

Abstract
Following the discovery of the antidepressant properties of ketamine, there has been a recent resurgence in the interest in this NMDA receptor antagonist. Although detailed animal models of the molecular mechanisms underlying ketamine's effects have emerged, there are few MEG/EEG studies examining the acute subanesthetic effects of ketamine infusion in man. We recorded 275 channel MEG in two experiments (n = 25 human males) examining the effects of subanesthetic ketamine infusion. MEG power spectra revealed a rich set of significant oscillatory changes compared with placebo sessions, including decreases in occipital, parietal, and anterior cingulate alpha power, increases in medial frontal theta power, and increases in parietal and cingulate cortex high gamma power. Each of these spectral effects demonstrated their own set of temporal dynamics. Dynamic causal modeling of frontoparietal connectivity changes with ketamine indicated a decrease in NMDA and AMPA-mediated frontal-to-parietal connectivity. AMPA-mediated connectivity changes were sustained for up to 50 min after ketamine infusion had ceased, by which time perceptual distortions were absent. The results also indicated a decrease in gain of parietal pyramidal cells, which was correlated with participants' self-reports of blissful state. Based on these results, we suggest that the antidepressant effects of ketamine may depend on its ability to change the balance of frontoparietal connectivity patterns.
SIGNIFICANCE STATEMENT In this paper, we found that subanesthetic doses of ketamine, similar to those used in antidepressant studies, increase anterior theta and gamma power but decrease posterior theta, delta, and alpha power, as revealed by magnetoencephalographic recordings. Dynamic causal modeling of frontoparietal connectivity changes with ketamine indicated a decrease in NMDA and AMPA-mediated frontal-to-parietal connectivity. AMPA-mediated connectivity changes were sustained for up to 50 min after ketamine infusion had ceased, by which time perceptual distortions were absent. The results also indicated a decrease in gain of parietal pyramidal cells, which was correlated with participants' self-reports of blissful state. The alterations in frontoparietal connectivity patterns we observe here may be important in generating the antidepressant response to ketamine.
Keywords: alpha rhythms, depression, dynamic causal modelling, gamma rhythms, ketamine, magnetoencephalography
Introduction
The now widely replicated observation that a single subanaesthetic dose of ketamine can have rapid antidepressant effects (Berman et al., 2000; Zarate et al., 2006b; Murrough et al., 2013) has caused a re-evaluation of the neurobiology of major depressive disorder. Although it is unclear whether ketamine could be safely adopted for general clinical practice (Krystal et al., 2013; Rush, 2013), ketamine's rapid efficacy highlights the importance of glutamatergic systems in major depression and at the same time serves as a useful interventional compound for studying the molecular basis of depression.
Used as both an anesthetic and drug of abuse, the phencyclidine derivative ketamine has a “rich” pharmacology: in addition to its principal mechanism of action as a noncompetitive antagonist of NMDA receptors, it exhibits action at muscarinic, σ, μ-opioid receptors, and α and β adrenergic receptors; and blocks HCN1 channels as well as the serotonin and norepinephrine transporters (Bergman, 1999; Chen et al., 2009; Stahl, 2013). Despite this multitude of effects, animal models suggest that the antidepressant actions of ketamine are mediated by its antagonism of NMDA receptors. According to these models, blockade of NMDA receptors by ketamine results in increased AMPA-mediated glutamatergic signaling, which in turn triggers the activation of intracellular synaptogenic pathways (Li et al., 2010; Autry et al., 2011). This activation includes an increase in the mTOR signaling pathway, which is necessary for the antidepressant response (Li et al., 2010). It is possible that ketamine modulates neurotransmission through additional pathways given the failure of the antagonist memantine to generate anti-depressant effects (Zarate et al., 2006a). However, NMDA-mediated effects are important as evidenced by positive antidepressive effects with gaseous nitrous oxide (Nagele et al., 2015) and AZD-6765 (Sanacora et al., 2014), both NMDA antagonists. It has been demonstrated with MEG that patients who respond to ketamine exhibit enhanced sensory evoked responses providing evidence in humans that synaptic potentiation may mediate the antidepressant response to ketamine (Cornwell et al., 2012).
A number of brain imaging studies demonstrate that major depressive disorder is characterized by hyperactivity in the brain's default mode network (Greicius et al., 2007; Sheline et al., 2009), whose major hubs include the medial prefrontal and posterior cingulate/precuneus cortices (Andrews-Hanna et al., 2010). It has been suggested that antidepressants may work in part by reducing activity in this network (Posner et al., 2013; Wang et al., 2015), and indeed one study has shown that subanesthetic ketamine infusion reduces default mode network activity in healthy participants 1 d after administration. Several MEG studies have demonstrated that patients with lowest task-based connectivity and activity in the anterior cingulate cortex show the largest antidepressant response to ketamine (Salvadore et al., 2009, 2010). These studies again highlight importance of connectivity alterations in depression and moreover the usefulness of MEG as a tool to probe electrophysiological connectivity in the context of ketamine and depression.
Based on these facts, in the present studies we hypothesized that during subanesthetic ketamine administration there would be a rapid reduction in frontoparietal connectivity. To quantify connectivity we combined MEG recordings of resting-state oscillatory activity with dynamic causal modeling (DCM) estimates of effective connectivity, and used a crossover design with subanesthetic ketamine and saline infusions. In DCM, biologically plausible biophysical models are fit to empirical data to allow estimates of synaptic responses within specified neuronal ensembles (Moran et al., 2011b), the effective connectivity between ensembles in interconnected brain regions (Friston et al., 2012), and how these connectivity parameters are altered under pharmacological challenge (Moran et al., 2015). Thus, DCM provides a potentially powerful translational bridge between animal models and human neurophysiology, where these parameters are not directly measurable. Here, we used source reconstruction to estimate the activity from two principal hubs of the default mode, the medial prefrontal cortex and the precuneus, and estimated the changes in effective connectivity within and between these regions during and after ketamine infusion.
Materials and Methods
Experiment 1
Participants and experimental procedure.
Experiment 1 included 19 healthy male participants (mean age = 25.8 years, range = 19–39). All participants gave informed consent to participate in the study, which was approved by a UK National Health Service research ethics committee. Volunteer exclusion criteria included any current or previous psychiatric disorder, determined with the Mini-International Neuropsychiatric Interview (MINI), current recreational or prescription drug use, contraindications for MEG/MRI, and needle phobia. Inclusion criteria for participation were that volunteers be of age 18–45, nonsmokers, American Society of Anesthesiologists Physical Status 1, and have a body mass index of 18–30 kg/m2.
Participants underwent two scans on separate days, one using a placebo saline infusion and one using ketamine. In each scan session, 5 min of resting MEG was recorded, and then infusion commenced. Each participant received an initial bolus of 0.25 mg/kg delivered over ∼1 min, followed by maintenance infusion at a rate of 0.375 mg/kg/h. Ten minutes of resting MEG were recorded following the start of the infusion. At this point, participants were asked to provide a rating of their subjective “high” on a scale between 0 and 40, using a two-digit button box. Measures of saccadic eye movement velocity were then taken. After this, participants commenced a series of tasks, data for which are reported previously (Shaw et al., 2015).
MEG recordings.
For the MEG recordings, participants lay in supine position. Participants' pulse rate and blood oxygenation level were continually monitored throughout the experiment via a probe over their left hand index finger with the intravenous cannula inserted on the back of the left wrist. Participants' wore a nasal cannula attached to medical oxygen, which was to be used in case of an emergency.
Whole-head MEG recordings were made using a CTF 275-channel radial gradiometer system sampled at 1200 Hz (0–300 Hz bandpass). An additional 29 reference channels were recorded for noise cancellation purposes and the primary sensors were analyzed as synthetic third-order gradiometers (Vrba and Robinson, 2001). Three of the 275 channels were turned off due to excessive sensor noise. In addition to the MEG channels, we recorded participants' ECG: horizontal and vertical electrooculograms (EOG), as well as electromyograms (EMG) from bilateral frontalis and temporalis muscles. Monocular pupilometry data were obtained using an iViewX-MEG system (SensoMotoric Instruments) sampled at 250 Hz.
Data preprocessing.
All MEG recordings were initially high-pass filtered at 1 Hz, and segmented into epochs of 2 s in length (450 epochs with 150 preinfusion). Each epoch was then visually inspected, and those with gross artifacts (e.g., head movements, jaw clenches) were removed from the analysis. An automated algorithm was used to remove further epochs contaminated with muscle artifacts. In this algorithm, a set of 30 gradiometer sensors were predefined at the edge of the MEG dewar, as these are most likely to be contaminated by muscle artifacts (Muthukumaraswamy, 2013). Using Hanning-windowed Fourier transformations, we calculated the mean spectral power for these sensors in the 105–145 Hz frequency band (Muthukumaraswamy, 2013) for each epoch. If the resulting power averaged across these sensors exceeded 10 fT2 then that epoch was eliminated from subsequent analysis On the remaining epochs we then performed independent component analysis (ICA) as implemented in Fieldtrip/EEGLAB (Delorme and Makeig, 2004) to identify and remove ocular, muscular and cardiac artifacts from the data. Any components that showed a correlation (r > 0.10) in the time domain with the EOG/EMG electrodes were automatically removed. Likewise, any components that showed correlations (r > 0.10) with similarly filtered EOG/EMG channels after being bandpass filtered in the range 105–145 Hz were removed. Visual inspection was also used to remove artifact components. All subsequent analyses were performed on the ICA cleaned datasets.
Frequency analysis: sensor space.
Using the FieldTrip toolbox (Oostenveld et al., 2011) we converted our MEG data to planar gradient configuration, and then conducted a frequency analysis of the individual vector directions. Frequency analysis was conducted using Hanning windowed fast Fourier transforms between 1 and 100 Hz at 0.5 Hz frequency intervals and then the planar directions combined to give local maxima under the sensors. Analysis of sensor-level MEG data in a planar gradient (spatial-derivative) configuration has the advantage of easy interpretability, because field maps can be interpreted as having a source directly underneath field maxima (Bastiaansen and Knösche, 2000). For statistical analysis, we divided individual spectra into the following frequency bands: delta (1–4 Hz), theta (4–8 Hz), alpha (8–13 Hz), beta (13–30 Hz), low gamma (30–49 Hz), and high gamma (51–99 Hz; Muthukumaraswamy et al., 2013). The preintervention baseline spectra were subtracted from each postintervention spectra and the differences between intervention and placebo tested using permutation testing of t statistics at each postintervention time point (Nichols and Holmes, 2002; Maris and Oostenveld, 2007). The Type 1 error rate was controlled using cluster randomization analysis with an initial cluster-forming threshold of p = 0.05 repeated >5000 permutations. The same spectral analysis technique was applied to the EMG channels in the 55–95 Hz band to check for possible muscle artifact contamination and to the EOG channels in the 1–20 Hz band to check for possible ocular artifacts.
Source localization.
To localize drug-induced changes in oscillatory power, we used the beamformer algorithm synthetic aperture magnetometry (SAM; Robinson and Vrba, 1999). Global covariance matrices were calculated for the following six bandpass-filtered versions of the datasets: delta (1–4 Hz), theta (4–8 Hz), alpha (8–13 Hz), beta (13–30 Hz), low gamma (30–49 Hz), and high gamma (51–99 Hz). Based on these covariance matrices, using the beamformer algorithm, a set of beamformer weights was computed for all voxels in the brain at 4 mm isotropic voxel resolution. A multiple local-spheres (Huang et al., 1999) volume conductor model was derived by fitting spheres to the brain surface extracted by the FSL Brain Extraction Tool (Smith, 2002). For SAM imaging, virtual sensors were constructed at each beamformer voxel and Student's t images of source power changes computed for postinfusion versus preinfusion epochs. Volumetric group statistical analyses were conducted as previously described (Muthukumaraswamy et al., 2013). Five thousand permutations were calculated for each statistical test conducted with a 5 mm Gaussian smoothing kernel applied to the variance maps. We computed a paired t test for the ketamine versus placebo images to reveal the drug interaction effect: t images were thresholded at p = 0.05 (cluster corrected).
Dynamic causal modeling of effective connectivity.
Based on the results, we selected two regions-of-interest within the typical default-mode regions, for connectivity analysis. These were extracted from virtual sensors derived from local maxima in individual source localization images. Maxima were selected at image peaks near the medial prefrontal cortex showing increases in theta activity, and near parietoccipital cortex showing alpha power reductions. Cross-spectral densities were estimated for 1–45 Hz for both preinfusion and postinfusion avoid any contamination of subsequent modeling with (50 Hz) line noise artifacts. These spectral densities formed the data features of our connectivity analysis. DCM uses a biophysical model of neuronal responses based on neural mass models (David et al., 2006) to predict electrophysiological data. The neural mass model we used (Fig. 1a) was comprised of four populations of neurons (superficial pyramidal cells, deep pyramidal cells, spiny stellate cells, and inhibitory interneurons). Intrinsic (within area) excitatory connections are mediated by two excitatory receptor types, AMPA and NMDA and intrinsic inhibitory connections use GABAA receptors. Each receptor type is furnished with its own time constants and dynamics (Fig. 1b,c). In the case of NMDA-mediated receptor activity, this is additionally controlled by a nonlinear sigmoid to reflect the voltage-dependent magnesium ion block. Extrinsic (between area) connections are mediated by two connection types: “feedforward” connections, which project from superficial pyramidal cells to stellate cells, and “feedback” connections, which project from deep pyramidal neurons to superficial layers (interneurons and superficial pyramidal cells). We initially tested three models of extrinsic connectivity (see Results) using data from all participants and compared the log model evidences to determine the most likely model. To constrain the parameter set, we specified a set of four connectivity parameters that could potentially be modulated by ketamine: AMPA-mediated frontal-to-parietal connectivity, NMDA-mediated frontal-to-parietal connectivity, AMPA-mediated parietal-to-frontal connectivity, and NMDA-mediated parietal-to-frontal connectivity. We further specified a gain parameter for each neuronal subpopulation to account for within ensemble changes in excitability. These gain parameters are “lumped” parameters that encapsulate a variety of potential gain-control mechanisms, including both NMDA and non-NMDA mediated effects of ketamine. All DCM analyses used conductance-based neural mass models as implemented in the “cmm_nmda” model available in the spm12 (http://www.fil.ion.ucl.ac.uk/spm/) software. The generative models were then inverted (fitted to each individual's prefrontal and parietal responses) to obtain posterior densities over connectivity and gain parameters, initializing the parameter estimates with those obtained from the grand-average analysis. Specifically, a variational Bayesian scheme (variational Laplace) was used to approximate the posterior density over parameters by maximizing the negative free energy, a bound on the log-model evidence (Friston et al., 2007; Moran et al., 2011a).
Figure 1.

a, Construction of the neural mass model from the winning model tested (Model 3). Two connected neural masses comprised the winning model, each with four subpopulations of neurons (superficial pyramidal cells, deep pyramidal cells, spiny stellate cells, and inhibitory interneurons). Intrinsic to each mass, excitatory connections are illustrated with black arrows, and intrinsic inhibitory connections are represented with red arrows. Right, Intrinsic excitatory connections are mediated by two excitatory receptor types (AMPA and NMDA), whereas intrinsic inhibitory connections use GABAA receptors. Each receptor type is furnished with its own time constants and dynamics, and in the case of NMDA receptors, this includes the nonlinear behavior caused by a voltage-gated magnesium ion block. Each population of cells also contains an intrinsic gain parameter. In Model 3, between-region connectivity was modeled as lateral connections (nonhierarchical; Felleman and Van Essen, 1991; David et al., 2006) with full reciprocal connections between regions (Kobayashi and Amaral, 2003, 2007; Andrews-Hanna et al., 2010). Connections arise from both superficial and deep pyramidal cells and transmit to layer IV and layers I–III, respectively. Empirical data used for each population was derived from virtual sensors in the prefrontal cortex and precuneal areas of each participant. To explain differences between preketamine and postketamine infusion cross-spectral densities, we allowed six parameters to exhibit modulation by ketamine; NMDA mediated frontal and posterior connectivity, AMPA mediated frontal and posterior connectivity and a gain parameter (representing the excitability) for each mass (E, F). b, Equations used in the generation of model dynamics with key parameter priors and implementation of the voltage gate on the NMDA receptor (Durstewitz et al., 2000) given in c. This model is the standard cmm_nmda model implemented in the spm_fx_cmm_nmda.m file, freely available in the spm12 software (http://www.fil.ion.ucl.ac.uk/spm/).
Independent component network analysis of functional connectivity.
For analysis of functional connectivity, agnostic to cell type or synaptic mechanisms, we also used the methodology described by Brookes et al. (2011) and in our previous work (Muthukumaraswamy et al., 2013). For each participant and frequency band, beamformer weights were computed on an 8 mm grid and beamformer time courses were then generated at every voxel and normalized by an estimate of the projected noise amplitude at that voxel. The Hilbert transform was applied to each voxel time course and the absolute value computed to generate an amplitude envelope of the oscillatory signals in each frequency band. The data at each voxel was down-sampled to an effective sampling rate of 1 Hz, transformed to standard (MNI) space using FLIRT in FSL and data from all subjects were concatenated in the time dimension across subjects. Temporal ICA was applied to the concatenated datasets (separately for all six frequency bands) using the fast-ICA algorithm. Prewhitening was applied to reduce the dimensionality of the source space Hilbert envelope signals to 20 principal components before ICA (Hyvärinen and Oja, 2000; Brookes et al., 2011; Hall et al., 2013). Fifteen independent components were derived for each frequency band. From these components we identified those that matched previous reports (Brookes et al., 2011). For the identified components, we computed the SD of the component time course for each subject and condition. Differences in SD of the independent component time course between placebo and ketamine were assessed using t tests using a Bonferroni correction value of 15.
Experiment 2
Participants and experimental procedure.
Experiment 2 included six healthy male participants (mean age = 27.2 years, range = 20–36) subject to the same exclusion/inclusion criteria as Experiment 1. In Experiment 2, 10 min of resting MEG was recorded, then infusion commenced with a bolus of 0.25 mg /kg. This was followed by 20 min of maintenance infusion (0.375 mg/kg/h). MEG was recorded continuously for a further 50 min, making the sessions 80 min long in total. Participants were asked to lie still, with their eyes open and provide a rating of their subjective “high” on a scale between 0 and 40 at 2 min intervals using a two-digit button box.
MEG recordings and data preprocessing.
Identical to Experiment 1, except here there were 2400 epochs (300 preinfusion). Epochs contaminated with activity from the button press were excluded (10 seconds of every 2 min).
Source localization, virtual sensor generation, and pharmacodynamics.
For Experiment 2, the same preprocessing and source analysis steps were used to identify regions-of-interest in the early time window of ketamine infusion. In addition to the two regions identified in Experiment 1, a third parietal gamma source was identified. The location of this source was somewhat variable in individuals and frequently spatially overlapped with the alpha power change locations. Based on the identified regions of interest, we mapped the temporal evolution of power by generating the band-limited power in 2 min time bins (between-subjective ratings) for the entire 80 min. To map the time course of DCM parameter changes in in the experiment we took the same two functional regions (posterior alpha/ prefrontal theta) and applied the winning connectivity model from Experiment 1 (Model 3). We divided the 70 min postrecording into seven, 10 min time windows and extracted the six parameters as in Experiment 1 for each time window. To relate the time course of DCM parameters to the spectral evolution of power a general linear model approach was used to control for within participant effects.
Results
Experiment 1
As a quality check of our in-scanner infusions, consistent with known physiological effects, ketamine caused increases in heart rate, blood pressure, and pupil diameter, and decreases in saccadic eye-movement velocity, indicative of sedation (Fig. 2). Similarly, participants' post hoc reports revealed large perturbations from normal consciousness as assessed with the 5D-ASC (Dittrich, 1998; Studerus et al., 2010); participants reported subjective states of disembodiment, hallucinations, and blissful feelings.
Figure 2.

Basic physiological measures before and after infusion of either ketamine or placebo. Blue is ketamine; red is placebo. a–c, As expected participants demonstrated significant increases in systolic (F(1,18) = 23.7, p < 0.001) and diastolic (F(1,18) = 12.1, p = 0.003) blood pressure, as well as heart rate ((F(1,18) = 16.0, p < 0.001). d–f, Participants also showed standard physiological responses of sedation, including increases in pupil diameter (F(1,18) = 27.1, p < 0.001) and saccadic eye movement (SEM) velocity (F(1,18) = 6.83, p < 0.02), but not saccadic eye movement onset latency (F(1,18) = 0.39, p = 0.34). After the 10 min MEG recording following the onset of infusion, the mean subjective intensity rating of participants was 13.2/20 (SE = 1.08) compared with 0 after placebo. Error bars are the standard error of the mean.
Estimates of ketamine-induced spectral changes were performed in both sensor space (Fig. 3) and source space (Fig. 4) using the beamformer source localization technique SAM. Both these figures display the contrast of ketamine after-versus-before infusion compared with placebo after-versus-before infusion with the peak spatial locations from the source localization presented in Table 1. Comparison of Figures 3 and 4 reveal highly consistent spatial patterns of power changes with decreases in source power seen in the delta (1–4 Hz), posterior theta (4–8 Hz), alpha (8–13 Hz), and beta (13–30 Hz) frequency bands. Conversely, in anterior theta (4–8 Hz), low (30–49 Hz), and high gamma (51–99 Hz) bands, increases in source power were seen.
Figure 3.

Statistical analysis of planar gradiometer configured MEG data for ketamine and placebo. Spectra for preinfusion data recordings were subtracted from the postinfusion data and then contrasts performed. Red indicates relatively more power following the drug and blue indicates relatively less power. Units are t statistics. Significant sensor clusters are marked such that dark circles correspond to p < 0.01 and crosses to p < 0.05.
Figure 4.

Statistical parametric maps showing the locations of significant (p < 0.05, corrected) changes in source oscillatory power in the six frequency bands that span from 1 to 99 Hz. Contrasts of spectral power represent the difference of ketamine after-and-before infusion versus placebo after-and-before infusion. Red colors represent increases in source power and yellow colors represent decreases.
Table 1.
Local maxima of source power changes (Talairach coordinates) corresponding to the images in Figure 4
| X | Y | Z | t | |
|---|---|---|---|---|
| Delta | ||||
| Cuneus (R) | 11 | −87 | 31 | −9.98 |
| Middle occipital gyrus (R) | −25 | −87 | 17 | −9.92 |
| Lingual gyrus (R) | 19 | −67 | −5 | −8.47 |
| Angular gyrus (R) | 53 | −61 | 33 | −8.21 |
| Superior parietal lobule (R) | 29 | 67 | 53 | −7.92 |
| Inferior parietal lobule (R) | 49 | 51 | 53 | −7.44 |
| Superior temporal gyrus (L) | −47 | −11 | 7 | −7.27 |
| Middle occipital gyrus (L) | −45 | −67 | −17 | −7.24 |
| Superior parietal lobule (L) | −13 | −59 | 63 | −6.69 |
| Insula (R) | 43 | −17 | 13 | −6.46 |
| Inferior frontal gyrus (L) | −35 | 31 | 3 | −5.68 |
| Middle temporal gyrus (R) | 43 | −1 | −33 | −5.08 |
| Inferior frontal gyrus (R) | 49 | 37 | −17 | −4.32 |
| Theta | ||||
| Middle occipital gyrus (R) | 21 | −97 | 3 | −4.91 |
| Middle occipital gyrus (L) | −19 | −95 | 1 | −4.30 |
| Superior frontal gyrus (L) | −9 | 61 | −7 | 3.71 |
| Medial frontal gyrus (L) | −3 | 45 | 29 | 3.62 |
| Superior frontal gyrus (R) | 23 | 65 | −5 | 3.58 |
| Alpha | ||||
| Cuneus (L) | −17 | −81 | 17 | −10.0 |
| Middle occipital gyrus (R) | 49 | −71 | 3 | −9.60 |
| Middle temporal gyrus (R) | 57 | −39 | −3 | −8.93 |
| Middle occipital gyrus (L) | −47 | −71 | −13 | −8.67 |
| Middle temporal gyrus (L) | −47 | −53 | 5 | −8.65 |
| Superior temporal gyrus (L) | −61 | −59 | 15 | −8.44 |
| Middle temporal gyrus (R) | 33 | −67 | 27 | −8.43 |
| Precuneus (L) | −11 | −81 | 53 | −8.23 |
| Inferior parietal lobule (L) | −43 | −71 | 45 | −8.21 |
| Superior parietal lobule (R) | 41 | −57 | 57 | −7.22 |
| Postcentral gyrus (L) | −53 | −11 | 23 | −6.44 |
| Anterior cingulate gyrus (L) | −1 | 21 | −3 | −6.13 |
| Precentral gyrus (R) | 49 | −1 | 53 | −4.66 |
| Beta | ||||
| Superior temporal gyrus (L) | −59 | −61 | 15 | −11.9 |
| Inferior temporal gyrus (L) | −47 | −65 | −5 | −11.5 |
| Middle occipital gyrus (L) | −25 | −75 | 5 | −11.3 |
| Fusiform gyrus (R) | 23 | −59 | −13 | −10.5 |
| Middle occipital gyrus (R) | 27 | −75 | 11 | −10.2 |
| Low gamma | ||||
| Inferior frontal gyrus (R) | 31 | 31 | 7 | 9.52 |
| Superior temporal gyrus (R) | 61 | −57 | 19 | 6.10 |
| Middle temporal gyrus (L) | −65 | −55 | 5 | 5.64 |
| Cuneus (L) | −3 | −91 | 21 | 4.10 |
| High gamma | ||||
| Superior parietal lobule (L) | −7 | −73 | 59 | 15.6 |
| Precuneus (R) | 13 | −71 | 63 | 15.5 |
| Anterior cingulate gyrus (R) | 17 | 37 | 11 | 13.2 |
| Cingulate gyrus (R) | 17 | −11 | 43 | 13.1 |
| Anterior cingulate gyrus (L) | −23 | 25 | 15 | 13.1 |
| Cuneus (R) | 13 | −99 | −7 | 9.27 |
| Inferior occipital gyrus (L) | −39 | −85 | −17 | 8.03 |
We performed spectral analysis of the peripheral EOG and EMG channels that we recorded in Experiment 1. These analyses (Fig. 5) revealed that the anterior theta frequency power increases were not due to differential eye movement patterns. Similarly, we confirmed that the increases in gamma-band activity were not due to peripheral facial electromyographic activity. Additionally, we found no differences in pre/postscan head position, which could explain the differences (Wilcoxon test, p = 0.36, median ketamine = 2.2 mm, median placebo = 1.9 mm).
Figure 5.
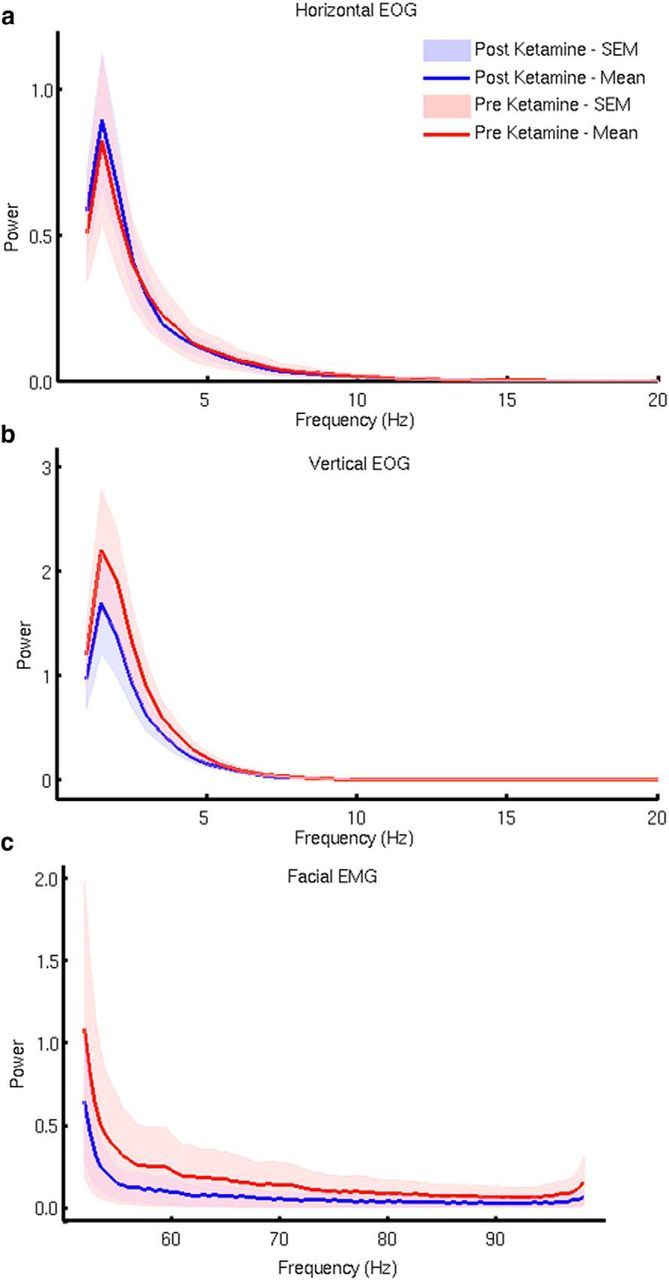
Spectral analysis of the vertical and horizontal EOGs and facial EMGs before and after ketamine infusion. Because of the location of frontal theta increases in the medial prefrontal cortex, we wished to confirm that theta differences were not potentially due to differential eye movements between the conditions. Spectral analysis of the two EOG channels (a, b) revealed no difference between ketamine and placebo that could account for the increase in medial prefrontal cortex theta power. Similarly, because of the increase in gamma-band amplitudes in the MEG data, we found no evidence from this analysis (c) that these were due to an increase in facial EMG activity.
Effective connectivity analysis
We began our DCM analysis of frontoparietal effective connectivity by comparing three potential models of connectivity. In Model 1, the two sources (precuneus, prefrontal cortex) were connected by posterior-to-anterior feedforward connections and anterior-to-posterior feedback connections (Fig. 1a, arrows C and B). In Model 2, these connections were reversed such that feedforward connections were anterior-to-posterior and feedback connections posterior-to-anterior (Fig. 1a, arrows A and D). Models 1 and 2 both represent hierarchically connected areas, whereas Model 3 contains fully reciprocated connectivity with all connections present (A–D). After fitting the single trial data of all concatenated individuals we compared the log model evidence and determined that Model 3 was the winning model (model evidences; Model 1 = 458, Model 2 = 435, Model 3 = 463). The log- model evidence values take into account the complexity of models with different numbers of parameters. This complexity term is the Kullback–Leibler divergence between prior and posterior parameter space and takes account of effective degrees-of-freedom introduced by new parameters (Penny et al., 2010). The grand-averaged cross-spectral densities for both modeled and empirical data are displayed in Figure 6a; these show good fits of the models to the empirical data. As example data features in the cross-conjugate space (Fig. 6b), beta band coherence and source lag appear altered with ketamine. The posterior estimates from the grand-average were then used to initialize parameter fits for individual spectra. A correlation (Fig. 6c) was observed between participants' self report of blissful state and the decrease in parietal gain (ρ = −0.53, p = 0.02), but not the other DCM parameters (all p > 0.05). In terms of connectivity parameters, Wilcoxon signed rank tests showed significant modulation of the NMDA-mediated backward connectivity (p = 3.1e−6), AMPA-mediated backward connectivity (p = 0.017), but not NMDA-mediated forward connectivity (p > 0.05) or AMPA-mediated forward connectivity (p > 0.05). A significant decrease in the gain of the parietal population was also found (p = 8.9e−6), but no difference in the frontal population gain (p > 0.05; Fig. 6d).
Figure 6.

a, Cross-spectral densities and model fits. Grand-averaged data (n = 19) were computed for preketamine (solid black line) and postketamine (solid blue line) for the frequency range 1–45 Hz. Grand-averaged DCM model fits are represented by the dashed lines. b, Example source space data features including frequency dependence coherence and time-lag between sources. c, Correlation of participants' rating of “blissful experience” and posterior gain modulation. Participants' ratings of “bliss” were computed from 5D-ASC subscales as devised by Studerus et al. (2010) and scaled to a percentage of the maximum possible score. A significant negative correlation was found between participants ratings of bliss and gain reduction in the parietal lobe. Spearman correlation tests were used to reduce outlier effects and control for non-normality in the data. d, Parameters modulations by ketamine for each of the modulation parameters (A–F). Sign rank tests were used to reduce outlier effects and control for non-normality in the data. A Bonferroni adjustment of six to the presented significance values to control the familywise error rate (6 parameters). Error bars are the standard error of the mean.
Functional connectivity analysis
Finally, we performed a functional connectivity analysis on the band limited source power envelopes (Fig. 7). A number of simple networks including visual parietal and motor networks were significantly altered by ketamine in the alpha and beta frequency bands. Although a number of frontoparietal networks were identified in the lower frequency bands, we did not observe modulation of these by ketamine, even if we removed the conservative Bonferroni correction.
Figure 7.

Results of functional connectivity analyses showing the localization of MEG resting-state network significantly altered by ketamine, which consisted of bilateral parietal (a), motor (b), and occipital (c) networks. Images show absolute ICA weights (in a.u.) thresholded at a value of 0.2. All modulations were decreases in the activity of the networks derived by independent component analysis of the band-power limited source waveforms. Although frontoparietal networks were identified in delta, theta, alpha, and beta frequency bands, these networks were not significantly modulated by ketamine. A Bonferroni adjustment of 15 (15 components per frequency band) is applied to the presented significance values.
Experiment 2
Pharmacodynamics
We temporally mapped the pharmacodynamics effects in a further set of participants. Notably, the subjective effects of ketamine had abated ∼40 min after the infusion had been ceased (Fig. 8c). The three areas and frequency bands chosen for further analysis each displayed distinct temporal dynamics, with theta band showing only a transient increase in power. Similar to the psychological effects, the increase in posterior gamma band power ceased ∼40 min after infusion had ceased. Alpha power, however (Fig. 8f), showed sustained power decreases that had still not returned to baseline 50 min after the infusion. In these data, we applied the same connectivity model (Model 3) that we used to track the temporal dynamics of effective connectivity changes. The effects on connectivity in these independent data were very similar to those in Experiment 1. The AMPA-mediated connectivity and the parietal gain were still decreased 50 min after the termination of infusion (Fig. 9). To examine potential relationships between the DCM parameter time courses and the power envelopes (three power envelopes, 6 DCM parameters, Bonferroni correction = 18) we used a general linear model analysis controlling for random subject effects. We found that the posterior gain parameter tended to follow the alpha envelope (t = 10.8, p = 1e−11), whereas the anterior gain followed the theta envelope (t = 3.6, p = 0.014). Notably, the connectivity parameters did not track the envelope parameters (all p > 0.05) even when uncorrected for multiple comparisons.
Figure 8.

Time-course dynamics for data collected in Experiment 2 (80 min resting state). a, The infusion protocol used is illustrated starting with an initial bolus followed by slow infusion. b, Estimated time course plasma concentration using the two-compartment model of Clements and Nimmo (1981). The pharmacokinetic parameters for this model were originally obtained using participants of a similar age/sex/health status to ours. c, Subjective ratings of drug “effect intensity” given by participants at 2 min intervals. d–f, Time courses of band-limited virtual sensor power for specific time-frequency regions of interest computed as percentage change from the preinfusion time period (0–10 min). Shaded areas represent the standard error of the mean.
Figure 9.

Time course of parameter modulations for the six DCM (a–f) parameters tested in Experiment 2 (Fig. 1). The time period of ketamine infusion is shaded red. The parameters were generated by comparing the spectra of the preinfusion window with the postinfusion period binned into 10 min windows. Error bars are the standard error of the mean.
Discussion
In these experiments, we found that subanesthetic infusions of ketamine caused a decrease in posterior alpha-band power, an increase in prefrontal theta power, as well as widespread increases in gamma-band power. These oscillatory changes were accompanied by temporally sustained reductions in frontoparietal effective connectivity, which we assessed using dynamic causal modeling. Previous studies have investigated the anesthetic effects on the EEG of ketamine-induced unconsciousness and have shown that 2 mg/kg of ketamine also causes reductions in directed frontoparietal connectivity (Lee et al., 2013; Blain-Moraes et al., 2014). Our data demonstrate these reductions occur at doses well below those needed to induce loss of consciousness, suggesting that reduced frontoparietal connectivity need not be associated with loss of consciousness (Boly et al., 2012). In contrast to EEG/MEG connectivity studies of subanesthetic ketamine doses, fMRI studies have shown mixed results with regard to functional connectivity. One fMRI study using a similar dosing protocol to ours reported globally increased hyperconnectivity (Driesen et al., 2013). Similarly, a study in rats found dose-dependent increases in hippocampal-prefrontal cortex connectivity following subanesthetic ketamine infusion (Gass et al., 2014). Conversely, a different study reported increases in connectivity in visual networks but decreases in affective networks (Niesters et al., 2012). Finally, others have reported decreases in functional connectivity in the default mode, cognitive control, and affective networks 24 h after ketamine infusion (Scheidegger et al., 2012). It is important to bear in mind that fMRI measures of functional connectivity are highly dependent on hemodynamic and vascular factors (Murphy et al., 2013). At subanesthetic doses, ketamine is well known to cause relatively large systemic changes in blood pressure and heart rate (Fig. 2; Pandit et al., 1980). Both of these factors can confound fMRI resting state studies (Murphy et al., 2013), which makes it surprising that few fMRI studies of ketamine have attempted to apply physiological correction methods. Alternatively, the differences in results between fMRI and EEG/MEG studies may be due to the fact that fMRI measures are potentially sensitive to a more diverse range of neural processes, whereas MEG/EEG are highly tuned to measuring the summation of synchronized postsynaptic potentials at pyramidal cells (Lopes da Silva, 2011).
In the present studies, we observed that the reduction in parietal gain was correlated with participants' rating of “blissful state”. This gain parameter is a “lumped” parameter and could potentially reflect either the intrinsic action of ketamine at NMDA receptors within the parietal pyramidal cell ensemble, or many of the non-NMDA affinities of ketamine, such as antagonism of HCN channels (Chen et al., 2009; see Introduction). Regardless, the correlation of parietal gain changes with positive subjective experiences is somewhat similar to the results of our previous study, where decreases in parietal activity caused by the mixed 5HT2A receptor agonist psilocybin were correlated with increases in participants' ratings of ego dissolution and magical thinking (Muthukumaraswamy et al., 2013). Although many studies have focused on the psychotomimetic properties of ketamine, fewer have focused on the euphoric feelings that people experience during ketamine sedation. At subanesthetic doses, most participants report that they experience feelings of euphoria and that the hallucinations induced are generally pleasant; they also report that they would be happy to repeat the same treatment again (Pandit et al., 1980). Caution is advised in interpreting these results, however, because of self-selection in the study populations.
The pharmacodynamics results that we report here are consistent with previous EEG anesthesia studies, which have shown similar transient increases in theta immediately after ketamine infusion with more sustained posterior alpha-band decreases (Kochs et al., 1996). Further, the increase in gamma-band activity and its time course, here in humans, is strikingly similar to the increases in rat EEG with ketamine and MK-801(Pinault, 2008; Wood et al., 2012). Importantly, the effects we observe are present in the resting MEG/EEG, whereas some previous descriptions of ketamine gamma/delta frequency-changes are based on evoked response paradigms (Hong et al., 2010) or task-induced oscillatory changes (Shaw et al., 2015), which may have different generative mechanisms and are limited mostly to sensory cortices. This widespread “aberrant” (Pinault, 2008) gamma-band activity in the neocortex could potentially be a mechanism that drives the decrease in interpopulation connectivity. Increased gamma-band activity may represent pharmacologically-induced noise at pyramidal cells assemblies, which disrupts information processing and the ability of these cell assemblies to send and/or receive long-range signals efficiently (Moran et al., 2015).
It is notable in this experiment that significant modulations of frontoparietal effective connectivity were observed using the DCM approach, however, no modulations of frontoparietal functional connectivity were observed when using a technique based on bandpass power correlations (Brookes et al., 2011). This probably reflects different signal elements to which each technique is sensitive. In particular, in the computation of the bandpass power envelopes, all phase information from the signals is discarded and further the envelopes are usually heavily down-sampled. DCM for steady-state responses considers a broader scope of data features including phase (cross-spectral density) information. In Experiment 2, the DCM connectivity parameters did not track the time course of the amplitude envelopes, whereas the gain parameters did. This suggests that the connectivity parameters are independent of signal-to-noise ratio (Friston, 2011), which can confound some estimates of functional connectivity, such as phase locking factors (Muthukumaraswamy and Singh, 2011).
The ability of ketamine to reduce resting state effective connectivity has potentially important implications for the treatment of depression. Sheline et al. (2009) have demonstrated that the default mode network is overactive in major depression, and that it has reduced ability to “switch off” during task performance. Similarly, increased resting state functional connectivity in the subgenual cingulate and thalamus is increased in depression, which is correlated with the length of depressive episode (Greicius et al., 2007). Overactivity of the default network in depression is correlated with behavioral measures of rumination and brooding (Berman et al., 2011). Quantitative electroencephalography has also shown increased occipital-to-parietal coherence in the alpha and theta frequency bands in major depressive disorder (Leuchter et al., 2012). Further, following treatment with SSRI's, functional connectivity of the default mode network is reduced; this effect correlates with a reduction in depressive symptoms (Wang et al., 2015). Together, these results suggest that, at least in a subset of patients, the major depressive disorder is characterized by hyperconnectivity of the default mode network. The rapid therapeutic action of ketamine may be due to its ability to reduce this hyperconnectivity, a hypothesis that requires testing.
Current molecular models of depression based largely on animal data have emphasized the importance of synaptogenic mechanisms in mediating the antidepressant effects of ketamine (Duman et al., 2012). In these models, blockade of NMDA receptors by ketamine causes increased activity at AMPA receptors, which causes increased activity of the mTOR signaling pathway, which in turn triggers synthesis of synaptic proteins, such as Arc and synapsin. Indeed, low doses of ketamine cause rapid increases in spine numbers on layer V pyramidal cells of the prefrontal cortex (Li et al., 2010). However, it could be argued that activation of the mTOR pathway may not be sufficient as an explanation of the antidepressant effects of ketamine. It is well known that a number of drugs of abuse that do not have antidepressant effects, including THC, morphine, alcohol, and cocaine, also activate the mTOR signaling pathway (Neasta et al., 2014). Indeed activation of the mTOR pathways may be a mechanism underlying addictive behaviors (Neasta et al., 2014), but given its general role in mediating long-term plasticity (Costa-Mattioli and Monteggia, 2013), it may serve the more general function of adapting the brain to remaining in its current dynamical state. We therefore speculatively propose that the antidepressant actions of ketamine may depend on both its ability to reduce hyperconnectivity and its activation of synaptogenic mechanisms. This can cause long-term stabilization of these changes allowing brain networks to restabilize in a different part of state-space.
Footnotes
This work was supported by the MRC/EPSRC funded UK MEG Partnership Grant MR/K005464/1, and S.D.M. is supported by a New Zealand Royal Society Rutherford Discovery Fellowship.
The authors declare no competing financial interests.
References
- Andrews-Hanna JR, Reidler JS, Sepulcre J, Poulin R, Buckner RL. Functional-anatomic fractionation of the brain's default network. Neuron. 2010;65:550–562. doi: 10.1016/j.neuron.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaansen MC, Knösche TR. Tangential derivative mapping of axial MEG applied to event-related desynchronization research. Clin Neurophysiol. 2000;111:1300–1305. doi: 10.1016/S1388-2457(00)00272-8. [DOI] [PubMed] [Google Scholar]
- Bergman SA. Ketamine: review of its pharmacology and its use in pediatric anesthesia. Anesth Prog. 1999;46:10–20. [PMC free article] [PubMed] [Google Scholar]
- Berman MG, Peltier S, Nee DE, Kross E, Deldin PJ, Jonides J. Depression, rumination and the default network. Soc Cogn Affect Neurosci. 2011;6:548–555. doi: 10.1093/scan/nsq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Blain-Moraes S, Lee U, Ku S, Noh G, Mashour GA. Electroencephalographic effects of ketamine on power, cross-frequency coupling, and connectivity in the alpha bandwidth. Front Syst Neurosci. 2014;8:114. doi: 10.3389/fnsys.2014.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boly M, Moran R, Murphy M, Boveroux P, Bruno MA, Noirhomme Q, Ledoux D, Bonhomme V, Brichant JF, Tononi G, Laureys S, Friston K. Connectivity changes underlying spectral EEG changes during propofol-induced loss of consciousness. J Neurosci. 2012;32:7082–7090. doi: 10.1523/JNEUROSCI.3769-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes MJ, Woolrich M, Luckhoo H, Price D, Hale JR, Stephenson MC, Barnes GR, Smith SM, Morris PG. Investigating the electrophysiological basis of resting state networks using magnetoencephalography. Proc Natl Acad Sci U S A. 2011;108:16783–16788. doi: 10.1073/pnas.1112685108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shu S, Bayliss DA. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J Neurosci. 2009;29:600–609. doi: 10.1523/JNEUROSCI.3481-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JA, Nimmo WS. Pharmacokinetics and analgesic effect of ketamine in man. Br J Anaesth. 1981;53:27–30. doi: 10.1093/bja/53.1.27. [DOI] [PubMed] [Google Scholar]
- Cornwell BR, Salvadore G, Furey M, Marquardt CA, Brutsche NE, Grillon C, Zarate CA., Jr Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:555–561. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16:1537–1543. doi: 10.1038/nn.3546. [DOI] [PubMed] [Google Scholar]
- David O, Kiebel SJ, Harrison LM, Mattout J, Kilner JM, Friston KJ. Dynamic causal modeling of evoked responses in EEG and MEG. Neuroimage. 2006;30:1255–1272. doi: 10.1016/j.neuroimage.2005.10.045. [DOI] [PubMed] [Google Scholar]
- Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Dittrich A. The standardized psychometric assessment of altered states of consciousness (ASCs) in humans. Pharmacopsychiatry. 1998;31:80–84. doi: 10.1055/s-2007-979351. [DOI] [PubMed] [Google Scholar]
- Driesen NR, McCarthy G, Bhagwagar Z, Bloch M, Calhoun V, D'Souza DC, Gueorguieva R, He G, Ramachandran R, Suckow RF, Anticevic A, Morgan PT, Krystal JH. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry. 2013;18:1199–1204. doi: 10.1038/mp.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durstewitz D, Seamans JK, Sejnowski TJ. Neurocomputational models of working memory. Nat Neurosci. 2000;3:1184–1191. doi: 10.1038/81460. [DOI] [PubMed] [Google Scholar]
- Felleman DJ, Van Essen DC. Distributed hierarchical processing in the primate cerebral cortex. Cereb Cortex. 1991;1:1–47. doi: 10.1093/cercor/1.1.1. [DOI] [PubMed] [Google Scholar]
- Friston KJ. Functional and effective connectivity: a review. Brain Connect. 2011;1:13–36. doi: 10.1089/brain.2011.0008. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Bastos A, Litvak V, Stephan KE, Fries P, Moran RJ. DCM for complex-valued data: cross-spectra, coherence and phase-delays. Neuroimage. 2012;59:439–455. doi: 10.1016/j.neuroimage.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston K, Mattout J, Trujillo-Barreto N, Ashburner J, Penny W. Variational free energy and the Laplace approximation. Neuroimage. 2007;34:220–234. doi: 10.1016/j.neuroimage.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Gass N, Schwarz AJ, Sartorius A, Schenker E, Risterucci C, Spedding M, Zheng L, Meyer-Lindenberg A, Weber-Fahr W. Sub-anesthetic ketamine modulates intrinsic BOLD connectivity within the hippocampal-prefrontal circuit in the rat. Neuropsychopharmacology. 2014;39:895–906. doi: 10.1038/npp.2013.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Flores BH, Menon V, Glover GH, Solvason HB, Kenna H, Reiss AL, Schatzberg AF. Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry. 2007;62:429–437. doi: 10.1016/j.biopsych.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall EL, Woolrich MW, Thomaz CE, Morris PG, Brookes MJ. Using variance information in magnetoencephalography measures of functional connectivity. Neuroimage. 2013;67:203–212. doi: 10.1016/j.neuroimage.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Hong LE, Summerfelt A, Buchanan RW, O'Donnell P, Thaker GK, Weiler MA, Lahti AC. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology. 2010;35:632–640. doi: 10.1038/npp.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MX, Mosher JC, Leahy RM. A sensor-weighted overlapping-sphere head model and exhaustive head model comparison for MEG. Phys Med Biol. 1999;44:423–440. doi: 10.1088/0031-9155/44/2/010. [DOI] [PubMed] [Google Scholar]
- Hyvärinen A, Oja E. Independent component analysis: algorithms and applications. Neural Netw. 2000;13:411–430. doi: 10.1016/S0893-6080(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Amaral DG. Macaque monkey retrosplenial cortex: II. Cortical afferents. J Comp Neurol. 2003;466:48–79. doi: 10.1002/cne.10883. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Amaral DG. Macaque monkey retrosplenial cortex: III. Cortical efferents. J Comp Neurol. 2007;502:810–833. doi: 10.1002/cne.21346. [DOI] [PubMed] [Google Scholar]
- Kochs E, Scharein E, Möllenberg O, Bromm B, Schulte am Esch J. Analgesic efficacy of low-dose ketamine: somatosensory-evoked responses in relation to subjective pain ratings. Anesthesiology. 1996;85:304–314. doi: 10.1097/00000542-199608000-00012. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73:1133–1141. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee U, Ku S, Noh G, Baek S, Choi B, Mashour GA. Disruption of frontal-parietal communication by ketamine, propofol, and sevoflurane. Anesthesiology. 2013;118:1264–1275. doi: 10.1097/ALN.0b013e31829103f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchter AF, Cook IA, Hunter AM, Cai C, Horvath S. Resting-state quantitative electroencephalography reveals increased neurophysiologic connectivity in depression. PloS One. 2012;7:e32508. doi: 10.1371/journal.pone.0032508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Silva F. Biophysical aspects of EEG and magnetoencephalogram generation. In: Schomer DL, Lopes da Silva F, editors. Niedermeyer's electroencephalography. Ed 6. Philadelphia: Lippincott Williams and Wilkins; 2011. [Google Scholar]
- Maris E, Oostenveld R. Nonparametric statistical testing of EEG- and MEG-data. J Neurosci Methods. 2007;164:177–190. doi: 10.1016/j.jneumeth.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Moran RJ, Stephan KE, Dolan RJ, Friston KJ. Consistent spectral predictors for dynamic causal models of steady-state responses. Neuroimage. 2011a;55:1694–1708. doi: 10.1016/j.neuroimage.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran RJ, Symmonds M, Stephan KE, Friston KJ, Dolan RJ. An in vivo assay of synaptic function mediating human cognition. Curr Biol. 2011b;21:1320–1325. doi: 10.1016/j.cub.2011.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran RJ, Jones MW, Blockeel AJ, Adams RA, Stephan KE, Friston KJ. Losing control under ketamine: suppressed cortico-hippocampal drive following acute ketamine in rats. Neuropsychopharmacology. 2015;40:268–277. doi: 10.1038/npp.2014.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K, Birn RM, Bandettini PA. Resting-state fMRI confounds and cleanup. Neuroimage. 2013;80:349–359. doi: 10.1016/j.neuroimage.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, Iqbal S, Pillemer S, Foulkes A, Shah A, Charney DS, Mathew SJ. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–1142. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumaraswamy SD. High-frequency brain activity and muscle artifacts in MEG/EEG: a review and recommendations. Front Hum Neurosci. 2013;7:138. doi: 10.3389/fnhum.2013.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumaraswamy SD, Singh KD. A cautionary note on the interpretation of phase-locking estimates with concurrent changes in power. Clin Neurophysiol. 2011;122:2324–2325. doi: 10.1016/j.clinph.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Muthukumaraswamy SD, Carhart-Harris RL, Moran RJ, Brookes MJ, Williams TM, Errtizoe D, Sessa B, Papadopoulos A, Bolstridge M, Singh KD, Feilding A, Friston KJ, Nutt DJ. Broadband cortical desynchronization underlies the human psychedelic state. J Neurosci. 2013;33:15171–15183. doi: 10.1523/JNEUROSCI.2063-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele P, Duma A, Kopec M, Gebara MA, Parsoei A, Walker M, Janski A, Panagopoulos VN, Cristancho P, Miller JP, Zorumski CF, Conway CR. Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol Psychiatry. 2015;78:10–18. doi: 10.1016/j.biopsych.2014.11.016. [DOI] [PubMed] [Google Scholar]
- Neasta J, Barak S, Hamida SB, Ron D. mTOR complex 1: a key player in neuroadaptations induced by drugs of abuse. J Neurochem. 2014;130:172–184. doi: 10.1111/jnc.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols TE, Holmes AP. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp. 2002;15:1–25. doi: 10.1002/hbm.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesters M, Khalili-Mahani N, Martini C, Aarts L, van Gerven J, van Buchem MA, Dahan A, Rombouts S. Effect of subanesthetic ketamine on intrinsic functional brain connectivity: a placebo-controlled functional magnetic resonance imaging study in healthy male volunteers. Anesthesiology. 2012;117:868–877. doi: 10.1097/ALN.0b013e31826a0db3. [DOI] [PubMed] [Google Scholar]
- Oostenveld R, Fries P, Maris E, Schoffelen JM. FieldTrip: open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Comput Intell Neurosci. 2011;2011:156869. doi: 10.1155/2011/156869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit SK, Kothary SP, Kumar SM. Low dose intravenous infusion technique with ketamine: amnesic, analgesic and sedative effects in human volunteers. Anaesthesia. 1980;35:669–675. doi: 10.1111/j.1365-2044.1980.tb03882.x. [DOI] [PubMed] [Google Scholar]
- Penny WD, Stephan KE, Daunizeau J, Rosa MJ, Friston KJ, Schofield TM, Leff AP. Comparing families of dynamic causal models. PLoS Comput Biol. 2010;6:e1000709. doi: 10.1371/journal.pcbi.1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry. 2008;63:730–735. doi: 10.1016/j.biopsych.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Posner J, Hellerstein DJ, Gat I, Mechling A, Klahr K, Wang Z, McGrath PJ, Stewart JW, Peterson BS. Antidepressants normalize the default mode network in patients with dysthymia. JAMA Psychiatry. 2013;70:373–382. doi: 10.1001/jamapsychiatry.2013.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SE, Vrba J. Functional neuroimaging by synthetic aperture magnetometry (SAM) In: Yoshimoto T, Kotani M, Kuriki S, Karibe H, Nakasato N, editors. Recent advances in biomagnetism. Sendai: Tohoku UP; 1999. pp. 302–305. [Google Scholar]
- Rush AJ. Ketamine for treatment-resistant depression: ready or not for clinical use? Am J Psychiatry. 2013;170:1079–1081. doi: 10.1176/appi.ajp.2013.13081034. [DOI] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Colon-Rosario V, Coppola R, Grillon C, Zarate CA, Jr, Manji HK. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65:289–295. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Sambataro F, Latov D, Colon-Rosario V, Carver F, Holroyd T, DiazGranados N, Machado-Vieira R, Grillon C, Drevets WC, Zarate CA., Jr Anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task predict rapid antidepressant response to ketamine. Neuropsychopharmacology. 2010;35:1415–1422. doi: 10.1038/npp.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, Quirk MC. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014;19:978–985. doi: 10.1038/mp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidegger M, Walter M, Lehmann M, Metzger C, Grimm S, Boeker H, Boesiger P, Henning A, Seifritz E. Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS One. 2012;7:e44799. doi: 10.1371/journal.pone.0044799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AD, Saxena N, Jackson L, Hall JE, Singh KD, Muthukumaraswamy SD. Ketamine amplifies induced gamma frequency oscillations in the human cerebral cortex. Eur Neuropsychopharmacology. 2015 doi: 10.1016/j.euroneuro.2015.04.012. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Barch DM, Price JL, Rundle MM, Vaishnavi SN, Snyder AZ, Mintun MA, Wang S, Coalson RS, Raichle ME. The default mode network and self-referential processes in depression. Proc Natl Acad Sci U S A. 2009;106:1942–1947. doi: 10.1073/pnas.0812686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM. Fast robust automated brain extraction. Hum Brain Mapp. 2002;17:143–155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SM. Mechanism of action of ketamine. CNS Spectr. 2013;18:171–174. doi: 10.1017/S109285291300045X. [DOI] [PubMed] [Google Scholar]
- Studerus E, Gamma A, Vollenweider FX. Psychometric evaluation of the altered states of consciousness rating scale (OAV) PloS One. 2010;5:e12412. doi: 10.1371/journal.pone.0012412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrba J, Robinson SE. Signal processing in magnetoencephalography. Methods. 2001;25:249–271. doi: 10.1006/meth.2001.1238. [DOI] [PubMed] [Google Scholar]
- Wang L, Xia M, Li K, Zeng Y, Su Y, Dai W, Zhang Q, Jin Z, Mitchell PB, Yu X, He Y, Si T. The effects of antidepressant treatment on resting-state functional brain networks in patients with major depressive disorder. Hum Brain Mapp. 2015;36:768–778. doi: 10.1002/hbm.22663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J, Kim Y, Moghaddam B. Disruption of prefrontal cortex large scale neuronal activity by different classes of psychotomimetic drugs. J Neurosci. 2012;32:3022–3031. doi: 10.1523/JNEUROSCI.6377-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, Manji HK, Charney DS. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006a;163:153–155. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006b;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]