. However, after 7 months of age UbG76V-GFP-Syb2 mice became progressively paralyzed; the paralysis first appeared in the hindlimb and then progressed to the rest of the body. By 8 months of age, UbG76V-GFP-Syb2 transgenic mice were totally paralyzed; they could not support their own weight or walk, and the majority of the UbG76V-GFP-Syb2 mice died by 12 months of age. In contrast, GFP-Syb2 transgenic mice were viable and indistinguishable from nontransgenic control mice (Fig. 4A,B). Furthermore, UbG76V-GFP-Syb2 mice exhibited marked astrogliosis, indicated by increases in GFAP staining, compared with nontransgenic control or GFP-Syb2 mice (Fig. 4C).
Figure 4.

Neuron-specific expression of UbG76V-GFP-Syb2 leads to adult-onset paralysis and progressive degeneration of motor nerve terminals. A, external phenotype of an 8-month-old UbG76V-GFP-Syb2 transgenic mouse, compared with a nontransgenic littermate control mouse. The UbG76V-GFP-Syb2 transgenic mouse is paralyzed at this age. B, Kaplan–Meier survival curve of UbG76V-GFP-Syb2 (N = 131), GFP-Syb2 (N = 118), and nontransgenic control mice (N = 63). The majority of the UbG76V-GFP-Syb2 mice died within 12 months of age. C, Cross sections of ventral spinal cords immunostained with anti-GFAP antibodies. GFAP-positive astrocytes (arrowheads) are markedly increased in UbG76V-GFP-Syb2 mice, compared with GFP-Syb2 or non-Tg control mice. Dashed lines mark the boundary of the ventral horn. D, Quantitative analysis of the NMJs in the fourth lumbrical muscles from hemizygous UbG76V-GFP-Syb2 mice, compared with age-matched littermate control mice. The numbers of the NMJs obtained from three pairs of mice at each age are as follows: 1 month (control, n = 356; UbG76V-GFP-Syb2, n = 420); 3 months (control, n = 411; UbG76V-GFP-Syb2, n = 327); 5 months (control, n = 301; UbG76V-GFP-Syb2, n = 357); 8 months (control, n = 325; UbG76V-GFP-Syb2, n = 320). E, More than 60% of end-plates (EPs) in UbG76V-GFP-Syb2 mice at 5 months of age, and >90% at 8 months of age, are either partially denervated or totally denervated. In contrast, 100% of the NMJs in control mice are fully innervated at 1, 3, 5, and 8 months of age. F, Motor neuron morphology (black arrowheads) revealed by Toluidine Blue-stained semithin (1 μm) sections of the ventral horn of lumbar spinal cord from UbG76V-GFP-Syb2 and nontransgenic littermate control mice (8 months of age). G, Quantification of motor neuron numbers from transverse sections (at 10 μm thickness) of lumbar spinal segments (L3–L5). At 6 months of age, motor neuron numbers (n = 3006 ± 35, N = 3 mice) in UbG76V-GFP-Syb2 mice were similar to non-Tg control mice (n = 2875 ± 120, N = 3 mice). At 8 months of age, motor number numbers were significantly (p = 0.0012) decreased in UbG76V-GFP-Syb2 mice (n = 1933 ± 106, N = 3 mice), compared with age-matched non-Tg control mice (n = 2715 ± 35, N = 3 mice). H, I, Cross section of lumbrical muscles from non-Tg control and UbG76V-GFP-Syb2 mice (8 months of age): black arrowhead in I points to a cluster of atrophic muscle fibers in UbG76V-GFP-Syb2 mice. J–O, The NMJ morphology in homozygous GFP-Syb2 and UbG76V-GFP-Syb2 mice (7 weeks of age). A small bundle of teased thigh muscle fibers (J, M, phase contrast) from GFP-Syb2 (J–L) and UbG76V-GFP-Syb2 mice (M–O) were labeled with Texas Red-conjugated α-bungarotoxin (L, O). The nerve terminals were identified by the presence of GFP fluorescence (K, N). In GFP-Syb2 mice, motor nerve terminals appear in a typical “pretzel-like” shape (arrow in K). In contrast, in UbG76V-GFP-Syb2 mice, nerve terminals appear shrunken (1 in N) or swollen (2 and 3 in N); muscle fibers appear atrophic (* in M), and two of five muscle fibers are completely denervated (4 and 5 in O). Scale bars: C, 200 μm; E, 50 μm; F, 20 μm; H, I, 50 μm; J–O, 50 μm.
To further characterize the phenotype in UbG76V-GFP-Syb2 mice, we systematically analyzed the NMJs in the fourth lumbrical muscle in hemizygous UbG76V-GFP-Syb2 mice at 1, 3, 5, and 8 months of age, and compared them to littermate nontransgenic control mice. At 1 month of age, UbG76V-GFP-Syb2 and nontransgenic control mice exhibited 100% fully innervated muscle fibers, and their NMJs were indistinguishable (control, n = 356; UbG76V-GFP-Syb2, n = 420). However, at later stages (3–8 months of age), UbG76V-GFP-Syb2 mice exhibited progressive muscle denervation. At 3 months of age, only a small fraction of the muscles (3 of 327 muscles) in UbG76V-GFP-Syb2 mice was affected with either partial or total denervation. At 5 months of age, 38.64 ± 15.12% of the muscle fibers in UbG76V-GFP-Syb2 mice (n = 357) were partially denervated, 26.84 ± 4.99% were totally denervated, and 34.5% remained fully innervated. At 8 months of age, 27.77 ± 3.07% muscle fibers in UbG76V-GFP-Syb2 mice were partially denervated, 62.77 ± 3.53% were totally denervated, and <10% of the muscle fibers (9.46 ± 0.80%) remained fully innervated. In contrast, 100% of the muscle fibers in nontransgenic control mice were fully innervated at 3, 5, and 8 months of age (Fig. 4D,E). Interestingly, in contrast to hemizygous UbG76V-GFP-Syb2 mice that exhibited late-onset paralysis and progressive muscle denervation, as described above, homozygous UbG76V-GFP-Syb2 mice became paralyzed by 7 weeks of age. Further examination of the NMJs revealed that muscle denervation occurred in these 7-week-old homozygous UbG76V-GFP-Syb2 mice (Fig. 4M–O), suggesting a possible gene dose effect of UbG76V-GFP-Syb2 on the degeneration of motor nerve terminals.
To examine motor neurons, we analyzed Nissl-stained sections of the lumbar spinal segments (L3–L5), and counted the number of motor neurons in UbG76V-GFP-Syb2 and nontransgenic littermate control mice at 6 and 8 months of age. At 6 months of age, when significant muscle denervation had occurred, motor neuron numbers in UbG76V-GFP-Syb2 mice (n = 3006 ± 35, N = 3 mice) were comparable to those observed in their nontransgenic littermate controls (n = 2875 ± 120, N = 3 mice; Fig. 4G). At 8 months of age, however, motor neuron numbers were significantly (p = 0.0012) reduced in UbG76V-GFP-Syb2 mice (n = 1933 ± 106, N = 3 mice), compared with nontransgenic littermate controls (n = 2715 ± 35, N = 3 mice; Fig. 4G).
Despite a reduction in motor neuron numbers in 8-month-old UbG76V-GFP-Syb2 mice, the morphology of the motor neurons that survived at this age appeared indistinguishable from those in control mice, as revealed by both light microscopy and electron microscopy. Toluidine Blue-stained semithin (1 μm) sections showed that the morphology of motor neurons was similar between UbG76V-GFP-Syb2 and control mice (Fig. 4F). The ultrastructure of motor neurons was also similar between UbG76V-GFP-Syb2 and control mice; motor neuron somata contained a large round-shaped nucleus and an abundance of cellular organelles, including mitochondria, Golgi bodies, endoplasmic reticulum, ribosomes, and lysosomes (Fig. 5C,D). Furthermore, synapses on motor neuron somata also appeared normal (Fig. 5E,G). Together, these data demonstrate that denervation at the NMJs precedes the loss of motor neuron cell bodies.
Figure 5.
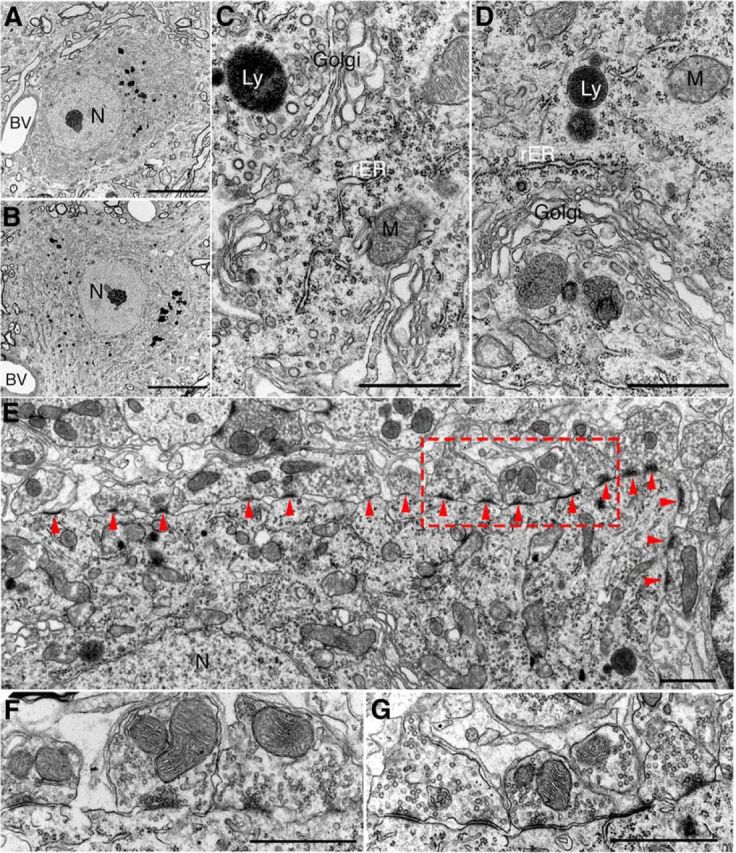
Ultrastructure of motor neuron in UbG76V-GFP-Syb2 mice. A, B, Representative electron micrographs (low-power view) of an example of lumbar spinal motor neuron from nontransgenic control mice (A) and UbG76V-GFP-Syb2 mice (B, 8 months of age); both somata contain a large round-shaped nucleus (N) with a prominent nucleolus and organelle-rich cytoplasm. Blood vessels (BV) are frequently observed. C, D, High-power electron micrographs of motor neuron soma from control (C) and UbG76V-GFP-Syb2 mice (D), showing subcellular organelles including mitochondria (M), Golgi apparatus (Golgi), rough endoplasmic reticulum (rER), and lysosomes (Ly). E–G, Synapses on motor neuron in UbG76V-GFP-Syb2 mice (E, G) and control mice (F; 8 months of age). Numerous synapses (arrowheads) are found on the plasma membrane of motor neuron soma in UbG76V-GFP-Syb2 mice (E, low-power view; G, high-magnification view of the region in E marked by the rectangular area) showing features similar to those of the synapses on motor neuron soma in control mice (F). Scale bars: A, B, 10 μm; C, D, 1 μm; E, 1 μm; F, G, 1 μm.
Progressive dysfunction of neuromuscular synaptic transmission in UbG76V-GFP-Syb2 mice
To determine the function of the NMJs in UbG76V-GFP-Syb2 mice, we performed electrophysiological analyses. We systematically measured the spontaneous miniature end-plate potentials (mEPPs) and evoked end-plated potentials (EPPs) at the NMJs in lumbrical muscles at 1, 3, 5, 8, and 10 months of age (Fig. 6). At 1 month of age, mEPP frequency was similar between UbG76V-GFP-Syb2 mice and their nontransgenic littermate controls. At 3 months of age, the mEPP frequency in UbG76V-GFP-Syb2 mice was significantly (p < 0.05) lower than that found in age-matched controls. Strikingly, mEPP frequencies were progressively reduced with age: 20% reduction at 5 months, 57% at 8 months, and 78% at 10 months (Fig. 6C). In contrast to this marked reduction in mEPP frequency, mEPP amplitudes in UbG76V-GFP-Syb2 mice were maintained at levels similar to those observed in nontransgenic littermate control mice (Fig. 6D). The change in mEPP frequency without a change in mEPP amplitude suggests that these impairments in synaptic transmission are likely due to defects in presynaptic nerve terminals.
Figure 6.

Age-dependent impairment of neuromuscular synaptic transmission in UbG76V-GFP-Syb2 mice. A, B, Sample traces of mEPPs (A) and EPPs (B) from control and UbG76V-GFP-Syb2 mice (1 and 8 months of age). C, D, Quantification of mEPP frequency (C) and amplitude (D) in control and UbG76V-GFP-Syb2 mice at 1, 3, 5, 8, and 10 months of age. After 3 months of age, mEPP frequency is significantly decreased in UbG76V-GFP-Syb2 mice compared with age-matched controls. p = 0.0218 (3 months of age); p = 0.0144 (5 months of age); p = 0.0382 (8 months of age); p = 0.0035 (10 months of age). In contrast, mEPP amplitudes remained similar between control and UbG76V-GFP-Syb2 mice. E, F, Quantification of EPP amplitudes (E) and quantal content (F). After 5 months of age, EPP amplitudes are significantly reduced in UbG76V-GFP-Syb2 mice, compared with controls. p = 0.0156 (5 months of age); p = 0.0055 (8 months of age); p = 0.0049 (10 months of age). F, Quantal content is also significantly decreased at 5, 8, and 10 months of age in UbG76V-GFP-Syb2 mice compared with controls. p = 0.0413 (5 months of age); p = 0.0135 (8 months of age); p = 0.0030 (10 months of age). The number of muscle fibers and mice analyzed: control (n = 44 muscle fibers, N = 4 mice); and UbG76V-GFP-Syb2 mice (n = 28 muscle fibers, N = 3 mice).
Consistent with the reduction in spontaneous synaptic activity, EPPs and quantal content were also progressively reduced at 5, 8, and 10 months of age in UbG76V-GFP-Syb mice (Figs. 6B,E,F). The application of trains of stimuli with increasing frequencies further revealed synaptic transmission failures in UbG76V-GFP-Syb2 mice (Fig. 7). To quantitatively compare the number of successful EPP responses to each stimulus, we counted the number of EPPs and divided this number by the number of stimuli to obtain the rate of successful response. In contrast to age-matched control mice, which exhibited a 100% response rate at all frequencies tested, the EPP response rate decreased progressively in 8-month-old UbG76V-GFP-Syb2 mice as the frequency of repetitive stimulation increased (Fig. 7B,C). This decrease in EPP response rate to repetitive stimulation was also age dependent. Synaptic transmission failure was detected in UbG76V-GFP-Syb2 transgenic mice at 3 months of age, and became more pronounced at later stages (5, 8, 10, and 12 months of age; Fig. 7D). Together, these results demonstrate that neuromuscular synapses in UbG76V-GFP-Syb2 mice failed to sustain high-frequency, repetitive synaptic transmission in an age-dependent and use-dependent manner.
Figure 7.

Use-dependent impairment of neuromuscular synaptic transmission in UbG76V-GFP-Syb2 mice. A, B, Sample EPP traces evoked by a 1-s-duration train containing an increasing frequency of stimuli (10, 30, 50, 70, 100, and 150 Hz). Traces in each column are recorded from the same muscle cell. In control mice (A), EPPs follow closely with increasing frequency of stimulation from 10–150 Hz. In UbG76V-GFP-Syb2 mice, EPPs fail to follow at high frequencies of stimulation; transmission failure first appears at 30 Hz (B, arrowheads point to stimulus artifact). C, Quantification of success rates (the percentage of EPP responses as a function of the frequency of stimulation) between control mice (open bar) and UbG76V-GFP-Syb2 transgenic mice (filled bar) at 10, 30, 50, 70, 100, and 150 Hz. With increasing stimulation frequency, the rates of successful synaptic transmission are significantly decreased in UbG76V-GFP-Syb2 mice. Control mice: n = 15 muscle fibers, N = 3 mice; UbG76V-GFP-Syb2 mice: n = 18 muscle fibers, N = 3 mice. p = 0.0107 (30 Hz); p = 0.0135 (50 Hz); p = 0.0132 (70 Hz); p = 0.0014 (100 Hz); and 1.367 × 10−8 (150 Hz). D, Quantification of success rates as a function of age in response to a 1 s train at 100 Hz (1 s) in control mice (open bar) and UbG76V-GFP-Syb2 mice (filled bars). The number of muscles (n) and the number of mice (N) analyzed at each age group are listed as follows: 1-month-old group: control mice, n = 15, N = 3; UbG76V-GFP-Syb2 mice, n = 15, N = 3; 3-month-old group: control mice, n = 15, N = 3; UbG76V-GFP-Syb2 mice, n = 14, N = 3, p = 0.005; 5-month-old group: control mice, n = 14, N = 3; UbG76V-GFP-Syb2 mice, n = 15, N = 3, p = 0.0089; 8-month-old group: control mice, n = 12, N = 3; UbG76V-GFP-Syb2 mice, n = 12, N = 3, p = 5.493 × 10−6; 10-month-old group: control mice, n = 15, N = 3; UbG76V-GFP-Syb2 mice, n = 15, N = 3, p = 3.891 × 10−7; 12-month-old group: control mice, n = 12, N = 3; UbG76V-GFP-Syb2 mice, n = 12, N = 3, p = 7.693 × 10−8). Calibration: the line underneath the 30 Hz example applies to both the 10 and 30 Hz traces; the line underneath the 150 Hz example applies to 50, 70, 100, and 150 Hz panels (traces represent the first 200 ms of the recording).
Ubiquitin, deubiquitinating enzyme and the proteasome activity in UbG76V-GFP-Syb2 mice
How does UbG76V-GFP-Syb2 lead to synaptic dysfunction and degeneration? Since UbG76V-GFP carries multiple lysine residues for ubiquitination (Dantuma et al., 2000; Lindsten et al., 2003), it raises the possibility that UbG76V-GFP-Syb2 may serve as a substrate for the UPS and impede the UPS in neurons. Indeed, changes in proteasome activity are implicated in SNARE-dependent neurodegeneration (Sharma et al., 2012b), and the loss of deubiquitination enzymes (DUBs), such as UchL1 (Saigoh et al., 1999; Chen et al., 2010; Bilguvar et al., 2013) or Usp14 (Wilson et al., 2002; Chen et al., 2009) leads to neurodegeneration. This raises the possibility that UbG76V-GFP-Syb2 might cause nonspecific global changes in the UPS system due to the presence of UbG76V-GFP in the fusion protein. To test this idea, we performed the following experiments.
First, we examined ubiquitin and deubiquitinating enzyme levels as well as proteasomal function in UbG76V-GFP-Syb2 mice. We isolated spinal cord homogenates from nontransgenic and UbG76V-GFP-Syb2 mice (8 months of age), and probed with anti-polyubiquitin antibodies. We found that both polyubiquitination and free ubiquitin levels appeared comparable between UbG76V-GFP-Syb2 and control mice (Fig. 8A). In addition, proteasomal function, as measured by the enzymatic activity of the proteasome, including ChT-L, trypsin-like, and PGPH activities, was similar between UbG76V-GFP-Syb2 and control mice (Fig. 8B). Furthermore, the expression of UchL1 and Usp14 in UbG76V-GFP-Syb2 mice was comparable to that observed in control or GFP-Syb2 mice (Fig. 8C–P). These results indicate that the expression of UbG76V-GFP-Syb2 in neurons did not cause nonspecific, global changes in the UPS system.
Figure 8.
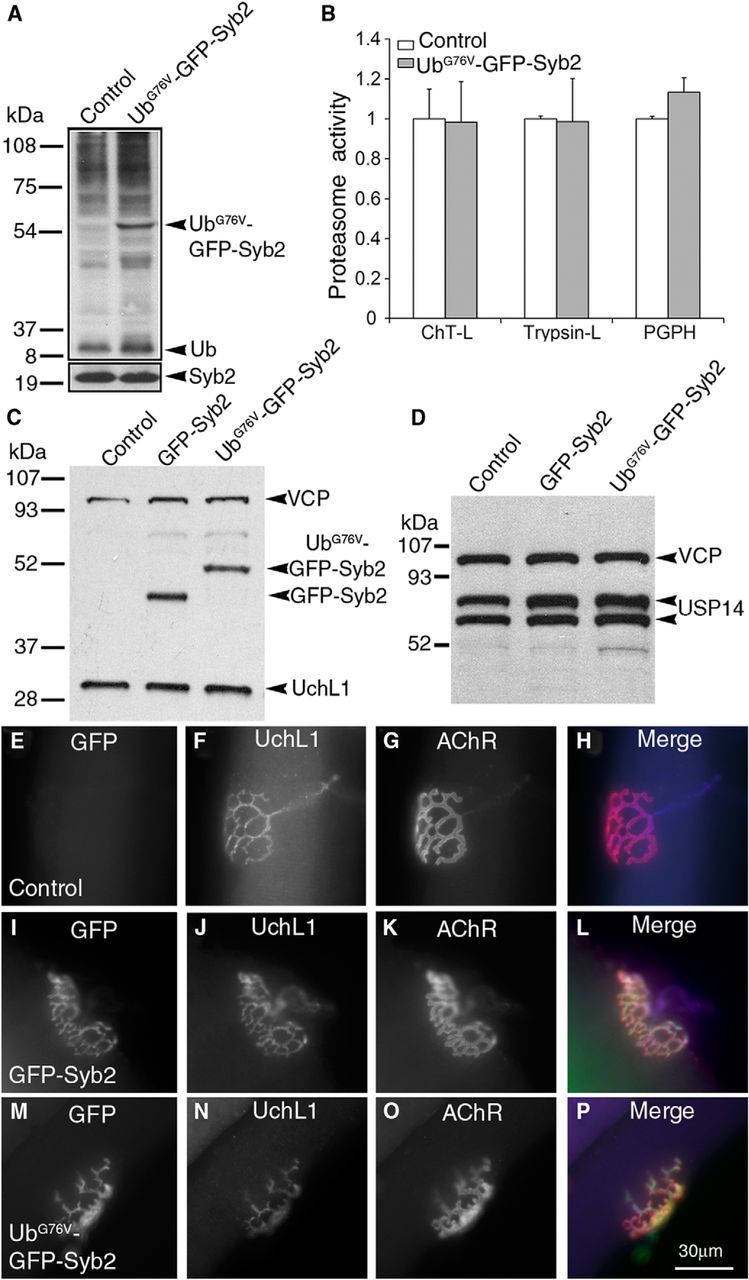
Analysis of ubiquitin, deubiquitinating enzymes, and proteasome activity. A, Spinal cord homogenates from control and UbG76V-GFP-Syb2 mice (8 months of age) separated by stepping gel (8% and 12%) and probed with anti-polyubiquitin antibodies. B, Proteasome activity, including ChT-L, trypsin-like, and PGPH activity is similar between control and UbG76V-GFP-Syb2 mice (N = 3 pairs, 8 months of age). C, Spinal cord homogenates from control, GFP-Syb2, and UbG76V-GFP-Syb2 mice (8 months of age) were probed with anti-GFP antibodies and then reprobed with anti-UchL1 antibodies (PGP9.5). UchL1 levels are similar among all three genotypes. D, Spinal cord homogenates from control, GFP-Syb2, and UbG76V-GFP-Syb2 mice (8 months of age) were probed using anti-Usp14 antibodies. Both isoforms of Usp14 are expressed at similar levels in all three genotypes. E–P, Teased thigh muscle fibers from control, GFP-Syb2, and UbG76V-GFP-Syb2 mice (5 months of age) were immunostained with anti-UchL1 antibodies and costained with α-bgt. UchL1 is localized at the NMJs in UbG76V-GFP-Syb2 mice (N), in a pattern similar to that seen in control mice (F) and GFP-Syb2 mice (J). Scale bars: (in P) E–P, 30 μm.
The NMJs are normal in UbG76V-GFP and UbG76V-GFP-Syntaxin1 transgenic mice
Next, we tested whether the overexpression of UbG76V-GFP in mice may affect neuromuscular synaptic function. We performed electrophysiological analyses of neuromuscular synapses in UbG76V-GFP mice that had been previously generated using a global β-actin promoter (Lindsten et al., 2003). We found that mEPP and EPP parameters as well as the capacity of neuromuscular synapses to sustain high-frequency stimulation in UbG76V-GFP mice (8 months old, N = 3) were indistinguishable from their nontransgenic littermate controls (8 months old, N = 3; Fig. 9). These electrophysiological data indicate that a global expression of UbG76V-GFP in mice does not affect neuromuscular function.
Figure 9.

Neuromuscular synaptic activity is normal in UbG76V-GFP transgenic mice. A, Sample traces of mEPPs recorded from the lumbrical muscles from control and UbG76V-GFP mice (8 months of age). B, Quantification of mEPP frequencies (UbG76V-GFP mice: 1.39 ± 0.05 Hz, n = 13; control mice: 1.36 ± 0.05 Hz, n = 15). No significant difference is found between UbG76V-GFP and control mice. C, EPPs recorded from the lumbrical muscles from UbG76V-GFP and control mice (8 months of age). D, Quantification of EPP amplitudes. EPP amplitudes are similar between UbG76V-GFP mice (25.82 ± 0.80 mV, n = 13 muscle fibers, N = 3 mice) and control mice (26.76 ± 0.61 mV, n = 15 muscle fibers, N = 3 mice). E, EPPs recorded from the lumbrical muscles in UbG76V-GFP and control mice (8 months of age) in response to a 1 s, 100 Hz train stimulation (only the first 20 EPPs are displayed). Similar responses are detected from UbG76V-GFP and from control mice.
Third, we tested whether UbG76V-GFP fused to a nonvesicular protein, such as Syntaxin1 (Syx1), might adversely affect neuromuscular synaptic function. We generated UbG76V-GFP-Syntaxin1 transgenic mice using the same neuron-specific Thy1 promoter used in UbG76V-GFP-Syb2 or GFP-Syb2 transgenic mice (Fig. 10A–C). Remarkably, UbG76V-GFP-Syntaxin1 mice were viable and had normal life spans with no signs of degeneration of motor neurons or of neuromuscular failure. In UbG76V-GFP-Syntaxin1 mice, GFP fluorescence was distributed along the nerve fibers and was highly concentrated at the motor nerve terminals (Fig. 10D). However, unlike UbG76V-GFP-Syb2, in which GFP puncta accumulate within motor neuron somata (Fig. 4), no GFP puncta were found in motor neurons in UbG76V-GFP-Syntaxin1 mice (Fig. 10E). Furthermore, electrophysiological analysis also showed that synaptic transmission at the NMJ in UbG76V-GFP-Syntaxin1 mice was indistinguishable from that control mice (Fig. 10F–J). Together, these data demonstrate that neither a global expression of UbG76V-GFP nor a neuron-specific overexpression of UbG76V-GFP-Syntaxin1 in mice caused synaptic degeneration.
Figure 10.

Normal synaptic activity at the NMJs in UbG76V-GFP-Syntaxin1 mice. A, A schematic diagram of the design of the construct for generating UbG76V-GFP-Syntaxin1 transgenic mice. Syx1, Syntaxin1. B, Western blot analysis of spinal cord homogenates from three lines of UbG76V-GFP-Syntaxin1 transgenic mice, probed with antibodies against syntaxin1. C, Western blot analysis of spinal cord homogenates, illustrating expression levels of transgene among UbG76V-GFP-Syntaxin1, UbG76V-GFP-Syb2, and GFP-Syb2 mice. D, The NMJ morphology in the triangularis sterni muscle of an 8-month-old UbG76V-GFP-Syntaxin1 transgenic mouse, labeled by anti-Syt2 antibodies and Texas-Red-conjugated α-bungarotoxin. Note that the nerve terminals are intensely labeled by GFP and Syt2. E, Frozen sections (12 μm thickness) of the ventral horn of spinal cords from UbG76V-GFP-Syntaxin1 mice (8 months of age), viewed under epifluorescence. GFP fluorescence is uniformly distributed. F, Sample traces of mEPPs recorded from the lumbrical muscles from UbG76V-GFP-Syntaxin1 and control mice (8 months of age). G, Quantification of mEPP frequency (UbG76V-GFP-Syntaxin1 mice: 1.26 ± 0.11 Hz, n = 22; control mice: 1.31 ± 0.14 Hz, n = 17) and amplitude (UbG76V-GFP-Syntaxin1 mice: 1.07 ± 0.10 mV, n = 22; control mice: 1.15 ± 0.08 mV, n = 17). Both mEPP frequency and amplitude are similar between UbG76V-GFP-Syntaxin1 and control mice. H, Sample traces of EPPs recorded in the lumbrical muscles from UbG76V-GFP-Syntaxin1 and control mice (8 months of age). I, Quantification of EPP amplitude. EPP amplitudes are similar between UbG76V-GFP-Syntaxin1 mice (27.29 ± 0.84 mV, n = 22) and control mice (26.99 ± 1.34 mV, n = 17). J, EPPs recorded from the lumbrical muscles from UbG76V-GFP-Syntaxin1 and control mice (8 months of age) in response to a 1 s, 100 Hz train stimulation (only the first 20 EPPs are displayed). Similar responses were detected from UbG76V-GFP-Syntaxin1 and control mice. Scale bars: D, 30 μm; E, 100 μm.
UbG76V-GFP-Syb2 interacts with SNAP-25 and Syntaxin1
We next asked whether the levels of synaptic proteins were altered in UbG76V-GFP-Syb2 mice, since the alteration of synaptic proteins may impact synaptic transmission. Quantitative immunoblotting analyses showed that synaptic proteins, including CSPα, synaptophysin, SNAP-25, Syb2, Syntaxin1, Munc18, complexin, synaptotagmin, synapsin, synaptojanin, synaptogyrin, SV2A, SV2B, Velis, Rab3a, Rab5a, and Rabphilin were detected at similar levels in UbG76V-GFP-Syb2 and control mice (Fig. 11A).
Figure 11.

UbG76V-GFP-Syb2 interacts with SNARE protein Syntaxin1 and SNAP-25. A, Quantitative immunoblotting analyses comparing the levels of synaptic protein expression in spinal cord homogenates of UbG76V-GFP-Syb2 and nontransgenic control mice (8 months of age, three pairs of each genotype). Synaptic proteins are expressed at similar levels between UbG76V-GFP-Syb2 and non-Tg control mice. B, Spinal cord homogenates from UbG76V-GFP, GFP-Syb2, UbG76V-GFP-Syb2, and control mice (8 months of age) were immunoprecipitated with anti-GFP antibodies, and then probed with anti-syntaxin1 and anti-SNAP-25 antibodies. Both syntaxin1 and SNAP-25 were coimmunoprecipitated from GFP-Syb2 and UbG76V-GFP-Syb2 mice, but not from UbG76V-GFP or nontransgenic control mice. C, Syntaxin1 immunoblotting detected a high-molecular-weight SNARE core complex in lysate from HEK293 cells that expressed SNAP-25 and Syntxin1A together with UbG76V-GFP-Syb2 (lane 4, unboiled), but not with UbG76V-GFP or a UbG76V-GFP-Syb2 mutant that was mutated in its SNARE motif. The SNARE core complex is sensitive to boiling (lane 3). Lanes 1, 3, 5: boiled; lanes 2, 4, 6: unboiled.
We next tested whether UbG76V-GFP-Syb2 interacted with other SNARE proteins such as SNAP-25 and Syntaxin1. Coimmunoprecipitation analysis showed that both GFP-Syb2 and UbG76V-GFP-Syb2, but not UbG76V-GFP, interacted with Syntaxin1 and SNAP-25 (Fig. 11B). Furthermore, SDS-resistant SNARE complexes were detected in the lysate of HEK293 cells cotransfected with SNAP-25, Syntaxin1 and UbG76V-GFP-Syb2. However, no SNARE complex was detected in cells expressing SNAP-25 and Syntaxin1 with UbG76V-GFP or with the UbG76V-GFP-Syb2 mutant that was mutated in its SNARE motif (Fig. 11C). Together, these data indicate that UbG76V-GFP-Syb2 is capable of interacting with SNARE proteins SNAP-25 and Syntaxin1.
Ultrastructural defects of presynaptic motor nerve terminals in UbG76V-GFP-Syb2 mice
To further determine whether physiological dysfunction of the NMJs in UbG76V-GFP-Syb2 mice was associated with alteration of the ultrastructure of motor nerve terminals, we performed EM analyses of the NMJs in UbG76V-GFP-Syb2 and control mice at 3, 5, and 8 months of age. At 3 months of age, presynaptic motor nerve terminals in UbG76V-GFP-Syb2 mice appeared similar to those of their littermate control mice (Fig. 12A–C), except for a small number of nerve terminals (5 of 39) that contained reduced numbers of synaptic vesicles. At 5 months of age, 14 of 30 NMJs in UbG76V-GFP-Syb2 mice contained partially denervated synaptic sites—sites where postsynaptic junctional folds were devoid of nerve terminals but were, in many cases, directly apposed by terminal Schwann cells. Nearly 30% of the nerve terminals (16 of 46) contained markedly reduced numbers of synaptic vesicles at this age. At 8 months of age, the density of synaptic vesicles in the majority of the nerve terminals (n = 55 nerve terminals, N = 5 mice) was significantly reduced (p < 0.05) compared with those of the control mice (n = 61 nerve terminals, N = 3 mice; Fig. 13, Table 1). Intriguingly, a significant number of nerve terminals (37 of 55) in UbG76V-GFP-Syb2 transgenic mice accumulated a large number of tubulovesicular profiles (Fig. 13D), and some postsynaptic sites were devoid of nerve terminals—instead, postsynaptic sites were occupied by Schwann cells (Fig. 13F–H). Consistent with the observations from light microscopic analysis, these EM studies demonstrated denervation of postsynaptic sites and reduction of synaptic vesicle density at the NMJs in UbG76V-GFP-Syb2 mice.
Figure 12.

Ultrastructure of motor nerve terminals in 3-month-old UbG76V-GFP-Syb2 mice. A, B, Examples of normal NMJs from control mice (A) and UbG76V-GFP-Syb2 mice (B). The nerve terminal (Nt) appears electron translucent and is filled with an abundance of synaptic vesicles (SVs) and mitochondria (M). C, An example of an impaired motor nerve terminal from UbG76V-GFP-Syb2 mice. The nerve terminal in this NMJ appears amorphous (*) and contains a reduced number of synaptic vesicle. White arrowheads point to junctional folds at the postsynaptic muscle membrane. Scale bar: (in C) A–C, 1 μm.
Figure 13.

Ultrastructural defects of motor nerve terminals in UbG76V-GFP-Syb2 mice (8 months of age). A, B, Examples of normal NMJs in control mice (A) and UbG76V-GFP-Syb2 mice (B). C, An example of an abnormal NMJ from UbG76V-GFP-Syb2 mice. This NMJ contains two nerve terminals (marked by 1 and 2), which are further magnified in D and E. The density of synaptic vesicles is markedly reduced; the axoplasm appears amorphous [denoted by an asterisk (*)] and is characterized by the accumulation of abnormal tubulovesicular structures (D, white arrow). F–H, Examples of degenerated NMJs: a nerve terminal engulfed by a Schwann cell process (F, black arrowhead), and synapses devoid of motor nerve terminals (G, H). Synaptic sites are marked by the presence of postsynaptic junctional folds (F–H, black arrow). nt, Nerve terminal; SC, Schwann cell. Scale bars: A–C, E–H, 1 μm; D, 0.5 μm.
Table 1.
Morphometric analysis of synaptic ultrastructure
| Genotype | Non-Tg control mice (n = 61, N = 3) | UbG76V-GFP-Syb2 mice (n = 55, N = 5) |
|---|---|---|
| Nerve terminal area (μm2) | 2.92 ± 0.29 | 3.44 ± 0.48 |
| Nerve terminal perimeter (μm) | 6.76 ± 0.34 | 7.81 ± 0.63 |
| Presynaptic contact length (μm) | 3.35 ± 0.21 | 2.93 ± 0.32 |
| Presynaptic/perimeter ratio (%) | 48.0 ± 1.3 | 35.3 ± 2.0* |
| Synaptic vesicle number (per nerve terminal) | 175 ± 14.6 | 120 ± 17.9† |
| Synaptic vesicle density (per μm2) | 67.6 ± 4.1 | 41.7 ± 3.9‡ |
Values are reported as the mean ± SEM. Synaptic contact ratio and synaptic vesicle density were significantly decreased in UbG76V-GFP-Syb2 mice (8 months of age) compared with littermate control mice. n, Number of nerve terminals; N, number of mice.
*p = 4.0753 × 10−7.
†p = 0.0177.
‡p = 1.5886 × 10−5.
Defects of synaptic vesicle endocytosis and membrane trafficking in UbG76V-GFP-Syb2 mice
The striking reduction of synaptic vesicle numbers at nerve terminals in UbG76V-GFP-Syb2 mice may be caused by an impairment of synaptic vesicle endocytosis and membrane trafficking. To test this possibility, we performed HRP uptake experiments in isolated synaptosomes and live-cell imaging experiments of synaptic terminals of primary hippocampal neurons expressing UbG76V-GFP-Syb2.
First, we isolated synaptosomes from three pairs of UbG76V-GFP-Syb2 mice and their nontransgenic littermate controls (8 months of age). Synaptosome preparations were incubated with or without 60 mm KCl for 15 min in the presence of HRP. After reaction with DAB, the preparations were processed for EM. Under EM, the numbers of HRP-positive synaptic vesicles were counted in each synaptosome. We found that, under basal conditions, the number of HRP-positive SVs was similar among synaptosomes isolated from UbG76V-GFP-Syb2 and control mice (Fig. 14A,C). However, upon stimulation with 60 mm KCl, the number of HRP-positive SVs was significantly reduced in UbG76V-GFP-Syb2-derived synaptosomes compared with those of control synaptosomes (Fig. 14B,D,E).
Figure 14.

Impairments in synaptic vesicle endocytosis and membrane trafficking in UbG76V-GFP-Syb2 mice. A–D, EM images of synaptosomes isolated from control (A, B) and UbG76V-GFP-Syb2 mice (C, D). A and C show examples of synaptosomes at rest; B and D show examples of synaptosomes containing HRP-labeled SVs (arrowheads) after incubation with 60 mm KCl for 15 min. E, Bar graphs showing the average number of HRP-positive SVs per synaptosome after depolarization with 60 mm KCl for 15 min. A total of 215 synaptosomes from UbG76V-GFP-Syb2 mice (N = 3) and 209 synaptosomes from control mice (N = 3) were analyzed. The average numbers of HRP-positive SVs per synaptosome from UbG76V-GFP-Syb2 mice are significantly (p = 0.0493) reduced in UbG76V-GFP-Syb2 mice (6.24 ± 1.66 vesicles, N = 3 mice) compared with control mice (11.42 ± 1.55 vesicles, N = 3 mice). F, Schematic diagram of GFP-Syb2-mOrange and UbG76V-GFP-Syb2-mOrange. G, Average traces of fluorescence changes in synaptic boutons expressing GFP-Syb2-mOrange (n = 8) and UbG76V-GFP-Syb2-mOrange (n = 9). H, Analysis from the same experiments as in G. The peak fluorescence generated during 20 Hz stimulation is normalized to the peak fluorescence generated after NH4Cl treatment. The average peak values in synaptic boutons expressing UbG76V-GFP-Syb2-mOrange are significantly (p = 0.0067) lower than those expressing GFP-Syb2-mOrange. Scale bar: A–D (in C), 300 nm.
Next, we performed live imaging experiments to measure membrane trafficking in the synapses of hippocampal neurons expressing either GFP-Syb2 or UbG76V-GFP-Syb2, both tagged with pH-sensitive mOrange, to visualize their trafficking in isolation from the GFP fluorescence (Ramirez et al., 2012; Fig. 14F). We observed similar kinetics of vesicle trafficking between UbG76V-GFP-Syb2-mOrange and GFP-Syb2-mOrange, measured by the time to reach maximum fluorescence by stimulation, and by decay time to baseline values. Specifically, the time constants (t1/2) in synaptic boutons expressing UbG76V-GFP-Syb2-mOrange were 10.9 ± 1.5 s (rise time) and 30.1 ± 7.9 s (decay time), which were similar to those expressing GFP-Syb2-mOrange (rise time, 8.9 ± 3.3 s; decay time, 25.5 ± 11.3 s; p > 0.3, no statistical difference). However, baseline fluorescence was significantly lower (p = 0.011) in synaptic boutons expressing UbG76V-GFP-Syb2-mOrange (79.3 ± 4.2 a.u., n = 9) than those expressing GFP-Syb2-mOrange (106.6 ± 8.3 a.u., n = 8), indicating that a lower fraction of UbG76V-GFP-Syb2-mOrange-tagged vesicles was available for trafficking. In agreement with this observation, synaptic boutons in neurons expressing UbG76V-GFP-Syb2-mOrange displayed significantly reduced levels of vesicle trafficking, as indicated by a lower ratio of fluorescence during stimulation normalized to the fluorescence after NH4Cl application, which alkalinizes all acidic vesicles and uncovers the total pool of mOrange-tagged protein (Fig. 14G,H). These data demonstrate impaired vesicle trafficking in synapses expressing UbG76V-GFP-Syb2 compared with those expressing GFP-Syb2.
Discussion
Our initial motivation for the present study was to generate an in vivo mouse model for monitoring the function of the ubiquitin proteasome system in synapses, as Ub/Ub conjugates are present at both central synapses (Chapman et al., 1992, 1994) and peripheral synapses (Serdaroglu et al., 1992), as well as in a broad spectrum of neurodegenerative diseases (for review, see Ciechanover and Brundin, 2003; Dennissen et al., 2012; Dantuma and Bott, 2014). We have generated transgenic mice expressing fusion proteins (UbG76V-GFP-Syb2, GFP-Syb2, or UbG76V-GFP-Syntaxin1) under the control of a neuron-specific Thy-1 promoter. Surprisingly, UbG76V-GFP-Syb2 transgenic mice develop adult-onset degeneration of the presynaptic motor nerve terminals and premature death, whereas transgenic mice expressing either GFP-Syb2 or UbG76V-GFP-Syntaxin1 are normal and viable. In UbG76V-GFP-Syb2 mice, synaptic vesicle endocytosis and membrane trafficking are impaired, and motor nerve terminals become progressively dysfunctional. Thus, UbG76V-GFP-Syb2 may likely exert its role “locally” at the nerve terminal in UbG76V-GFP-Syb2 transgenic mice.
How overexpression of UbG76V-GFP-Syb2 leads to degeneration of motor nerve terminals remains to be further elucidated. Our data indicate that overexpressing UbG76V-GFP-Syb2 in neurons does not alter the expression levels of ubiquitin or DUBs, or proteasome activity in general. These results are consistent with those of a number of previous studies. First, transgenic mice overexpressing either wild-type or mutated ubiquitin are viable and normal (Tsirigotis et al., 2001). Thus, global alteration of ubiquitin expression itself does not appear sufficient to cause neurodegeneration. Second, transgenic mice overexpressing UbG76V-GFP are also grossly normal (Lindsten et al., 2003) and exhibit no neuromuscular abnormalities (present study). Finally, transgenic mice overexpressing UbG76V-GFP-Syntaxin1 are normal. Since both UbG76V-GFP-Syb2 and UbG76V-GFP-Syntaxin1 carry an N-terminal fusion of UbG76V-GFP, the markedly different phenotype between UbG76V-GFP-Syb2 and UbG76V-GFP-Syntaxin1 mice indicates that synaptic degeneration in UbG76V-GFP-Syb2 mice is unlikely due to nonspecific global changes in the UPS system.
Overexpression of either UbG76V or GFP-Syb2 in mice does not affect the NMJs, but a combination of both UbG76V and GFP-Syb2 (UbG76V-GFP-Syb2) produces drastic effects on the motor nerve terminals. Biochemical analyses indicate that UbG76V-GFP-Syb2 appears predominantly as a single band (Fig. 1B), suggesting that UbG76V-GFP-Syb2 mainly exists in a monoubiquitinated form. Since monoubiquitination regulates protein targeting (Hicke and Dunn, 2003), it is possible that UbG76V-GFP-Syb2 may be mis-sorted and/or mistargeted. Indeed, numerous GFP-positive puncta are present in motor neuron somata in UbG76V-GFP-Syb2 mice, but not in GFP-Syb2 mice. These GFP-positive puncta are closely associated with cis-Golgi, probably localized to trans-Golgi and/or the endoplasmic reticulum. This suggests a possibility of abnormal trafficking of UbG76V-GFP-Syb2, relative to endogenous Syb2 or GFP-Syb2. In addition, the GFP puncta detected in motor neuron somata in UbG76V-GFP-Syb2 mice are reminiscent of those observed in transgenic mice expressing a mutant G85R SOD1-yellow fluorescent protein that results in progressive motor neuron degeneration (Wang et al., 2009). This also raises a possibility that UbG76V-GFP-Syb2 may have been misfolded in the motor neuron cell body, ultimately resulting in neurodegeneration.
Synaptobrevins, including Syb1 and Syb2, are expressed in spinal motor neurons (Trimble et al., 1988; Baumert et al., 1989; Elferink et al., 1989; Südhof et al., 1989; Li et al., 1996; Jacobsson et al., 1998; Raptis et al., 2005) and are essential for survival (Schoch et al., 2001; Nystuen et al., 2007; Liu et al., 2011). Syb2 is critically involved in synaptic vesicle exocytosis (Schoch et al., 2001; Deák et al., 2006) and endocytosis (Sampo et al., 2003; Deák et al., 2004; Xu et al., 2013; Zhang et al., 2013). In Drosophila, the loss of neuronal synaptobrevin leads to neurodegeneration (Haberman et al., 2012). Therefore, it is conceivable that the fusion protein UbG76V-GFP-Syb2 competes with both Syb1 and Syb2, and interferes with their normal function by incorporating them into the SNARE complex with other SNARE proteins, including SNAP-25 and Syntaxin1. Thus, it is plausible that the incorporation of UbG76V-GFP-Syb2 into the SNARE complexes disrupts normal SNARE function, leading to a progressive loss of synaptic vesicle. Consistent with this hypothesis, previous studies have shown that perturbation of the N-terminal region of synaptobrevin inhibits neurotransmitter release (Cornille et al., 1995) and blocks membrane retrieval in calyx synapses (Hosoi et al., 2009).
Further evidence that a deficit in synaptic function underlies the neurodegenerative phenotype observed in UbG76V-GFP-Syb2 mice comes from EM data in the present study, which shows an accumulation of tubulovesicular structures in the motor nerve terminals in UbG76V-GFP-Syb2 mice. These tubulovesicular structures might be precursors involved in synaptic vesicle biogenesis (Nakata et al., 1998), and their accumulation would implicate impairments of synaptic vesicle biogenesis. Alternatively, the accumulation of tubulovesicular structures may indicate a possible defect in synaptic vesicle recycling/endocytosis, as similar ultrastructural defects are reported in shibire mutant flies (Koenig and Ikeda, 1989) and dynamin mutant mice (Ferguson et al., 2007; Hayashi et al., 2008; Lou et al., 2008, 2012; Park et al., 2013). Furthermore, these ultrastructural defects are also present in the nerve terminals in mutant mice deficient in the deubiquitinating enzyme UChL1 (Chen et al., 2010); in rats treated with dithiobiuret, a toxic organosulfur compound (Xu et al., 2002); and in humans and animals afflicted with transmissible spongiform encephalopathies (Liberski and Jeffrey, 2004).
Our data demonstrate that synaptic degeneration in UbG76V-GFP-Syb2 mice progresses through age, a feature that is common to many neurodegenerative disorders. It remains unclear what mechanism underlies this age-dependent progressive degeneration. In UbG76V-GFP-Syb2 mice, the majority of the NMJs do not degenerate until after 5 months of age, even though UbG76V-GFP-Syb2 is expressed as early as P0. The slow, chronic effect of UbG76V-GFP-Syb2 on synaptic degeneration could be attributed to the relatively low levels of transgene expression in hemizygous UbG76V-GFP-Syb2 mice. For example, the phenotype may accumulate over time and reach a “threshold” level. Indeed, synaptic degeneration occurs much more acutely (at ∼7 weeks) in homozygous UbG76V-GFP-Syb2 mice compared with hemizygous UbG76V-GFP-Syb2 mice (at ∼7 months of age), indicating that higher levels of transgene expression could exacerbate the degeneration of the motor nerve terminals in these transgenic mice.
Loss of presynaptic motor nerve terminals at the NMJ occurs in amyotrophic lateral sclerosis (ALS) patients as well as in mouse models of ALS, leading to muscle denervation (Kennel et al., 1996; Bruijn et al., 1997, 1998; Williamson and Cleveland, 1999; Frey et al., 2000; Fischer et al., 2004; Schaefer et al., 2005; Gould et al., 2006; Vinsant et al., 2013). The mechanisms underlying presynaptic degeneration remain largely unknown. We believe that UbG76V-GFP-Syb2 mice may serve as a model for further elucidating the underlying cellular and molecular mechanisms of synaptic degeneration.
Footnotes
This work was supported by grants (to W.L.) from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS-055028), the Robert Packard Center for ALS Research at Johns Hopkins, and the Cain Foundation in Medical Research. We thank Dr. M. Goedert (MRC Laboratory, Cambridge, UK) for providing the Thy1 vector; Dr. Nico Dantuma (Karolinska Institute, Stockholm, Sweden) for providing the UbG76V-GFP vector and UbG76V-GFP transgenic mice; and Daniel Padgett for technical assistance. We also thank Drs. Wanjin Hong, Thomas Gould, Nico Dantuma, Wesley Thompson, and U.J. McMahan for their input and comments.
References
- Archer BT, 3rd, Ozçelik T, Jahn R, Francke U, Südhof TC. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. J Biol Chem. 1990;265:17267–17273. [PubMed] [Google Scholar]
- Bachmair A, Varshavsky A. The degradation signal in a short-lived protein. Cell. 1989;56:1019–1032. doi: 10.1016/0092-8674(89)90635-1. [DOI] [PubMed] [Google Scholar]
- Baumert M, Maycox PR, Navone F, De Camilli P, Jahn R. Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J. 1989;8:379–384. doi: 10.1002/j.1460-2075.1989.tb03388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, Delil S, Caglayan AO, Baranoski JF, Erturk O, Yalcinkaya C, Karacorlu M, Dincer A, Johnson MH, Mane S, Chandra SS, Louvi A, Boggon TJ, Lifton RP, Horwich AL, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:3489–3494. doi: 10.1073/pnas.1222732110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF, Martinelli DC, Südhof TC. The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc Natl Acad Sci U S A. 2011;108:2534–2539. doi: 10.1073/pnas.1019577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd IA, Martin AR. The end-plate potential in mammalian muscle. J Physiol. 1956;132:74–91. doi: 10.1113/jphysiol.1956.sp005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/S0896-6273(00)80272-X. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Butz S, Okamoto M, Südhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/S0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/S0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chapman AP, Courtney SC, Smith SJ, Rider CC, Beesley PW. Ubiquitin immunoreactivity of multiple polypeptides in rat brain synaptic membranes. Biochem Soc Trans. 1992;20:155S. doi: 10.1042/bst020155s. [DOI] [PubMed] [Google Scholar]
- Chapman AP, Smith SJ, Rider CC, Beesley PW. Multiple ubiquitin conjugates are present in rat brain synaptic membranes and postsynaptic densities. Neurosci Lett. 1994;168:238–242. doi: 10.1016/0304-3940(94)90459-6. [DOI] [PubMed] [Google Scholar]
- Chen F, Sugiura Y, Myers KG, Liu Y, Lin W. Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc Natl Acad Sci U S A. 2010;107:1636–1641. doi: 10.1073/pnas.0911516107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Qin LN, Li XM, Walters BJ, Wilson JA, Mei L, Wilson SM. The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J Neurosci. 2009;29:10909–10919. doi: 10.1523/JNEUROSCI.2635-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/S0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Clarke PG, Oppenheim RW. Neuron death in vertebrate development: in vitro methods. Methods Cell Biol. 1995;46:277–321. doi: 10.1016/S0091-679X(08)61933-0. [DOI] [PubMed] [Google Scholar]
- Cornille F, Deloye F, Fournié-Zaluski MC, Roques BP, Poulain B. Inhibition of neurotransmitter release by synthetic proline-rich peptides shows that the N-terminal domain of vesicle-associated membrane protein/synaptobrevin is critical for neuro-exocytosis. J Biol Chem. 1995;270:16826–16832. doi: 10.1074/jbc.270.28.16826. [DOI] [PubMed] [Google Scholar]
- Dantuma NP, Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci. 2014;7:70. doi: 10.3389/fnmol.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- Deák F, Schoch S, Liu X, Südhof TC, Kavalali ET. Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol. 2004;6:1102–1108. doi: 10.1038/ncb1185. [DOI] [PubMed] [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Cameron R, Greengard P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J Cell Biol. 1983;96:1337–1354. doi: 10.1083/jcb.96.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennissen FJ, Kholod N, van Leeuwen FW. The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog Neurobiol. 2012;96:190–207. doi: 10.1016/j.pneurobio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elferink LA, Trimble WS, Scheller RH. Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J Biol Chem. 1989;264:11061–11064. [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wölfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, O'toole E, Flavell R, Cremona O, Miesenböck G, Ryan TA, De Camilli P. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fykse EM, Li C, Südhof TC. Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinases in vitro. J Neurosci. 1995;15:2385–2395. doi: 10.1523/JNEUROSCI.15-03-02385.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman A, Williamson WR, Epstein D, Wang D, Rina S, Meinertzhagen IA, Hiesinger PR. The synaptic vesicle SNARE neuronal Synaptobrevin promotes endolysosomal degradation and prevents neurodegeneration. J Cell Biol. 2012;196:261–276. doi: 10.1083/jcb.201108088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Rhee JS, Maximov A, Lin W, Hammer RE, Rosenmund C, Südhof TC. C-terminal ECFP fusion impairs synaptotagmin 1 function: crowding out synaptotagmin 1. J Biol Chem. 2005;280:5089–5100. doi: 10.1074/jbc.M408757200. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Raimondi A, O'Toole E, Paradise S, Collesi C, Cremona O, Ferguson SM, De Camilli P. Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. Proc Natl Acad Sci U S A. 2008;105:2175–2180. doi: 10.1073/pnas.0712171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo. Ed 2. Plainview, NY: Cold Spring Harbor Laboratory; 1994. [Google Scholar]
- Hosoi N, Holt M, Sakaba T. Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron. 2009;63:216–229. doi: 10.1016/j.neuron.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Hubbard JI, Llinas R, Quastel DMJ. Investigation of presynaptic function. Baltimore: Williams & Wilkins; 1969. [Google Scholar]
- Jacobsson G, Piehl F, Meister B. VAMP-1 and VAMP-2 gene expression in rat spinal motoneurones: differential regulation after neuronal injury. Eur J Neurosci. 1998;10:301–316. doi: 10.1046/j.1460-9568.1998.00050.x. [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Johnson ES, Ma PC, Ota IM, Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem. 1995;270:17442–17456. doi: 10.1074/jbc.270.29.17442. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AM, Zacks SI. The fine structure of motor endplate morphogenesis. J Cell Biol. 1969;42:154–169. doi: 10.1083/jcb.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: an electromyographic study. Neuroreport. 1996;7:1427–1431. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- Khvotchev M, Südhof TC. Proteolytic processing of amyloid-beta precursor protein by secretases does not require cell surface transport. J Biol Chem. 2004;279:47101–47108. doi: 10.1074/jbc.M408474200. [DOI] [PubMed] [Google Scholar]
- Koenig JH, Ikeda K. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. J Neurosci. 1989;9:3844–3860. doi: 10.1523/JNEUROSCI.09-11-03844.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Edelmann L, Jahn R, Dahlström A. Axonal transport and distribution of synaptobrevin I and II in the rat peripheral nervous system. J Neurosci. 1996;16:137–147. doi: 10.1523/JNEUROSCI.16-01-00137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberski PP, Jeffrey M. Tubulovesicular structures—the ultrastructural hallmark for transmissible spongiform encephalopathies or prion diseases. Folia Neuropathol. 2004;42(Suppl B):96–108. [PubMed] [Google Scholar]
- Liley AW. An investigation of spontaneous activity at the neuromuscular junction of the rat. J Physiol. 1956;132:650–666. doi: 10.1113/jphysiol.1956.sp005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten K, Uhlíková T, Konvalinka J, Masucci MG, Dantuma NP. Cell-based fluorescence assay for human immunodeficiency virus type 1 protease activity. Antimicrob Agents Chemother. 2001;45:2616–2622. doi: 10.1128/AAC.45.9.2616-2622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten K, Menéndez-Benito V, Masucci MG, Dantuma NP. A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol. 2003;21:897–902. doi: 10.1038/nbt851. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Lin W. The role of Synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. J Physiol. 2011;589:1603–1618. doi: 10.1113/jphysiol.2010.201939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Paradise S, Ferguson SM, De Camilli P. Selective saturation of slow endocytosis at a giant glutamatergic central synapse lacking dynamin 1. Proc Natl Acad Sci U S A. 2008;105:17555–17560. doi: 10.1073/pnas.0809621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Fan F, Messa M, Raimondi A, Wu Y, Looger LL, Ferguson SM, De Camilli P. Reduced release probability prevents vesicle depletion and transmission failure at dynamin mutant synapses. Proc Natl Acad Sci U S A. 2012;109:E515–E523. doi: 10.1073/pnas.1121626109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos MF, Mukherjee K, Chen X, Rizo J, Südhof TC. Evidence for SNARE zippering during Ca2+-triggered exocytosis in PC12 cells. Neuropharmacology. 2003;45:777–786. doi: 10.1016/S0028-3908(03)00318-6. [DOI] [PubMed] [Google Scholar]
- Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystuen AM, Schwendinger JK, Sachs AJ, Yang AW, Haider NB. A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics. 2007;8:1–10. doi: 10.1007/s10048-006-0068-7. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci. 2006;26:13493–13504. doi: 10.1523/JNEUROSCI.3519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Shen H, Liu L, Liu X, Ferguson SM, De Camilli P. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J Cell Sci. 2013;126:5305–5312. doi: 10.1242/jcs.138578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- Puttaparthi K, Wojcik C, Rajendran B, DeMartino GN, Elliott JL. Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes. J Neurochem. 2003;87:851–860. doi: 10.1046/j.1471-4159.2003.02028.x. [DOI] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET. Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron. 2012;73:121–134. doi: 10.1016/j.neuron.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raptis A, Torrejón-Escribano B, Gómez de Aranda I, Blasi J. Distribution of synaptobrevin/VAMP 1 and 2 in rat brain. J Chem Neuroanat. 2005;30:201–211. doi: 10.1016/j.jchemneu.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Rizo J, Südhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- Sampo B, Kaech S, Kunz S, Banker G. Two distinct mechanisms target membrane proteins to the axonal surface. Neuron. 2003;37:611–624. doi: 10.1016/S0896-6273(03)00058-8. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Serdaroglu P, Askanas V, Engel WK. Immunocytochemical localization of ubiquitin at human neuromuscular junctions. Neuropathol Appl Neurobiol. 1992;18:232–236. doi: 10.1111/j.1365-2990.1992.tb00785.x. [DOI] [PubMed] [Google Scholar]
- Sharma M, Burré J, Südhof TC. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- Sharma M, Burré J, Bronk P, Zhang Y, Xu W, Südhof TC. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012a;31:829–841. doi: 10.1038/emboj.2011.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Burré J, Südhof TC. Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci Transl Med. 2012b;4:147ra113. doi: 10.1126/scitranslmed.3004028. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Baumert M, Perin MS, Jahn R. A synaptic vesicle membrane protein is conserved from mammals to Drosophila. Neuron. 1989;2:1475–1481. doi: 10.1016/0896-6273(89)90193-1. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Trimble WS, Cowan DM, Scheller RH. VAMP-1: a synaptic vesicle-associated integral membrane protein. Proc Natl Acad Sci U S A. 1988;85:4538–4542. doi: 10.1073/pnas.85.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigotis M, Thurig S, Dube M, Vanderhyden BC, Zhang M, Gray DA. Analysis of ubiquitination in vivo using a transgenic mouse model. Biotechniques. 2001;31:120–126. 128–130. doi: 10.2144/01311rr03. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Vidal M, Morris R, Grosveld F, Spanopoulou E. Tissue-specific control elements of the Thy-1 gene. EMBO J. 1990;9:833–840. doi: 10.1002/j.1460-2075.1990.tb08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 2013;3:431–457. doi: 10.1002/brb3.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, 3rd, Perrin S, Feany MB, Horwich AL. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc Natl Acad Sci U S A. 2009;106:1392–1397. doi: 10.1073/pnas.0813045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Bhattacharyya B, Rachel RA, Coppola V, Tessarollo L, Householder DB, Fletcher CF, Miller RJ, Copeland NG, Jenkins NA. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat Genet. 2002;32:420–425. doi: 10.1038/ng1006. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol. 2001;64:393–429. doi: 10.1016/S0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- Xu J, Luo F, Zhang Z, Xue L, Wu XS, Chiang HC, Shin W, Wu LG. SNARE proteins synaptobrevin, SNAP-25, and syntaxin are involved in rapid and slow endocytosis at synapses. Cell Rep. 2013;3:1414–1421. doi: 10.1016/j.celrep.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Autio D, Rheuben MB, Atchison WD. Impairment of synaptic vesicle exocytosis and recycling during neuromuscular weakness produced in mice by 2,4-dithiobiuret. J Neurophysiol. 2002;88:3243–3258. doi: 10.1152/jn.00934.2001. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang D, Sun T, Xu J, Chiang HC, Shin W, Wu LG. The SNARE proteins SNAP25 and synaptobrevin are involved in endocytosis at hippocampal synapses. J Neurosci. 2013;33:9169–9175. doi: 10.1523/JNEUROSCI.0301-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]