

Abstract
The brainstem preBötzinger complex (preBötC) generates the rhythm underlying inspiratory breathing movements and its core interneurons are derived from Dbx1-expressing precursors. Recurrent synaptic excitation is required to initiate inspiratory bursts, but whether excitatory synaptic mechanisms also contribute to inspiratory–expiratory phase transition is unknown. Here, we examined the role of short-term synaptic depression using a rhythmically active neonatal mouse brainstem slice preparation. We show that afferent axonal projections to Dbx1 preBötC neurons undergo activity-dependent depression and we identify a refractory period (∼2 s) after endogenous inspiratory bursts that precludes light-evoked bursts in channelrhodopsin-expressing Dbx1 preBötC neurons. We demonstrate that the duration of the refractory period—but neither the cycle period nor the magnitude of endogenous inspiratory bursts—is sensitive to changes in extracellular Ca2+. Further, we show that postsynaptic factors are unlikely to explain the refractory period or its modulation by Ca2+. Our findings are consistent with the hypothesis that short-term synaptic depression in Dbx1 preBötC neurons influences the inspiratory–expiratory phase transition during respiratory rhythmogenesis.
SIGNIFICANCE STATEMENT Theories of breathing's neural origins have heretofore focused on intrinsically bursting “pacemaker” cells operating in conjunction with synaptic inhibition for phase transition and cycle timing. However, contemporary studies falsify an obligatory role for pacemaker-like neurons and synaptic inhibition, giving credence to burst-generating mechanisms based on recurrent excitation among glutamatergic interneurons of the respiratory kernel. Here, we investigated the role of short-term synaptic depression in inspiratory–expiratory phase transition. Until now, this role remained an untested prediction of mathematical models. The present data emphasize that synaptic properties of excitatory interneurons of the respiratory rhythmogenic kernel, derived from Dbx1-expressing precursors, may provide the core logic underlying the rhythm for breathing.
Keywords: breathing, central pattern generator, oscillation, respiration
Introduction
Breathing movements emanate from neural rhythms in the preBötzinger complex (preBötC) of the ventrolateral medulla (Smith et al., 1991; Feldman et al., 2013; Moore et al., 2013). The breathing cycle consists of an inspiratory phase in which preBötC neurons discharge bursts of spikes synchronously followed by a two-part expiratory phase in which preBötC neurons remain quiescent, at least during the initial postinspiratory stage (but may recover spontaneous activity during the second expiratory stage). Core rhythm-generating preBötC interneurons are derived from progenitors that express the homeobox gene Dbx1 (henceforth referred to as Dbx1 neurons), which are glutamatergic and interconnected bilaterally in the preBötC (Bouvier et al., 2010; Gray et al., 2010). AMPAR-mediated excitatory interactions are required to initiate inspiratory bursts (Funk et al., 1993; Wallén-Mackenzie et al., 2006), but there is no consensus regarding the cellular and synaptic mechanisms that terminate inspiratory bursts and lead to the quiescent postinspiratory phase of respiratory cycle (i.e., inspiratory–expiratory phase transition).
A longstanding view posits that the inspiratory–expiratory phase transition depends on respiratory circuits throughout the pons and medulla operating via postsynaptic inhibition (Richter, 1982; Bianchi et al., 1995). However, the obligatory role of inhibition is falsified by experiments that block chloride-mediated synaptic transmission in respiratory networks but do not significantly perturb respiratory rhythm in vitro (Brockhaus and Ballanyi, 1998; Ren and Greer, 2006; Feldman et al., 2013) or in vivo (Janczewski et al., 2013; Sherman et al., 2015). One alternative mechanism that does not depend on distributed inhibitory circuits is the “group pacemaker,” which instead focuses on collective activity among preBötC neurons. According to this model, recurrent excitatory synaptic activity initiates inspiratory bursts (for which there is strong evidence: Rekling and Feldman, 1998; Pace et al., 2007; Carroll and Ramirez, 2013; Carroll et al., 2013) and short-term synaptic depression promotes burst termination and inspiratory–expiratory phase transition (but this latter part remains an untested model prediction: Rubin et al., 2009).
Using the group-pacemaker model as our conceptual framework, we investigated the role of short-term synaptic depression in respiratory rhythm generation. Our results in a reduced slice context support the group-pacemaker model and are consistent with the notion that presynaptic depression in Dbx1 preBötC neurons facilitates inspiratory burst termination and influences postinspiratory network activity.
Materials and Methods
The Institutional Animal Care and Use Committee at the College of William & Mary approved these protocols. We used female mice that express Cre recombinase fused to a tamoxifen-sensitive mutant form of the human estrogen receptor (CreERT2) in cells that express the Dbx1 gene Dbx1CreERT2. For optical stimulation experiments, female Cre-driver mice were mated with male reporter mice with a Rosa26 locus that was modified by targeted insertion of a loxP-flanked STOP cassette followed by a channelrhodopsin-tdTomato fusion gene (Rosa26hChR2(H134R)-tdTomato, stock no. 12567; The Jackson Laboratory). To record Dbx1 neurons, female Cre-driver mice were mated with male reporter mice that express Cre-dependent tdTomato (Rosa26tdTomato, stock no. 007905; Jackson Laboratory). Tamoxifen (22.5 mg/kg body mass) was administered to pregnant females at embryonic day 9.5. Cre-mediated recombination resulted in the expression of the hChR2-tdTomato or cytosolic tdTomato in neurons with progenitors that express Dbx1.
We anesthetized neonatal mice via hypothermia and dissected their neuraxes in artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.5 CaCl2, 1 MgSO4, 25 NaHCO3, 0.5 NaH2PO4, and 30 dextrose equilibrated with 95% O2/5% CO2, pH 7.4. This aCSF was varied in one set of experiments by substituting MgCl2 for MgSO4. Therefore, changes in CaCl2 were counterbalanced by MgCl2 to maintain the net divalent cation concentration and Cl− gradient.
Transverse medullary slices (500 μm thick) that expose the preBötC rostrally (Ruangkittisakul et al., 2014) were perfused with 26°C aCSF at 5 ml/min. Extracellular K+ was elevated to 9 mm to sustain respiratory rhythm and motor output, which we recorded from hypoglossal (XII) nerve rootlets. The XII discharge was amplified (2000×), band-pass filtered (300–1000 Hz), and then full-wave rectified and smoothed for display.
We obtained whole-cell patch-clamp recordings under visual control using bright-field imaging on a fixed-stage microscope. Dbx1 neurons were identified by epifluorescence (Cy3 filter set). Patch pipettes (4–6 MΩ) were fabricated from capillary glass (1.50 mm outer diameter, 0.86 mm inner diameter). Patch pipette solution contained the following (in mm): 140 potassium gluconate, 5 NaCl, 0.1 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na3-GTP, and 50 μm Alexa Fluor 488 hydrazide dye (Invitrogen). Membrane potential was amplified (100×) and low-pass filtered (1 kHz) using a current-clamp amplifier before being digitally acquired at 10 kHz.
A glass micropipette connected to a stimulus isolation unit (3 μA for 5 ms) was used to stimulate midline-crossing axons. Evoked EPSPs with amplitudes that exceeded baseline potential fluctuations by >2 SD were considered for analysis. The preBötC contralateral to the whole-cell recording site was illuminated using a 100-μm-diameter fiber coupled to a 100 mW 473 nm laser (IkeCool). AMPA (100 μm, Sigma-Aldrich) was applied to Dbx1 preBötC neurons using a patch pipette connected to a pressure ejection system (5 pulses at 5 psi for 5 ms each; Toohey). Midline electrical stimulations and AMPA application experiments were performed in 3 mm extracellular K+ to decrease baseline membrane potential fluctuations and to minimize contributions from spontaneous EPSPs (sEPSPs).
We measured the peak amplitude and area of inspiratory bursts in Dbx1 preBötC neurons by digitally smoothing the membrane potential trajectory to eliminate spikes but preserve the underlying envelope of depolarization. Ionotropic receptor antagonists were applied at these concentrations (in μm): 10 CNQX, 20 dl-APV, 5 picrotoxin, and 5 strychnine (Sigma-Aldrich).
We report all measurements as mean ± SD. SPSS software (IBM) compared group means and probability distributions for statistical hypothesis testing. Paired-sample t tests evaluated mean differences between two groups and the Kolmogorov–Smirnov test compared cumulative probability distributions. Nonparametric Friedman tests evaluated mean differences in repeated-measures experiments when data did not meet the assumptions of normality and homogeneity of variance required for parametric analysis.
Results
Dbx1 Cre-driver mice (Dbx1CreERT2) were coupled with two different flox-stop reporter strains to selectively record and optically manipulate Dbx1 neurons in transverse brainstem slices that provide optimal experimental access to the preBötC. We identified Dbx1 neurons by native fluorescence in the region of the slice ventral to the semicompact division of the nucleus ambiguous and orthogonal to the dorsal boundary of the principal loop of the inferior olive, which corresponds to the rostral face of the preBötC (Fig. 1A; Ruangkittisakul et al., 2014). Dbx1 preBötC neurons, dialyzed with Alexa Fluor 488 through the patch pipette (Fig. 1B), discharged inspiratory bursts in phase with inspiratory-related XII nerve output (Fig. 1C).
Figure 1.

Rhythmically active slices expose Dbx1 preBötC neurons. A, Rostral slice surface from a Dbx1CreERT2;Rosa26tdTomato mouse pup showing hypoglossal motor nucleus (XII), semicompact division of the nucleus ambiguus (scNA), and the principal loop of the inferior olive (IOloop) sites colocalized with the preBötC (left). Dotted box marks the preBötC. B, Whole-cell recordings in Dbx1CreERT2;Rosa26tdTomato (top) and Dbx1CreERT2;Rosa26thChR2-tdTomato (bottom) mouse slices. Shown are tdTomato (left), Dodt contrast microscopy (middle), and Alexa Fluor 488 introduced via patch pipette (right). C, Inspiratory bursts in the Dbx1 preBötC neuron from B (top) with XII motor output.
Dbx1 preBötC interneurons project commissural axons to form synaptic connections with the contralateral preBötC (Bouvier et al., 2010). To determine whether excitatory synapses onto Dbx1 preBötC neurons undergo activity-dependent depression, we repetitively stimulated commissural axons during whole-cell recordings in Dbx1CreERT2;Rosa26tdTomato mouse slices. We observed a progressive amplitude reduction of evoked EPSPs (Fig. 2A, n = 8 Dbx1 neurons, 3 trials/neuron). Furthermore, the number of failures increased during each trial such that the last five pulses experienced a 40–50% failure rate (Fig. 2B). These data indicate that excitatory synapses among Dbx1 preBötC neurons undergo activity-dependent depression even when activated at a rate of 5 Hz, which is lower than typical intraburst spike frequency of 15–50 Hz.
Figure 2.

Activity-dependent synaptic depression in Dbx1 preBötC neurons. A, Evoked EPSPs in response to 5 Hz electrical stimulation of midline-crossing axons (top); group data from eight Dbx1 preBötC neurons (time synced) are also shown (bottom). Red circles show EPSP amplitudes; black bars show mean ± SD. B, Relative frequency of failures to evoke an EPSP as a function of pulse number.
We reasoned that, if short-term synaptic depression influences inspiratory–expiratory phase transition, then its effects would be measurable at the network level immediately after an endogenous inspiratory burst. First, we tested whether inspiratory-like bursts could be triggered by light in Dbx1CreERT2;Rosa26hChR-tdTomato mouse slices. We evoked bursts using 100 ms pulses of blue light delivered to the preBötC contralateral to the whole-cell recording site. Evoked bursts were reversibly blocked by the AMPAR antagonist CNQX, but not by coapplication of the NMDA receptor antagonist AP5 with the GABAA and glycine receptor antagonists picrotoxin and strychnine (Fig. 3A). These data demonstrate that evoked bursts depend on AMPAR-mediated synaptic interactions, akin to endogenous inspiratory bursts (Funk et al., 1993; Wallén-Mackenzie et al., 2006), whereas NMDA receptors and chloride-mediated inhibition are dispensable (Brockhaus and Ballanyi, 1998; Ren and Greer, 2006; Feldman et al., 2013; Janczewski et al., 2013; Sherman et al., 2015).
Figure 3.
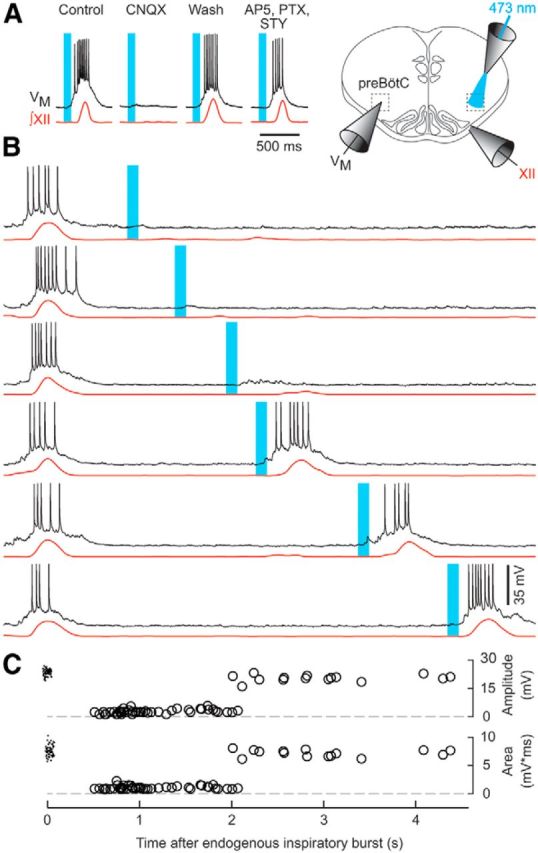
Light-evoked inspiratory-like bursts in Dbx1CreERT2;Rosa26hChR2-tdTomato mouse slices. A, Pharmacology of evoked bursts. Time calibration applies to A. B, Laser pulses delivered at increasing intervals after endogenous inspiratory bursts. Voltage calibration applies to A and B. C, Burst amplitude and area plotted versus the time interval between the endogenous inspiratory burst and stimulus onset. Endogenous control bursts are plotted at the 0 s tick. Time calibration for B and C is the abscissa.
We applied transient light pulses at varying intervals after endogenous inspiratory bursts, which reliably evoked subsequent bursts for intervals >2 s. However, intervals <2 s failed to evoke a burst (Fig. 3B). In this context, we defined the refractory period as the minimum duration after an endogenous inspiratory burst necessary to evoke a subsequent inspiratory-like burst of amplitude ≥75% of the average endogenous inspiratory burst (Fig. 3C). The refractory period duration was significantly shorter than the endogenous burst period (1.94 ± 0.74 s vs 5.80 ± 1.49 s, n = 10 preparations, Student's t(8) = 7.62, p = 5.6E-6). These data suggest that an ∼2 s refractory period is a previously unrecognized component of the respiratory cycle in Dbx1 preBötC neurons. This refractory period is relevant during the postinspiratory phase (∼2 s in duration), but not the remaining 3–4 s of the expiratory phase in vitro.
Presynaptic and postsynaptic factors cause short-term synaptic depression and both could potentially contribute to the refractory period (Fig. 3). First, we examined postsynaptic AMPAR desensitization as a potential contributing factor to the refractory period by comparing the amplitude of sEPSPs that were measurable before and after endogenous inspiratory bursts in Dbx1 preBötC neurons (Fig. 4A, left). We observed no difference in the cumulative probability sEPSP amplitude histogram (Fig. 4A, right, n = 8 Dbx1 neurons, 10 cycles/neuron, Kolmogorov–Smirnov test statistic D = 0.081, p = 0.89), suggesting that AMPARs do not remain desensitized for seconds after endogenous inspiratory bursts and therefore do not contribute to the refractory period. Nevertheless, it is conceivable that the sEPSPs we measured might originate from tonic (noninspiratory) neurons upstream of the preBötC, the excitatory synaptic properties of which may differ from excitatory synapses among Dbx1 neurons in the preBötC. Therefore, to further assess the contribution of AMPAR desensitization to the refractory period, we pressure-ejected AMPA onto Dbx1 neuron dendrites in five-pulse trains and then measured transient postsynaptic depolarizations (Fig. 4B, left). Whether we compared the first with the second pulse (Fig. 4B, black symbols) or the first with the fifth pulse (presumably steady state, Fig. 4B, gray symbols), there was no systematic change in the amplitude of the postsynaptic response to AMPA at any pulse interval (n = 5 Dbx1 neurons, Friedman test, all p > 0.05). These data suggest that AMPAR desensitization probably does not account for the refractory period.
Figure 4.
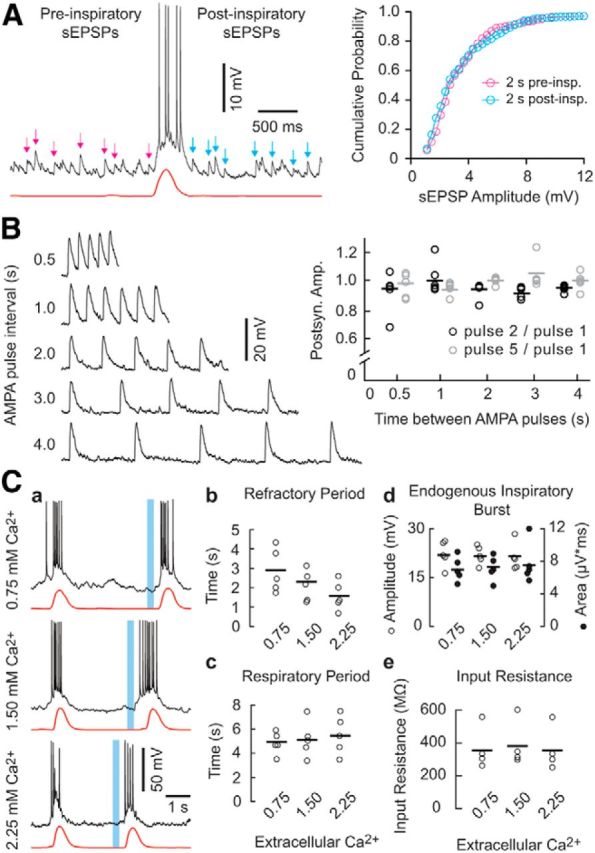
sEPSPs, dendritic AMPA pulses, and extracellular Ca2+ modulation of the refractory period. A, sEPSPs before (magenta) and after (cyan) an endogenous inspiratory burst and the cumulative probability histogram for sEPSP amplitude. Calibrations apply to A. B, Postsynaptic responses to repetitive dendritic AMPA pulses separated by 4, 3, 2, 1, or 0.5 s (left). The amplitude of the second (black) or fifth (gray) postsynaptic response, normalized to the first response, is plotted for each time interval (right). Bars show means. Voltage calibration applies to B; time calibration can be inferred by AMPA pulse interval timing. C, Minimum refractory period after an endogenous inspiratory burst (e.g., Fig. 3B) plotted for different [Ca2+]o (Ca). Voltage and time calibrations apply to Ca. Refractory period (Cb), respiratory cycle period (Cc), endogenous inspiratory burst amplitude and area (Cd), and input resistance (Ce) are plotted for each [Ca2+]o. Symbols show individual experiments; solid lines represent sample means.
We investigated presynaptic factors that could contribute to the refractory period. The most common form of short-term synaptic depression involves progressive depletion of the readily releasable pool of synaptic vesicles during bouts of intense neural activity. Because vesicle recycling machinery is Ca2+ sensitive, elevations in presynaptic Ca2+ accelerate the refilling of depleted terminals (Dittman and Regehr, 1998; Stevens and Wesseling, 1998; Yang and Xu-Friedman, 2008; Lipstein et al., 2013). Therefore, by manipulating extracellular Ca2+, we aimed to influence the amount of presynaptic Ca2+ that accumulates during an inspiratory burst and thus the rate of replenishment of expended synaptic vesicles. In 0.75 mm Ca2+, the refractory period measured 2.88 ± 1.18 s; in 1.50 mm Ca2+, it measured 2.22 ± 0.94 s; and in 2.25 mm Ca2+, it measured 1.66 ± 0.81 s (Fig. 4Ca). The duration of the refractory period is inversely proportional to the level of extracellular Ca2+ (Fig. 4Cb, n = 5, Friedman χ2 = 6.4, p = 0.04). In contrast, these same changes in extracellular Ca2+ caused no significant change in the respiratory cycle period (Fig. 4Cc, n = 5, Friedman χ2 = 0.7, p = 0.7) and did not affect the amplitude (n = 5 Dbx1 neurons, Friedman χ2 = 2.8, p = 0.25) or area (n = 5 Dbx1 neurons, Friedman χ2 = 1.6, p = 0.45) of endogenous inspiratory bursts (Fig. 4Cd). Ca2+ manipulations did not affect the input resistance of Dbx1 preBötC neurons (Fig. 4Ce, n = 4, Friedman χ2 = 0.5, p = 0.78). These data indicate that extracellular Ca2+ manipulations influence the duration of the refractory period, but do not affect excitability or basic intrinsic properties of Dbx1 preBötC neurons, suggesting that the refractory period and its modulation by extracellular Ca2+ cannot be attributed to postsynaptic factors.
Discussion
Breathing movements emanate from neural activity in a central pattern generator circuit for which core interneurons are known. Dbx1-derived preBötC neurons are bilaterally interconnected glutamatergic interneurons that express membrane properties consistent with rhythmogenic function. Dbx1 knock-out mice form no preBötC and suffer fatal asphyxia at birth (Bouvier et al., 2010; Gray et al., 2010). Therefore, elucidating the cellular and synaptic basis for rhythmogenesis in Dbx1 preBötC neurons is crucial for understanding the neural origins of the full breathing motor pattern.
We hypothesized that synaptic depression influences the phase transition from inspiratory to postinspiratory (i.e., early expiratory) activity in Dbx1 preBötC interneurons. To quantify short-term depression, we stimulated midline-crossing axons and observed a progressive diminution of evoked EPSP amplitude and increasing probability of synaptic failure (reaching 50% by the end of the bout). Because the decay time constant of evoked EPSPs was ∼100 ms, stimulation rates could not exceed 5 Hz without causing temporal summation and obscuring the analysis. Nonetheless, these data establish that excitatory synapses among Dbx1 preBötC neurons experience short-term depression when activated repetitively. We can infer that, in the context of rhythmogenesis, when Dbx1 preBötC neurons discharge 5–20 intraburst spikes at 15–50 Hz and thus more intensely activate excitatory transmission among the interconnected Dbx1 population, synaptic depression could decrease the magnitude of synaptic potentials by 50–70% and increase the probability of synaptic failure to 10–50%. Therefore, we surmise that synaptic depression could curtail recurrent excitation and thus contribute to inspiratory burst termination. However, this remains to be demonstrated.
Activity-dependent synaptic depression could contribute to terminating, not only bursts, but also “burstlets” in the preBötC, which are periodic inspiratory events of lower intensity that do not result in motor output, but have been proposed as a basic substrate of rhythmogenesis (Kam et al., 2013). preBötC neurons generate ∼6 spikes per burstlet at an average rate of 15 Hz (range 5–28 Hz, see Fig. 6 in Kam et al., 2013) and our results (Fig. 2) show that, at a minimum, synaptic potentials could decline by 40% and failure probability could rise to 10% within the constraints of these intraburstlet spiking parameters.
The mechanisms responsible for inspiratory burst termination and postinspiratory activity are vigorously debated. The group-pacemaker hypothesis posits that convolved synaptic and intrinsic properties of preBötC neurons mediate inspiratory–expiratory phase transition (Rekling et al., 1996; Rekling and Feldman, 1998). In a mathematical model of the group pacemaker, presynaptic depression can intervene during the inspiratory burst to curb recurrent excitation, which facilitates inspiratory burst termination and promotes the inspiratory–expiratory phase transition (Rubin et al., 2009). Simultaneously, activity-dependent outward currents, namely Na/K ATPase electrogenic pump current (Ipump), Na+-dependent K+ current (INa-K), and ATP-dependent K+ current (IK-ATP), play a complementary role in burst termination and phase transition. These outward currents in preBötC neurons have been identified experimentally (Del Negro et al., 2009; Krey et al., 2010), but the potential role for short-term synaptic depression remained an untested prediction of the group-pacemaker model. Ipump, INa-K, and IK-ATP relax within 15–300 ms after burst termination, which is too short a time to explain the refractory period in vitro that measured 2 s in response to hChR2activation but measured ∼1 s when a bolus of AMPA was administered unilaterally to the preBötC after inspiratory burst termination (Del Negro et al., 2009). This shorter refractory period is most likely attributable to the stronger stimulus. AMPA application depolarized preBötC neurons by 40 mV and then slowly decayed for 1 min, whereas hChR2 depolarized Dbx1 preBötC neurons <10 mV for exactly 100 ms. In both cases, the refractory period outlasts postsynaptic contributions of Ipump, INa-K, and IK-ATP. Therefore, we propose that activity-dependent outward currents and synaptic depression act in concert to influence inspiratory–expiratory phase transition, terminating the inspiratory burst and then causing a transient phase of postinspiratory quiescence in the network.
Could this mechanism identified in models and in vitro apply in vivo? We observed an ∼2 s refractory period after inspiratory bursts, which comprises 25–33% of the respiratory cycle period in vitro. By optogenetically stimulating the preBötC in adult rats in vivo, Alsahafi et al. (2015) recently documented a refractory period of 200–400 ms after inspiration, which comprises ∼25–60% of the breathing cycle period. The refractory period measured in vitro constitutes the same proportion of the cycle period as the refractory period measured in vivo, suggesting that the refractory period is a real phenomenon and not an artifact of the in vitro preparation. Therefore, the biophysical mechanisms that we identify in vitro could govern the inspiratory–expiratory phase transition in vivo as well.
The most common form of short-term synaptic depression involves presynaptic vesicle depletion during high-frequency activity. Manipulations of Ca2+, which affect release and recovery processes (Neher and Sakaba, 2008), modulated the refractory period during the postinspiratory phase but did not affect respiratory cycle period, inspiratory burst magnitude, or input resistance of Dbx1 preBötC neurons (Fig. 4). The readily releasable vesicle pool recovery time varies from hundreds of milliseconds to seconds depending on the experimental model (Stevens and Tsujimoto, 1995; Hosoi et al., 2007; Cohen and Segal, 2011). The refractory period in vitro and the refractory period Alsahafi et al. (2015) measured in vivo are within this range, suggesting that vesicle depletion is a viable explanation for the lack of synaptic efficacy after inspiratory bursts in vitro or inspiration in vivo.
AMPAR desensitization is unlikely to contribute on the time scale associated with the refractory period based on measurements of sEPSPs before and immediately after inspiratory bursts, as well as the lack of modulation of postsynaptic responses to repetitive AMPA pulses. However, it remains possible that we measured sEPSPs from a source extrinsic to the preBötC; for example, from an upstream tonic source of excitation to the preBötC with postsynaptic AMPARs that do not exhibit the same biophysical properties as AMPARs involved in recurrent excitation. Further, receptors may desensitize during bursts (and thus assist in burst termination) but then recover faster than could be measured using pressure-ejection techniques in which the decay kinetics of postsynaptic response precluded pulse rates in excess of 2 Hz. It is also possible that some portion of AMPARs targeted by local AMPA application are not inspiratory modulated, so we cannot rule out postsynaptic effects entirely.
Conceptual models of respiratory rhythm often feature obligatory roles for postsynaptic inhibition in inspiratory–expiratory phase transition. The present results indicate that excitatory synaptic dynamics of core rhythmogenic Dbx1 interneurons could influence inspiratory burst termination and the quiescent postinspiratory phase of the respiratory cycle, which should be added to existing frameworks for analysis and models of breathing's neural bases.
Footnotes
This work was supported by the National Institutes of Health (Grant R01 HL104127 to C.A.D.N.).
The authors declare no competing financial interests.
References
- Alsahafi Z, Dickson CT, Pagliardini S. Optogenetic excitation of preBötzinger complex neurons potently drives inspiratory activity in vivo. J Physiol. 2015 doi: 10.1113/JP270471. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi AL, Denavit-Saubié M, Champagnat J. Central control of breathing in mammals: neuronal circuitry, membrane properties, and neurotransmitters. Physiol Rev. 1995;75:1–45. doi: 10.1152/physrev.1995.75.1.1. [DOI] [PubMed] [Google Scholar]
- Bouvier J, Thoby-Brisson M, Renier N, Dubreuil V, Ericson J, Champagnat J, Pierani A, Chédotal A, Fortin G. Hindbrain interneurons and axon guidance signaling critical for breathing. Nat Neurosci. 2010;13:1066–1074. doi: 10.1038/nn.2622. [DOI] [PubMed] [Google Scholar]
- Brockhaus J, Ballanyi K. Synaptic inhibition in the isolated respiratory network of neonatal rats. Eur J Neurosci. 1998;10:3823–3839. doi: 10.1046/j.1460-9568.1998.00396.x. [DOI] [PubMed] [Google Scholar]
- Carroll MS, Ramirez JM. Cycle-by-cycle assembly of respiratory network activity is dynamic and stochastic. J Neurophysiol. 2013;109:296–305. doi: 10.1152/jn.00830.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MS, Viemari JC, Ramirez JM. Patterns of inspiratory phase-dependent activity in the in vitro respiratory network. J Neurophysiol. 2013;109:285–295. doi: 10.1152/jn.00619.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Segal M. Network bursts in hippocampal microcultures are terminated by exhaustion of vesicle pools. J Neurophysiol. 2011;106:2314–2321. doi: 10.1152/jn.00969.2010. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Kam K, Hayes JA, Feldman JL. Asymmetric control of inspiratory and expiratory phases by excitability in the respiratory network of neonatal mice in vitro. J Physiol. 2009;587:1217–1231. doi: 10.1113/jphysiol.2008.164079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA, Gray PA. Understanding the rhythm of breathing: so near, yet so far. Annu Rev Physiol. 2013;75:423–452. doi: 10.1146/annurev-physiol-040510-130049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk GD, Smith JC, Feldman JL. Generation and transmission of respiratory oscillations in medullary slices: role of excitatory amino acids. J Neurophysiol. 1993;70:1497–1515. doi: 10.1152/jn.1993.70.4.1497. [DOI] [PubMed] [Google Scholar]
- Gray PA, Hayes JA, Ling GY, Llona I, Tupal S, Picardo MC, Ross SE, Hirata T, Corbin JG, Eugenín J, Del Negro CA. Developmental origin of preBötzinger complex respiratory neurons. J Neurosci. 2010;30:14883–14895. doi: 10.1523/JNEUROSCI.4031-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Sakaba T, Neher E. Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J Neurosci. 2007;27:14286–14298. doi: 10.1523/JNEUROSCI.4122-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczewski WA, Tashima A, Hsu P, Cui Y, Feldman JL. Role of inhibition in respiratory pattern generation. J Neurosci. 2013;33:5454–5465. doi: 10.1523/JNEUROSCI.1595-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, Worrell JW, Ventalon C, Emiliani V, Feldman JL. Emergence of population bursts from simultaneous activation of small subsets of preBötzinger complex inspiratory neurons. J Neurosci. 2013;33:3332–3338. doi: 10.1523/JNEUROSCI.4574-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey RA, Goodreau AM, Arnold TB, Del Negro CA. Outward currents contributing to inspiratory burst termination in preBötzinger complex neurons of neonatal mice studied in vitro. Front Neural Circuits. 2010;4:124. doi: 10.3389/fncir.2010.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipstein N, Sakaba T, Cooper BH, Lin K-H, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E, Brose N. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca(2+)-calmodulin-Munc13-1 signaling. Neuron. 2013;79:82–96. doi: 10.1016/j.neuron.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Moore JD, Deschênes M, Furuta T, Huber D, Smear MC, Demers M, Kleinfeld D. Hierarchy of orofacial rhythms revealed through whisking and breathing. Nature. 2013;497:205–210. doi: 10.1038/nature12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Pace RW, Mackay DD, Feldman JL, Del Negro CA. Inspiratory bursts in the preBötzinger complex depend on a calcium-activated non-specific cation current linked to glutamate receptors in neonatal mice. J Physiol. 2007;582:113–125. doi: 10.1113/jphysiol.2007.133660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekling JC, Feldman JL. PreBötzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annu Rev Physiol. 1998;60:385–405. doi: 10.1146/annurev.physiol.60.1.385. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Champagnat J, Denavit-Saubié M. Electroresponsive properties and membrane potential trajectories of three types of inspiratory neurons in the newborn mouse brain stem in vitro. J Neurophysiol. 1996;75:795–810. doi: 10.1152/jn.1996.75.2.795. [DOI] [PubMed] [Google Scholar]
- Ren J, Greer JJ. Modulation of respiratory rhythmogenesis by chloride-mediated conductances during the perinatal period. J Neurosci. 2006;26:3721–3730. doi: 10.1523/JNEUROSCI.0026-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DW. Generation and maintenance of the respiratory rhythm. J Exp Biol. 1982;100:93–107. doi: 10.1242/jeb.100.1.93. [DOI] [PubMed] [Google Scholar]
- Ruangkittisakul A, Kottick A, Picardo MCD, Ballanyi K, Del Negro CA. Identification of the pre-Bötzinger complex inspiratory center in calibrated “sandwich” slices from newborn mice with fluorescent Dbx1 interneurons. Physiol Rep. 2014;2:e12111. doi: 10.14814/phy2.12111. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JE, Hayes JA, Mendenhall JL, Del Negro CA. Calcium-activated nonspecific cation current and synaptic depression promote network-dependent burst oscillations. Proc Natl Acad Sci U S A. 2009;106:2939–2944. doi: 10.1073/pnas.0808776106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman D, Worrell JW, Cui Y, Feldman JL. Optogenetic perturbation of preBötzinger complex inhibitory neurons modulates respiratory pattern. Nat Neurosci. 2015;18:408–414. doi: 10.1038/nn.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci U S A. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/S0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Wallén-Mackenzie A, Gezelius H, Thoby-Brisson M, Nygård A, Enjin A, Fujiyama F, Fortin G, Kullander K. Vesicular glutamate transporter 2 is required for central respiratory rhythm generation but not for locomotor central pattern generation. J Neurosci. 2006;26:12294–12307. doi: 10.1523/JNEUROSCI.3855-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Relative roles of different mechanisms of depression at the mouse endbulb of Held. J Neurophysiol. 2008;99:2510–2521. doi: 10.1152/jn.01293.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]