

ABSTRACT
TorsinA (also known as torsin-1A) is a membrane-embedded AAA+ ATPase that has an important role in the nuclear envelope lumen. However, most torsinA is localized in the peripheral endoplasmic reticulum (ER) lumen where it has a slow mobility that is incompatible with free equilibration between ER subdomains. We now find that nuclear-envelope-localized torsinA is present on the inner nuclear membrane (INM) and ask how torsinA reaches this subdomain. The ER system contains two transmembrane proteins, LAP1 and LULL1 (also known as TOR1AIP1 and TOR1AIP2, respectively), that reversibly co-assemble with and activate torsinA. Whereas LAP1 localizes on the INM, we show that LULL1 is in the peripheral ER and does not enter the INM. Paradoxically, interaction between torsinA and LULL1 in the ER targets torsinA to the INM. Native gel electrophoresis reveals torsinA oligomeric complexes that are destabilized by LULL1. Mutations in torsinA or LULL1 that inhibit ATPase activity reduce the access of torsinA to the INM. Furthermore, although LULL1 binds torsinA in the ER lumen, its effect on torsinA localization requires cytosolic-domain-mediated oligomerization. These data suggest that LULL1 oligomerizes to engage and transiently disassemble torsinA oligomers, and is thereby positioned to transduce cytoplasmic signals to the INM through torsinA.
KEY WORDS: AAA+ protein, Dystonia, Endoplasmic reticulum, Inner nuclear membrane, Torsin
Summary: Interaction between the torsinA AAA+ protein and the LULL1 activator of ATP hydrolysis in the endoplasmic reticulum controls torsinA entry to the inner nuclear membrane, its site of activity.
INTRODUCTION
Torsins are an animal-specific branch of the AAA+ ATPase superfamily that have developmentally essential functions in mouse, C. elegans and Drosophila (Basham and Rose, 2001; Goodchild et al., 2005; Wakabayashi-Ito et al., 2011). The importance of these proteins is further emphasized by the childhood-onset neurological disease of DYT1 dystonia, which is caused by a dominantly inherited loss-of-function point mutation in the best-studied torsin, torsinA (also known as torsin-1A, TOR1A) (Ozelius et al., 1997).
AAA+ ATPases typically use the energy of ATP hydrolysis to drive structural changes in substrates. The majority function as hexameric ring-shaped structures in which residues extending into a central pore interact with and transmit mechanical force to a target substrate. Although substrate(s) affected by torsins remain unidentified, it is clear that torsins possess fundamental AAA+ characteristics including motifs involved in nucleotide binding and hydrolysis (Kock et al., 2006) and assembly into hexameric rings (Jungwirth et al., 2010; Sosa et al., 2014; Vander Heyden et al., 2009). However, torsins also have distinctive features. Most, including torsinA, are embedded in the luminal leaflet of the endoplasmic reticulum (ER) membrane (Jungwirth et al., 2010; Vander Heyden et al., 2011). Importantly, torsinA binds two transmembrane proteins of the ER system, LAP1 (also known as TOR1AIP1) and LULL1 (also known as TOR1AIP2 and NET9) (Goodchild and Dauer, 2005; Kim et al., 2010; Naismith et al., 2009; Zhao et al., 2013; Zhu et al., 2010), that have recently been shown to adopt a partial AAA+ domain fold and co-assemble with torsinA as subunits of an active enzyme (Brown et al., 2014; Sosa et al., 2014). The luminal domains of LAP1 and LULL1 are homologous to each other, whereas the extraluminal domains diverge (Goodchild and Dauer, 2005). Like torsins, proteins similar to LAP1 and LULL1 have been identified across animal phyla and might have co-evolved to function together with torsins (Brown et al., 2014; Sosa et al., 2014).
The ER is a network of membrane tubules and sheets that extends throughout the cell body of eukaryotic cells. The nuclear envelope is the ER subdomain that surrounds the nucleus and consists of an inner (INM) and outer nuclear membrane (ONM) separated by a luminal perinuclear space. Although the ONM is continuous with the peripheral ER, it is only connected to the INM at sites of nuclear pore complex (NPC) insertion (Antonin et al., 2011; Burns and Wente, 2012). It is estimated that 50–100 proteins concentrate in the nuclear envelope and most are localized on the INM (de Las Heras et al., 2013; Schirmer et al., 2003). These INM proteins are translated and inserted into the ER or ONM and, although they can access the INM following open mitosis, the main route to the INM requires diffusion across the NPC-associated membrane. Importantly for dystonia, diffusion across this curved membrane is the only route to the INM in post-mitotic neurons. Protein mobility in the ER and ONM and the ability to diffuse across the curved NPC-associated membrane, past NPC components, will thus affect the INM proteome. Unhindered membrane protein diffusion to the INM is limited to proteins with extraluminal domains smaller than ∼60 kDa (Antonin et al., 2011; Burns and Wente, 2012; Laba et al., 2014; Ohba et al., 2004). INM proteins with larger extraluminal domains interact with importins for active transport from the ER to the INM in yeast (King et al., 2006), and energy-dependent movement of proteins to the INM in mammalian cells suggests comparable transport pathways (Ohba et al., 2004). Although less clearly defined, movement of membrane proteins into the INM is also sensitive to the size of luminal domains (Ohba et al., 2004), raising the possibility that proteins anchored on the inner leaflet of the membrane, such as torsins, will also be subject to size- or shape-based access restrictions.
There are multiple lines of evidence showing that there is a developmentally essential role for torsins at the nuclear envelope. Torsin loss in mouse, C. elegans and Drosophila as well as cultured mammalian cells causes nuclear envelope defects that involve both the INM and ONM (Goodchild et al., 2005; Jokhi et al., 2013; Naismith et al., 2004; VanGompel et al., 2015). Surprisingly, however, the majority of torsinA is distributed in the peripheral ER (Liang et al., 2014; Vander Heyden et al., 2011). Furthermore, ER-localized torsinA moves slowly and is excluded from the curved membranes of ER tubules and exit sites (Vander Heyden et al., 2011, 2009; Goodchild and Dauer, 2005), raising questions about whether and, if so, how torsinA accesses the INM.
We now explore this paradox and establish that nuclear-envelope-localized torsinA is in fact on the INM, and thus must move through the curved NPC-associated membrane. We find that the ER-localized torsinA activating protein, LULL1, but not the INM-localized LAP1, promotes torsinA redistribution to the INM. Key residues in both torsinA and LULL1 necessary for ATPase activity are required for torsinA to reach the INM. The cytosolic domain of LULL1 is also required and acts by inducing LULL1 oligomerization, likely increasing the efficiency of interaction with torsinA. Thus, although LAP1 and LULL1 both co-assemble with torsinA and stimulate torsin ATPase activity in vitro (Brown et al., 2014; Sosa et al., 2014; Zhao et al., 2013) they have unique effects on the distribution and availability of enzyme in the ER system. The fact that the required oligomerization of LULL1 is under control of its cytosolic domain provides a potential regulatory mechanism for cytosolic control of torsinA targeting to the INM.
RESULTS
Two differentially localized torsinA-activating factors
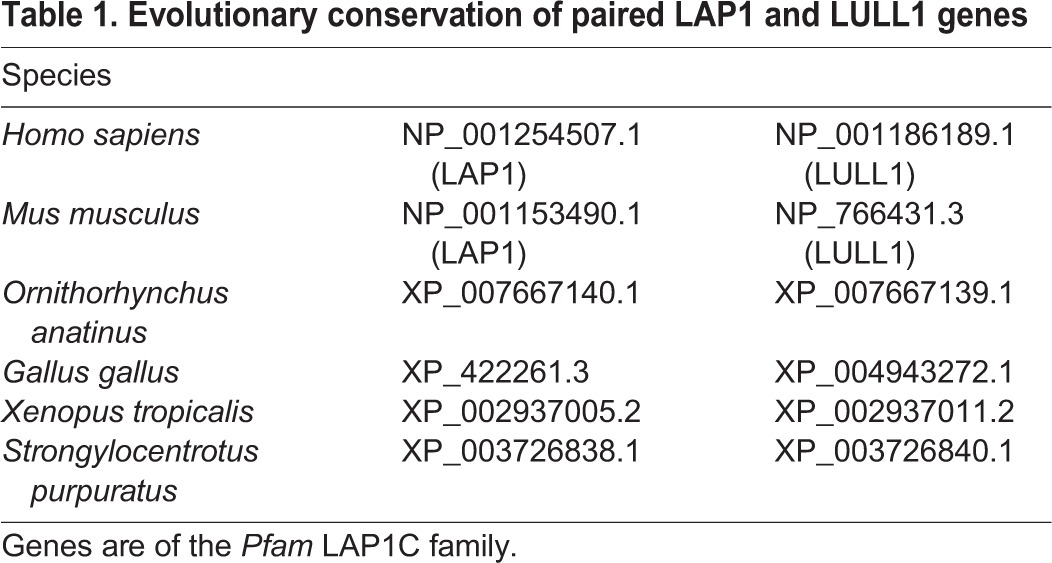
LAP1 and LULL1 have homologous torsin-interacting luminal domains but divergent extraluminal sequences (Fig. 1A). LAP1 is an INM-specific protein that appears to be required for nuclear-envelope-localized torsinA activity (Kim et al., 2010; Maison et al., 1997; Martin et al., 1995). In contrast, LULL1 localizes throughout the ER system, including in the nuclear envelope (Goodchild and Dauer, 2005; Naismith et al., 2009; Schirmer et al., 2003). The similarities between LAP1 and LULL1 (Brown et al., 2014; Sosa et al., 2014; Zhao et al., 2013) together with the fact that LULL1 increases, rather than decreases, torsinA in the nuclear envelope (Vander Heyden et al., 2009), raise the possibility that both LAP1 and LULL1 function as nuclear-envelope-specific torsin-activating factors. However, the evolutionary conservation of LAP1 and LULL1 gene pairs (Table 1) (Goodchild and Dauer, 2005), instead suggests that they are likely to play unique roles.
Fig. 1.

LAP1 and LULL1 have distinct localization in the ER system. (A) Schematic showing protein organization. (B,C) Differential permeabilization studies place LAP1 on the INM. An antibody recognizing the LAP1 nucleoplasmic domain detects nuclear-envelope-concentrated LAP1 in U2OS cells permeabilized with Triton X-100 but not digitonin, as is also observed for the control nuclear LAP2 protein. (D,E) Differential permeabilization studies place LULL1 in peripheral ER and ONM. An antibody recognizing the LULL1 cytoplasmic domain detects LULL1 throughout the cell following permeabilization with either Triton X-100 or digitonin. Nuclear LAP2 is only detected in Triton X-100 sample. (F,G) Rapamycin trapping demonstrates accumulation of control FRB–TM–GFP but not FRB–LULL1–mGFP in the INM. Images show (F) FRB–TM–GFP and (G) FRB–LULL1–mGFP in NIH-3T3 cells before (left panels) or after 90 min incubation (right panels) with rapamycin. Scale bars: 10 µm. (H) Quantification of the percentage of cells with nuclear-envelope-concentrated GFP (mean±s.e.m.). Data are from three transfections, each counting >50 cells.
Table 1.
Evolutionary conservation of paired LAP1 and LULL1 genes
To define differences between LAP1 and LULL1, we compared their subcellular localization using antibodies recognizing the extraluminal domains of each to immunostain cells permeabilized with digitonin (for selective permeabilization of the plasma membrane) or Triton X-100 (for full permeabilization and exposure of both cytosolic and nuclear epitopes). LAP1 immunoreactivity at the nuclear envelope was prominent in cells permeabilized with Triton X-100 but not in digitonin-permeabilized cells (Fig. 1B,C), consistent with its known INM localization (Maison et al., 1997; Martin et al., 1995). In contrast, LULL1 immunoreactivity was distributed throughout the cell and was similar in Triton-X-100- and digitonin-treated cells (Fig. 1D,E), indicating that this extraluminal domain is exposed to the cytoplasm. The lack of difference in LULL1 immunostaining at the nuclear envelope between Triton-X-100- and digitonin-permeabilized cells (Fig. 1D,E) further indicates that LULL1 is absent from the INM.
To determine whether LULL1 might nonetheless transiently access the INM (at a level not recognizable at steady state), we used a trapping system that accumulates protein that enters the INM by preventing its return to the ONM and ER (Ohba et al., 2004). We co-expressed the nuclear-localized FK-binding protein (FKBP) trap protein alongside LULL1–mGFP fused to FKBP12-rapamycin binding domain (FRB–LULL1–mGFP) (Fig. 1A). Rapamycin mediates strong binding between the nuclear FKBP trap protein and the FRB domain, thus immobilizing any FRB-tagged membrane protein that reaches the INM (Ohba et al., 2004; Putyrski and Schultz, 2012). As expected, we found that adding rapamycin led to the accumulation of a GFP-tagged FRB–transmembrane-domain reporter (FRB–TM–GFP) in the nuclear envelope (Fig. 1F,H). In contrast, treatment with rapamycin failed to reveal nuclear-envelope-localized FRB–LULL1–mGFP (Fig. 1G,H). Thus, despite homology to LAP1 and a mass permissive for diffusion through the nuclear pore membrane (Antonin et al., 2011), LULL1 does not normally reach the INM.
LULL1 promotes torsinA accumulation on the INM
TorsinA is excluded from regions of the ER that are highly curved, including tubules and exit sites (Vander Heyden et al., 2011). Given that interphase access of membrane proteins to the INM requires passage across the curved NPC-associated membrane (Antonin et al., 2011), we wondered whether torsinA reaches the INM, or instead resides primarily on the ONM. Indeed, candidate torsinA substrates have been described in both membranes (Nery et al., 2008; Vander Heyden et al., 2009). To assess the distribution of a luminal monotopic protein such as torsinA between ONM and INM, we needed a way to differentiate anchoring on membranes that are not resolved from each other by standard light microscopy. We therefore generated reporter proteins in which the torsinA N-terminal membrane-anchoring domain is replaced with a type II transmembrane domain and an extraluminal epitope tag that will be differentially accessible to antibody staining in selectively permeabilized cells depending on whether the protein is in the ONM facing the cytoplasm or the INM facing the nucleus (Fig. 2A; FLAG–TM–torsinA–mGFP). These torsinA reporters also include mGFP to enable direct visualization. We confirmed that FLAG–TM–torsinA–mGFP localizes throughout the ER and that anti-FLAG signal is detectable in digitonin-permeabilized cells, consistent with cytoplasmic localization and a correctly oriented transmembrane domain (supplementary material Fig. S1A,B).
Fig. 2.

TorsinA entry onto INM is enhanced by LULL1. (A) Schematic of torsinA reporter proteins. Note that GFP is appended to N- or C-terminus as shown, and we refer to these as GFP–torsinA and torsinA–GFP, respectively. (B,C) Differential permeabilization studies place torsinA-ΔE302/3 on the INM. U2OS cells co-expressing FLAG–TM–torsinA-ΔE302/3–mGFP and LULL1 (not imaged), permeabilized with (B) Triton X-100 or (C) digitonin. Direct GFP fluorescence (green) shows nuclear envelope enrichment of torsinA-ΔE302/3 whereas FLAG immunostaining (red) detects torsinA in the nuclear envelope only in Triton-X-100-permeabilized cells. Nuclear LAP2 is only detected following Triton X-100 permeabilization. (D,E) Linescan analysis showing detection of nuclear envelope enrichment. Scans were performed along lines shown in B and C, the background was subtracted and intensities were normalized for plotting. Green represents GFP, red represents anti-FLAG. (F,G) U2OS cells expressing FLAG–TM–torsinA-WT–mGFP and LULL1, treated as in B and C. (H) Effect of depleting LULL1 on localization of GFP–torsinA-ΔE302/3 in NIH-3T3 (upper row) and CAD cells (lower row) co-transfected with control or Lull1 siRNA. (I,J) Wild-type (I) or ΔE302/3 (J) FRB–TM–torsinA–mGFP in Lull+/+ or Lull1−/− MEFs after 0 or 90 min of rapamycin treatment. Asterisks highlight cells with nuclear-envelope-concentrated GFP signal. Scale bars: 10 µm. (K) Quantification of Lull+/+ (solid lines) or Lull1−/− MEFs (dashed lines) with nuclear-envelope-concentrated FRB–TM–torsinA–mGFP after indicated time with rapamycin. Data (mean±s.e.m.) are from four experiments and 50–100 GFP cells per genotype, torsin and time point. Green shows wild-type FRB–TM–torsinA–mGFP and red the ΔE302/3 mutant. Analysis of Lull1 genotype, time and torsinA mutation identified significant effects of time (P<0.02, F=5.25) and Lull1 (P<0.001, F=15.11) (univariate analysis of variance) on the percentage of cells with nuclear-envelope-concentrated torsin.
We then used FLAG–TM–torsinA–mGFP reporters to ask whether torsinA in the nuclear envelope is on the ONM or INM. We first examined the distribution of reporter containing the DYT1 dystonia ΔE302/3 mutation. This mutation removes one of a pair of glutamic acid residues from a C-terminal region of torsinA and often concentrates torsinA in the nuclear envelope (Giles et al., 2008; Goodchild and Dauer, 2004). This nuclear envelope concentration can be further enhanced by increasing LULL1 expression (Vander Heyden et al., 2009). We found that, as expected, the ΔE302/3 reporter was enriched in the nuclear envelope when examined by direct GFP fluorescence (Fig. 2B,C). In contrast, when examined by immunostaining, nuclear-envelope-concentrated anti-FLAG signal was far more apparent in Triton-X-100-than digitonin-permeabilized cells (Fig. 2B,C). Line scans highlight the differences in nuclear envelope signal (Fig. 2D,E). These data indicate that the FLAG epitope of this reporter is exposed in the nucleus and thus that nuclear-envelope-concentrated torsinA-ΔE302/3 is on the INM. We also examined wild-type torsinA shifted to the nuclear envelope by overexpressing LULL1 (Vander Heyden et al., 2009) and again found that nuclear-envelope-enriched direct GFP fluorescence is paralleled by anti-FLAG immunoreactivity in cells permeabilized with Triton X-100 but not digitonin, indicating that torsinA reaches the INM (Fig. 2F,G).
LULL1 but not LAP1 is essential for targeting torsinA to the INM
The above data demonstrate that torsinA accesses the INM with help from LULL1. We previously found that decreasing LULL1 by using short hairpin RNA (shRNA) reduces accumulation of torsinA-ΔE302/3 in the nuclear envelope of U2OS cells (Vander Heyden et al., 2009). To determine how general the requirement for LULL1 in targeting torsinA to the nuclear envelope is, we depleted it from cells in which torsinA-ΔE302/3 constitutively concentrates in the nuclear envelope. Once again, we found that reducing LULL1 shifts torsinA-ΔE302/3 away from the nuclear envelope (Fig. 2H; supplementary material Fig. S2A,B).
We next asked how LULL1 facilitates torsinA entry to the INM using the rapamycin INM-trap system. We previously found that increased LULL1 expression causes wild-type torsinA to relocalize from throughout the ER to the nuclear envelope over ∼45 min (Vander Heyden et al., 2009). We now asked how readily torsinA enters the INM in cells without excess LULL1 and detected relatively little FRB–torsinA at the INM even after 90 min of rapamycin (Fig. 2I; supplementary material Fig. S2C). We then asked how the complete absence of LULL1 affects torsinA entry to the INM. Although Lull1−/− cells still displayed a small but detectable rapamycin-dependent INM trapping of WT torsinA (Fig. 2I,K), the absence of LULL1 eliminated trapping of torsinA-ΔE302/3 on the INM (Fig. 2J,K). Notably, there was a significant effect of the Lull1 genotype on the proportion of cells with rapamycin-trapped torsinA in the INM (Fig. 2K).
These data show that LULL1 modulates torsinA entry to the INM, but do not rule out additional effects on retention of torsinA in the INM. After reaching the INM, many INM residents are trapped by binding to other nuclear-envelope-localized components (Antonin et al., 2011; Burns and Wente, 2012). We wondered whether LULL1 might somehow influence torsinA binding to its known INM partner, LAP1. We used small interfering RNA (siRNA) targeting the 3′ UTR of LAP1 to deplete all LAP1 splice variants (supplementary material Fig. S2D), but still found that increasing LULL1 redistributed wild-type torsinA from the ER to the nuclear envelope (Fig. 3A–C). These data are consistent with our earlier finding that nuclear envelope enrichment of torsinA-ΔE302/3 is not correlated with LAP1 expression (Jungwirth et al., 2011) and we conclude that LULL1 targets torsinA to the INM independently of LAP1.
Fig. 3.

LULL1 acts from the ER independently of LAP1 to promote torsinA relocalization. (A,B) U2OS cells stably expressing tetracycline-inducible LULL1–Myc (not imaged) and torsinA–mGFP (left panels) transfected with control (A) or LAP1 siRNA (B). Cells are immunostained to detect endogenous LAP1 (right panels). TorsinA–mGFP is localized throughout the ER under basal conditions (upper panels) but redistributes to the nuclear envelope following increased LULL1 expression (lower panels). LAP1 depletion is seen in (B) and confirmed by immunoblotting (supplementary material Fig. S2D), but does not change LULL1-mediated torsinA relocalization. (C) Quantification of results shown in A and B (mean±s.e.m.; n≥75). (D–F) ER-restricted PK–LULL1 relocalizes torsinA-ΔE302/3 to the nuclear envelope. Images of GFP–torsinA-ΔE302/3 (left panels) in CHO cells co-transfected with (D) control [chicken hepatic lectin transmembrane fused to Myc (Soullam and Worman, 1995)], or (E,F) the indicated Myc–LULL1 fusions (right panels; red). (G) ER-restricted LULL1 relocalizes torsinA–mGFP in transfected U2OS cells. (H) LULL1 and PK–LULL1 expression similarly increase the torsinA-ΔE302/3 nuclear envelope localization Index. Mean±s.e.m. are calculated for >20 CHO cells co-transfected with GFP–torsinA-ΔE302/3 and the indicated LULL1 or control transmembrane protein. ****P<0.001 difference in nuclear envelope localization index compared with cells expressing the control protein (one-way ANOVA with Bonferroni post-hoc analysis). Scale bars: 10 µm.
Although LULL1 resides in the peripheral ER and does not appear to access the INM (Fig. 1), we nevertheless wondered whether LULL1 within a torsinA complex might traffic with torsinA across the NPC-associated membrane. We therefore fused pyruvate kinase (PK, also known as PKM) to the N-terminus of LULL1 (Fig. 1A); PK is a large protein that has previously been shown to prevent transit to the INM (Ohba et al., 2004; Wu et al., 2002). PK–LULL1 is correctly targeted to the ER (supplementary material Fig. S2E), and as expected, rapamycin trapping failed to detect any FRB–PK–LULL1 entry to the INM (supplementary material Fig. S2F). Importantly, PK–LULL1 expression was as effective as wild type LULL1 in elevating the nuclear envelope concentration of wild-type and torsinA-ΔE302/3 (Fig. 3D–H), confirming that LULL1 increases torsinA on the INM without itself entering the INM.
TorsinA is present in high molecular mass complexes
How might interaction between torsinA and LULL1 in the ER increase the rate at which torsinA accesses the INM? Although torsinA is a ∼37 kDa protein, fluorescence recovery after photobleaching (FRAP) has revealed that torsinA–mGFP (∼62 kDa) diffuses at 0.09–0.13 µm2/s (Vander Heyden et al., 2009), which is faster than the translocon (∼1 MDa, ∼0.05 µm2/s) but slower than LULL1–mGFP (∼100 kDa, ∼0.45 µm2/s) (Vander Heyden et al., 2009). In fact, torsinA–mGFP moves as slowly in the ER as most proteins normally restricted to the INM (Goodchild and Dauer, 2005; Zuleger et al., 2011). We previously found that some torsinA is in an ∼250–300 kDa complex as detected by Blue-Native (BN)-PAGE (Jungwirth et al., 2010; Vander Heyden et al., 2009), consistent with assembly into a homo-hexamer or hetero-hexamer with LULL1 or LAP1 (Brown et al., 2014; Sosa et al., 2014; Zhao et al., 2013). Tethering to the membrane at six points could account for the slow mobility seen by FRAP. To ask whether yet larger oligomeric species of torsinA contribute to its slow mobility, we used electrophoresis conditions able to resolve structures up to ∼1.2 MDa (Edbauer et al., 2002). This revealed torsinA both in ∼250–300 kDa and larger species (Fig. 4A). Interestingly, the ΔE302/3 mutation increased the amount of torsinA in these larger species. To probe the oligomeric state of torsinA under physiological conditions [with a native ratio of torsinA to activating factor(s)], we examined torsinA in liver microsomes by BN-PAGE. This again detected a discrete high molecular mass species that does not correspond with total protein or calreticulin immunoreactivity that might indicate incomplete solubilization (Fig. 4B). We confirmed the presence of torsinA in this high molecular mass species using a second dimension of denaturing SDS-PAGE (Fig. 4C). These data suggest that torsinA forms multiple oligomeric complexes that are likely to contribute to its slow mobility in the ER system. An intrinsic tendency of torsinA to assemble into high molecular mass species has previously been noted in studies of purified torsinA, particularly ΔE302/3-torsinA (Sosa et al., 2014; Zhao et al., 2013).
Fig. 4.

Multimeric torsinA complexes are sensitive to LULL1 and ATPase activity. (A) Wild-type and torsinA-ΔE302/3 seen in high molecular mass structures by BN-PAGE. Panels show immunoblotting of 20 µg of 0.5% digitonin-solubilized cell lysate from NIH-3T3 cells transfected as indicated. (B) A high molecular mass torsinA species in mouse liver microsomes. 20 µg of 1% digitonin-solubilized microsomal protein separated on a 3.2–16% gradient BN-PAGE, with individual lanes stained for total protein or immunostained for torsinA and calreticulin. Anti-torsinA immunoreactivity is seen at ∼900 kDa. (C) Anti-torsinA immunoblotting following second dimension SDS-PAGE confirms that the 37 kDa torsinA is present in the high molecular mass immunoreactive species. (D) TorsinA-E171Q and LULL1 co-assemble into an ∼250 kDa species consistent with formation of a stable hetero-hexameric complex. Panels show anti-torsinA and anti-LULL1 immunoblotting of BN-PAGE separated lysates from U2OS cells transfected with torsinA-E171Q, LULL1–Myc, or both. Co-expressed torsinA-E171Q and LULL1 are present in an ∼250 kDa band that is absent when either one is overexpressed alone. (E) ATP hydrolysis destabilizes the LULL1–torsinA complex monitored by co-immunoprecipitation (IP) from NIH-3T3 cells stably expressing GFP–torsinA. Upper panel, lane one, shows total cellular LULL1 (Cell Lysate). Lanes 2–6 show the amount of LULL1 that co-immunoprecipitates with GFP–torsinA without added nucleotides, or upon the addition of 2 mM ATP, ADP, ATPγS or EDTA. The lower panel shows anti-GFP immunoblotting, which confirms immunoprecipitation of similar amounts of GFP–torsinA. (F) BN-PAGE shows that LULL1 expression decreases the ∼250–300 kDa species of torsinA resolved by BN-PAGE of digitonin-solubilized samples. Shown is anti-torsinA immunoblotting of BN-PAGE-separated lysates from U2OS cells transfected with wild-type torsinA alone or together with LULL1–Myc and solubilized in 1% digitonin. BN-PAGE separation is on a 7.5% gel. This experiment was repeated three times with the same result. (G) SDS-PAGE and immunoblotting shows torsinA and LULL1 in the samples resolved by BN-PAGE in F.
TorsinA oligomers are sensitive to ATPase activity and LULL1
To understand how LULL1 affects torsinA we now sought to confirm that torsinA and LULL1 coassemble in the ER membrane into the same type of hetero-hexameric assemblies seen in vitro with purified protein (Sosa et al., 2014; Zhao et al., 2013). We therefore examined complex(es) formed upon co-expression of LULL1–Myc and ATP-hydrolysis-deficient torsinA-E171Q. Indeed, we detect a LULL1 and torsinA-immunoreactive species by BN-PAGE that migrates as expected for a torsinA–LULL1 hetero-hexamer (Fig. 4D). We next considered that LULL1 increases INM entry of wild-type torsinA that, unlike torsinA-E171Q, is stimulated by LULL1 to hydrolyze ATP. We therefore asked how ATP hydrolysis affects binding between torsinA and LULL1 and, consistent with earlier mutagenesis studies (Goodchild and Dauer, 2005; Naismith et al., 2009), found that non-hydrolyzable ATPγS enhances co-immunoprecipitation of LULL1 with torsinA, compared to ATP or ADP (Fig. 4E). This confirms that ATP hydrolysis releases torsinA from LULL1, consistent with classic models of AAA+ protein function, and suggests two stages of engagement between LULL1 and torsinA – formation of ATPase-competent heterohexamers followed by ATPase-triggered dissociation. We therefore asked whether overexpressing LULL1 at a level that we know redistributes torsinA to the INM changes the observed torsinA oligomers, bearing in mind the limited sensitivity of BN-PAGE to detect transient changes. Significantly, we found a substantial decrease in the amount of ∼250–300 kDa torsinA-containing complex (Fig. 4F,G), demonstrating that LULL1 destabilizes torsinA complexes. Taken together, the large apparent size of torsinA complexes, slow torsinA mobility, and ability of LULL1 to decrease the abundance of torsinA oligomers led us to hypothesize that LULL1 facilitates torsinA entry onto the INM by, at least transiently, reducing the oligomeric size of torsinA.
LULL1- and torsinA-mediated ATPase activity is required to target torsinA to the INM
To further test this hypothesis, we asked whether ATP hydrolysis is required for LULL1 to concentrate torsinA on the INM. We had previously found that ATPase-deficient torsinA-E171Q is not relocalized to the nuclear envelope and instead remains with LULL1 in the peripheral ER (Vander Heyden et al., 2009). We now found that other mutations in torsinA that interfere with ATP hydrolysis – in sensor 1 and sensor 2 motifs (Kock et al., 2006) – also prevent LULL1-mediated redistribution (Fig. 5A–C). Thus, three different mutations that inhibit ATPase activity and the concomitant dissociation of the torsinA–LULL1 complex also inhibit LULL1-mediated targeting of torsinA to the INM.
Fig. 5.

LULL1-enhancement of torsinA INM localization requires ATPase activity. (A–C) ATP-hydrolysis-deficient forms of torsinA fail to relocalize to the INM when co-expressed with LULL1–Myc. Images show that wild-type torsinA–mGFP is in the nuclear envelope, whereas N208A or K320M mutants remain in the peripheral ER when transiently transfected into U2OS cells stably expressing LULL1–Myc. (D–F) The R449A arginine finger mutation in LULL1 inhibits torsinA-relocalizing activity. Images show U2OS cells stably expressing wild-type torsinA–mGFP (left panels) and transiently expressing the indicated LULL1–Myc (right panels). Scale bars: 10 µm. (G) LULL1 arginine finger mutations inhibit torsinA nuclear envelope concentration. Mean±s.e.m. from n>100 cells are shown per LULL1 mutant.
Interestingly, the recent crystal structure and modeling of the LAP1 and LULL1 luminal domain suggests an unusual functional interaction between LAP1 or LULL1 and torsinA. Although torsinA has a full AAA+ domain (Kock et al., 2006), it lacks candidate arginine finger residues typically necessary for ATP hydrolysis (Ogura et al., 2004). Intriguingly, the LAP1 luminal domain contains a cryptic AAA+ fold that is unable to bind nucleotide but presents an arginine residue that both modeling and in vitro testing suggest can promote ATP hydrolysis in hetero-oligomers of torsinA with LAP1 or LULL1 (Brown et al., 2014; Sosa et al., 2014). We therefore examined the importance of the putative arginine finger (R449) and surrounding residues in LULL1 for targeting torsinA to the INM. We find that torsinA relocalization tolerates several mutations in this region (D441A, S444A, L446R) (Fig. 5D–G), but mutating R449 (R449A) prevents LULL1-mediated relocalization of torsinA (Fig. 5F,G). In addition, we find that non-conservative changes in adjacent residues (I450R and L453R) also prevent torsinA relocalization (Fig. 5G). These results confirm a specific role for LULL1 R449 in relocalizing torsinA, supporting the proposal that this residue functions in trans between subunits as an arginine finger. Taken together, these data support a model in which ATP hydrolysis and the coincident release of torsinA from LULL1 increase torsinA mobility and access to the INM.
Essential role for LULL1 cytoplasmic domain mediated oligomerization in torsinA redistribution
The luminal domains of LAP1 and LULL1 are sufficient to bind torsinA and stimulate its ATPase activity (Brown et al., 2014; Sosa et al., 2014; Zhao et al., 2013). However, extraluminal domains are present in all torsin-activating proteins (Sosa et al., 2014). Although the extraluminal LAP1 domain binds nuclear lamins (Goodchild and Dauer, 2005; Kondo et al., 2002), the role of the LULL1 cytosolic domain is unknown. We investigated this using a panel of LULL1 deletion mutants (Fig. 6A). This revealed that LULL1-mediated torsinA relocalization tolerated deletion of the first 50, but not the first 100 amino acids of the 207-amino-acid LULL1 cytosolic domain (Fig. 6B–E). Intriguingly, we also found that torsinA-relocalizing activity was restored by replacing the cytoplasmic domain of LULL1 with the PK domain used in the size exclusion experiments above (Fig. 6A,F; supplementary material Fig. S3). There is no sequence homology between the LULL1 cytosolic domain and PK. However, PK is a well-characterized oligomer (Gupta and Bamezai, 2010; Sinning et al., 1993). We therefore wondered whether the LULL1 cytosolic domain might activate torsinA by promoting LULL1 oligomerization and productive heterohexamer assembly. Given that oligomer assembly is sensitive to protein concentration, this could explain why increasing LULL1 expression redistributes torsinA (Vander Heyden et al., 2009).
Fig. 6.

The LULL1 cytosolic domain causes LULL1 homo-oligomerization that is required for torsinA relocalization. (A) A series of truncated LULL1 mutants and fusion proteins to study domain function in torsinA relocalization. (B–D) TorsinA is concentrated on the INM of cells co-expressing full-length LULL1 (B) or LULL1 lacking only the first 50 amino acids of the cytosolic domain (D), but remains in the peripheral ER when expressed with LULL1 carrying larger cytosolic domain deletions (C). Images show the localization of GFP fluorescence in U2OS cells stably expressing wild-type torsinA–mGFP (left panels) and transfected with LULL1–Myc proteins (right panels). (E) LULL1-mediated torsinA relocalization requires regions of the LULL1 cytosolic domain. Mean±s.e.m. of the percentage of U2OS cells where wild-type torsinA–mGFP concentrated in the nuclear envelope in response to co-expression of full-length LULL1 or LULL1 deletion mutants. Only the first 50 amino acids of the LULL1 cytosolic domain were unnecessary for torsinA relocalizing activity. (F) A heterologous domain functionally compensates for deletion of the LULL1 cytoplasmic region. Columns show the mean±s.e.m. of the nuclear envelope localization index from n>20 CHO cells transfected with GFP–torsinA-ΔE302/3 and a transmembrane control or LULL1 proteins. ***P<0.001 for the nuclear envelope localization index compared with control (one-way ANOVA with Bonferroni post-hoc analysis). (G) LULL1 overexpression induces high molecular mass LULL1 complexes. Left panel shows that anti-LULL1 western blotting of BN-PAGE-separated U2OS whole-cell lysates detects a single species, and that overexpressed LULL1–Myc (after tetracycline induction) also exists in a higher molecular mass complex. (H) The chemical dimerizer AP20187 induces oligomerization of 2×FKBP LULL1 208-470. Anti-Myc western blotting of CN-PAGE-separated whole-cell lysates from U2OS cells stably expressing torsinA–mGFP and transfected with Myc-tagged 2×FKBP LULL1 208-470, in the absence or presence of the AP20187. (I) Addition of chemical dimerizer induces torsinA relocalization when co-expressed with otherwise inactive 2×FKBP LULL1 153-470. Images show U2OS cells stably expressing torsinA–mGFP (left panels) and transfected with Myc-tagged 2×Dmrb LULL1 153-470 LULL1 (right panels). Although the anti-GFP and Myc signals are both localized in the peripheral ER of untreated cells, AP20187 addition induces torsinA–mGFP relocalization to the INM. (J) The chemical dimerizer AP20187 significantly increases the torsinA-relocalizing activity of FKBP–LULL1 fusion proteins. Columns show the mean±s.e.m of the percentage of U2OS cells with nuclear-envelope-concentrated wild-type torsinA–mGFP in cells expressing either LULL1, 2×FKBP LULL1 153-470 or 2×FKBP LULL1 208-470, and in the presence or absence of AP20187. ****P<0.0001 difference in the percentage of cells with nuclear envelope concentrated torsinA (one-way ANOVA with Bonferroni post-hoc analysis). Scale bars: 10 µm.
We therefore asked whether LULL1, at the concentration needed to re-localize torsinA to the INM, assembles into oligomers. Indeed, we detected higher-order LULL1 oligomers when overexpressing LULL1 and not at the lower concentration natively present in U2OS cells (Fig. 6G). To specifically ask whether cytosolic-domain-mediated oligomerization is required for LULL1 to retarget torsinA to the INM, we replaced the LULL1 cytosolic region with a tandem repeat of a modified 12-kDa homodimerizing FKBP domain (Fig. 6A), which forms dimers and higher-order oligomers in the presence of rapamycin-analog dimerizers (Clackson et al., 1998; Schaupp et al., 2014). As expected, adding the rapamycin analog AP20187 generated higher-order LULL1 oligomers that we visualized by clear-native (CN)-PAGE (Fig. 6H). We then introduced 2×FKBP–LULL1-153-470 into cells expressing wild-type torsinA–mGFP. We observed that torsinA–mGFP maintained its normal distribution throughout the ER in cells expressing 2×FKBP–LULL1-153-470 or -208-470, but shifted to the nuclear envelope following addition of AP20187 (Fig. 6I,J). These data show that LULL1 homo-oligomerization, mediated by its cytosolic domain, is important for efficient redistribution of torsinA to the INM.
DISCUSSION
Although the majority of wild-type torsin proteins localize diffusely in the ER system (Basham and Rose, 2001; Goodchild and Dauer, 2004; Liang et al., 2014; Muraro and Moffat, 2006; Vander Heyden et al., 2009), the observations that ATP-trapped torsinA and disease-associated torsinA-ΔE302/3 concentrate in the nuclear envelope and are associated with defects in nuclear envelope morphology (Goodchild and Dauer, 2004; Goodchild et al., 2005; Jokhi et al., 2013; Kim et al., 2010; Liang et al., 2014; Naismith et al., 2004) attracted early and ongoing interest in the question of what torsinA does at the nuclear envelope. Indeed, recent studies implicate torsinA in large ribonucleoprotein export from the nucleus (Jokhi et al., 2013), herpes virus nuclear egress (Maric et al., 2011) and NPC insertion (VanGompel et al., 2015). What has attracted less attention, however, is the question of how torsinA concentrates in the nuclear envelope and especially whether and, if so, how, it reaches the INM. We now demonstrate that torsinA both reaches and concentrates on the INM. Although the ER and ONM are continuous, the INM is accessible during interphase only through NPC-associated membranes. Together with its slow mobility (Vander Heyden et al., 2009) and exclusion from curved membranes of the ER (Vander Heyden et al., 2011), it thus became clear that a significant barrier exists between the majority of torsinA and its subcellular site of activity. By examining factors that enable torsinA to enter the INM, we establish that LULL1 – recently shown to adopt a partial AAA+ domain fold – facilitates this entry in a reaction that requires its arginine finger motif and appears to at least transiently dissociate torsinA-containing complexes.
Several of our findings provide insight into how torsinA enters the INM and why this is facilitated by LULL1. Using native gel electrophoresis, we find oligomeric complexes of torsinA that can explain the restricted diffusion observed in vivo (Vander Heyden et al., 2009). We demonstrate that LULL1 both promotes formation of complexes containing torsinA and LULL1 (Fig. 4D) and drives their dissociation (Fig. 4F) in an activity-dependent reaction. These data support a model in which formation and subsequent disassembly of torsinA–LULL1 hetero-hexameric complexes releases monomeric torsinA from otherwise immobile high molecular mass structures to allow passage from the ER, across the NPC-associated membrane and onto the INM (Fig. 7).
Fig. 7.
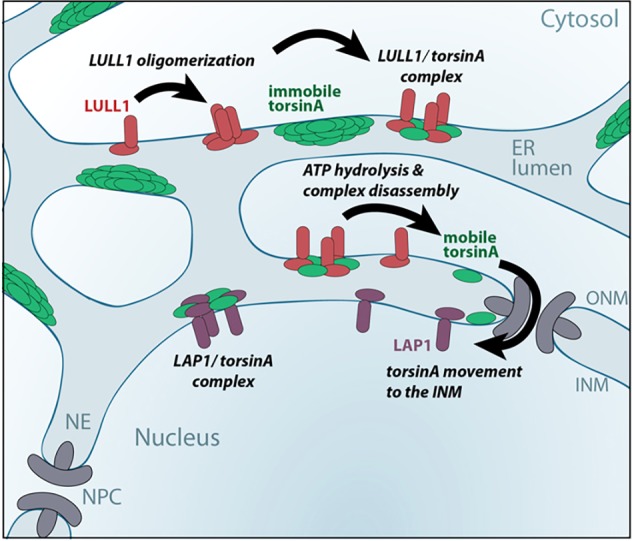
A model of how LULL1 relocalizes torsinA to the INM. In basal conditions the majority of torsinA is immobilized in the peripheral ER, depicted here as multimeric torsinA assemblies. Manipulations that induce LULL1 homo-oligomerization facilitate LULL1 co-assembly with torsinA to form hetero-hexameric ring structures. These torsinA–LULL1 hexamers have ATP-hydrolyzing activity (Brown et al., 2014; Sosa et al., 2014) and, typically, ATPase-induced AAA+ complex disassembly releases individual AAA+ proteins. Thus, our data suggest a model where that LULL1 oligomerization induces and disassembles LULL1–torsinA heterohexamers to release a short-lived diffusion-competent torsinA that might rebind LULL1, or reassemble into torsinA complexes, but also has the ability to access and traverse the curved NPC-associated membrane and enter the INM.
This model also explains why the LULL1 and LAP1 torsinA-activating proteins differentially affect localization of wild-type torsinA versus torsinA mutants with slowed ATP hydrolysis. Binding to LULL1 or LAP1 will recruit torsinA to their respective membrane if the rate of binding and complex assembly exceeds ATP-hydrolysis-driven complex disassembly. In contrast, when complex disassembly is rapid (e.g. when torsinA and oligomeric LULL1 co-assemble and efficiently induce ATP hydrolysis), potentially monomeric torsinA will be released to diffuse within the ER system including across the NPC-associated membrane and into the INM. In support of this, overexpressed LULL1 fails to colocalize with wild-type torsinA and facilitates trafficking into the INM, whereas ATPase-inhibited torsinA mutants bind and remain with LAP1 or LULL1 in their respective subcellular compartments (Goodchild and Dauer, 2005; Naismith et al., 2009). The fact that torsinA entering the INM after LULL1-driven complex dissociation tends to remain there rather than diffusing back to the ONM and peripheral ER might be the result of differences between LULL1 and LAP1 including the fact that LAP1 is a less potent activator of torsinA than LULL1 (Zhao et al., 2013) and is far less mobile than LULL1 (Zuleger et al., 2011; Vander Heyden et al., 2009). However, the picture underlying torsinA retention in the INM is complex and there is evidence that multiple factors, including additional INM-localized torsinA-binding partners, influence torsinA retention (Jungwirth et al., 2011).
This study also provides new insight into the cellular properties of the pathogenic torsinA-ΔE302/3 mutant. We show that the tendency to concentrate in the nuclear envelope that is a hallmark of this mutant reflects specific concentration in the INM. In addition, it was previously unclear whether the enhanced steady-state nuclear envelope-localization of torsinA-ΔE302/3 was due to enhanced entry or decreased exit from this domain; our data now suggest the mutation slows exit from the INM to the ER because inwards movement is not enhanced. What is more paradoxical is that torsinA-ΔE302/3 requires LULL1 to enter the nuclear envelope, yet biochemical analyses have consistently demonstrated that the ΔE302/3 mutation destabilizes binding to LULL1 and decreases ATPase activity (Naismith et al., 2009; Zhao et al., 2013). In fact, finding that torsinA-ΔE302/3 responds to LULL1 is in line with genetic demonstration that the mutant allele retains some activity (Liang et al., 2014). Our data suggest that the ΔE302/3 mutation decreases the LULL1-independent ability of torsinA to access the INM. Although we do not specifically explore the basis for LULL1-independent trafficking, we hypothesize that the normal equilibrium between torsinA monomer and oligomer is shifted by the ΔE302/3 mutation, making it more dependent on LULL1 for dissociation.
Importantly, the cytosolic domain of LULL1 is essential for its effects on torsinA migration to the INM and acts by inducing LULL1 oligomerization (Liang et al., 2014; Naismith et al., 2009; Zhao et al., 2013). This reveals a previously unknown role for this region in increasing the efficiency with which LULL1 engages and affects torsinA in the context of the ER membrane. Regulated assembly of active LULL1 multimers provides a pathway for transducing cytosolic signals to torsinA in the peripheral ER and then, through torsinA relocalization, to the INM. Although we found that the LULL1 cytosolic domain could be replaced by a heterologous multimerizing domain, there are undoubtedly important elements encoded in the endogenous protein structure which will in the future provide insight into the regulatory control of this process. Indeed, this cytosolic domain is a known target for phosphorylation (Dephoure et al., 2008) and conjugation of the ubiquitin-like FAT10 protein (Buchsbaum et al., 2012). It is estimated that the ER contains equivalent levels of torsinA and LULL1 (Beck et al., 2011; Brown et al., 2014; Goodchild and Dauer, 2005), and our induction of LULL1 expression leads to a situation where LULL1 levels exceed torsinA. We hypothesize that increased LULL1 levels stimulate torsinA relocalization because they promote LULL1 self-assembly and/or push the equilibrium towards torsinA binding LULL1. However, it is also possible that the extra LULL1 overpowers a constitutive inhibitory mechanism that otherwise suppresses efficient LULL1-mediated torsinA trafficking. Nevertheless, the sensitivity of torsinA to LULL1 levels and the role of LULL1 homo-multimerization are consistent with the need for more than one LULL1-supplied arginine finger for efficient hexamer ATPase activity and disassembly.
Our data clearly demonstrate that the amount of INM-localized torsinA depends on an ER-localized event and thus represents a novel example of an ER-licensing step controlling protein delivery to the INM. We do not determine whether LULL1 specifically facilitates torsinA diffusion though the curved NPC-associated membrane, or acts across the ER system. Biophysical studies assessing mobility of individual molecules will be needed to answer this question. Our study highlights events that regulate the ER licensing of torsinA movement to the INM; namely changes in LULL1 oligomerization and/or concentration. Rapid accumulation of torsinA in the INM might come into play when there are developmental and cell-type-specific needs for specifically localized torsinA activity (Goodchild et al., 2005; Liang et al., 2014). Given that previous work indicates that increased torsinA expression fails to increase (and can even inhibit) nuclear-envelope-localized torsinA activity (Kim et al., 2010), we hypothesize that LULL1-mediated ER-licensing is central to regulating torsinA activity at the INM in response to the physiological needs of the cell. Future work will build on these studies to define when cells change the localization of this developmentally essential enzyme and how it modifies the INM.
MATERIALS AND METHODS
Plasmids
Previously described plasmids include LULL1–Myc, LAP1C–Myc, torsinA-mGFP (Vander Heyden et al., 2009), torsinA-mGFP ATPase mutants (Naismith et al., 2009), Myc–LULL1, Myc–LAP1C, Myc–LULL1(210-470) (Goodchild and Dauer, 2005) and N-terminally tagged GFP–torsinA (Goodchild and Dauer, 2004). Transmembrane torsinA reporter constructs have a FLAG or Myc tag fused to residues 24–62 of chicken hepatic lectin (NM_205484; CHL) that includes its transmembrane domain (Soullam and Worman, 1995) replacing residues 1–40 of human torsinA. A C-terminal mGFP was added as previously described (Vander Heyden et al., 2009). FRB–TM–GFP and nuclear FKBP constructs are as described previously (Ohba et al., 2004). FRB–TM–torsinA–mGFP was constructed from FRB–TM–GFP by replacing GFP with sequence encoding torsinA–mGFP. FRB–LULL1–mGFP plasmids were generated by replacing TM–torsinA with LULL1 or PK–LULL1. Point mutations were introduced by QuikChange mutagenesis (Stratagene, La Jolla, CA). LULL1–Myc deletion mutants were made by PCR amplification of the desired fragments and ligation into pcDNA4/TO/MycHisB as described previously (Naismith et al., 2009). Other fusions to LULL1 [residues 17–476 of chicken pyruvate kinase (PK) (NCBI GenBank number AAA49021) (Soullam and Worman, 1995) and the Dmrb variant of FK506 binding protein (FKBP) (Clontech)] were introduced into an Nhe1 site as indicated in Myc–LULL1 (Goodchild and Dauer, 2005). Dmrb was inserted as a tandem dimer to enhance efficiency of oligomerization (Schaupp et al., 2014).
Cell culture
TorsinA–mGFP- and LULL1–Myc-expressing and untransfected U2OS cells have been described previously (Naismith et al., 2009; Vander Heyden et al., 2009). CAD, NIH-3T3 and CHO cells were cultured as described previously or recommended by the ATCC (Droggiti et al., 2011; Jungwirth et al., 2010). A cell line constitutively expressing torsinA–mGFP, while expressing LULL1–Myc under control of tetracycline was generated by introducing torsinA–mGFP into LULL1–Myc-inducible cells (Vander Heyden et al., 2009). Lull1+/+ and Lull1−/− mouse embryonic fibroblasts (MEFs) were produced as described previously (Kim et al., 2010), and immortalized by SV40 T-antigen transfection, selection with G418 and repeated passaging.
Transfections
Transient transfections were as follows. For U2OS, CAD and NIH-3T3 cells, Lipofectamine 2000 (Invitrogen) was used; for CHO cells, Lipofectamine LTX and Plus Reagent (Invitrogen) was used; and for MEFs electroporation with Neon™ system (Invitrogen) was used. siRNA against LAP1 (5′-CGUCUUUCCUCUAGUACUA-3′, 3′UTR) or against LULL1 (5′-GGACCUAUGGUUCCGUGUU-3′) were introduced using Dharmafect (GE Dharmacon) or Lipofectamine 2000 (Invitrogen), respectively. Knockdown of >90% of protein after 48 h was verified by western blotting.
Rapamycin dependent INM trap assay
The indicated plasmids were co-transfected at a 1:2 mass ratio (nuclear trap:test protein). NIH-3T3 cells were transferred 3 h after transfection cells onto collagen-coated coverslips, whereas MEFs were transferred 24 h after electroporation onto poly-L-lysine coated coverslips. Cells were cultured for 24 h before adding rapamycin (150 ng/ml, Sigma) for the time indicated. Cells were fixed and immunostained for GFP as described previously (Jungwirth et al., 2010). Trapping of FRB constructs in INM is quantified as the percentage of cells (from >50 individual cells) with clear GFP enrichment around nucleus.
Immunofluorescence
Cells were fixed and stained as described previously (Jungwirth et al., 2011; Vander Heyden et al., 2009). For selective permeabilization, fixed cells were incubated in buffer containing 0.1% Triton X-100 or 0.001% digitonin for 10 min on ice. Cells were imaged as indicated on (1) a Leica SP5 confocal with 63×1.4 NA objective, (2) an Olympus IX-81 with 60×1.42 NA objective (Olympus USA) and a FLASH 2.8 camera (Hamamatsu) using DAPI (EX387/11, 409DM), GFP (EX473/31, 495DM, EM520/35), mCherry (EX562/40, 593LP, EM641/75), and Cy5 (EX628/40, 660DM, EM692/40) filters or (3) a Nikon Eclipse Ti using Plan Fluor 100×1.3 NA objective, DS-QiMc camera and Chroma DAPI (31000), FITC (31001) and TRITC (31002) filters. Quantification of enrichment in the nuclear envelope used images with the focal plane midway through the nucleus as previously described (Jungwirth et al., 2011; Vander Heyden et al., 2009).
Cell and tissue preparation for BN-PAGE and CN-PAGE
Mouse liver microsomes were prepared and solubilized at ∼5 mg/ml for 30 min in BN-PAGE buffer containing 1% digitonin as described previously (Jungwirth et al., 2010) with insoluble material removed by two 20 min 100,000 g centrifugations. Cultured cell lysates were prepared in BN-PAGE or CN-PAGE buffer and subjected to electrophoresis and immunoblotting as described previously (Jungwirth et al., 2011; Vander Heyden et al., 2009; Wittig et al., 2006). Following electrophoresis, gels were equilibrated in transfer buffer for immunoblotting except individual lanes that were subject to Coomassie staining or second dimension SDS-PAGE.
Immunoprecipitations
For immunoprecipitation, cells were solubilized in buffer (50 mM imidazole-HCl pH 7.5, 5% glycerol, 0.5% digitonin, 4 mM MgCl2, 50 mM NaCl, protease inhibitor cocktail, 2 mM PMSF and nucleotides as indicated). Insoluble material was removed by centrifugation at 20,000 g. Lysates were incubated for 2 h with anti-GFP conjugated AminoLink resin (Pierce), washed and eluted with non-reducing 2× Laemmli buffer. Elutes and lysates were subjected to SDS-PAGE and immunoblotting.
Antibodies
Rabbit antibody recognizing the cytoplasmic domain of human LULL1 (residues 1–217) is as described previously (Vander Heyden et al., 2009) whereas that recognizing the nucleoplasmic domain of LAP1C (residues 1–217) was raised against GST–LAP1C, and affinity purified as described previously (Vander Heyden et al., 2009) and specificity confirmed using siRNA-treated cells (supplementary material Fig. S2D). Other antibodies used include rabbit anti-FLAG (Sigma, F7425), mouse anti-LAP2 (BD611074), mouse anti-Myc (Developmental Studies, clone 9E10), and rabbit anti-calreticulin (Sigma) antibodies. Rabbit antibodies recognizing GFP, torsinA and LULL1 are as previously described (Goodchild and Dauer, 2005; Jungwirth et al., 2010).
Supplementary Material
Acknowledgements
We wish to thank Dr Larry Gerace for plasmids encoding the rapamycin-based INM trapping system and Dr Howard Worman for plasmids CHL and PK. We acknowledge the Developmental Studies Hybridoma Bank for the anti-Myc mouse monoclonal antibody. We also want to thank Michael Jungwirth and Danielle Jeong for their help in quantifying fluorescent images.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
R.E.G. and P.I.H. designed and performed experiments, analyzed data and wrote the manuscript. A.L.B., K.H., T.V.N., K.B. and M.L.D. designed, performed and analyzed experiments. W.T.D. and C.-C.L. shared reagents.
Funding
Work in the Goodchild Laboratory was supported by the Foundation for Dystonia Research and the Dystonia Medical Research Foundation; and in the Hanson Laboratory by the National Institutes of Health [grant number R01 NS050717]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.167452/-/DC1
References
- Antonin W., Ungricht R. and Kutay U. (2011). Traversing the NPC along the pore membrane: targeting of membrane proteins to the INM. Nucleus 2, 87-91. 10.4161/nucl.2.2.14637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basham S. E. and Rose L. S. (2001). The Caenorhabditis elegans polarity gene ooc-5 encodes a Torsin-related protein of the AAA ATPase superfamily. Development 128, 4645-4656. [DOI] [PubMed] [Google Scholar]
- Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., Herzog F., Rinner O., Ellenberg J. and Aebersold R. (2011). The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549 10.1038/msb.2011.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. S. H., Zhao C., Chase A. R., Wang J. and Schlieker C. (2014). The mechanism of Torsin ATPase activation. Proc. Natl. Acad. Sci. USA 111, E4822-E4831. 10.1073/pnas.1415271111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum S., Bercovich B., Ziv T. and Ciechanover A. (2012). Modification of the inflammatory mediator LRRFIP2 by the ubiquitin-like protein FAT10 inhibits its activity during cellular response to LPS. Biochem. Biophys. Res. Commun. 428, 11-16. 10.1016/j.bbrc.2012.09.110 [DOI] [PubMed] [Google Scholar]
- Burns L. T. and Wente S. R. (2012). Trafficking to uncharted territory of the nuclear envelope. Curr. Opin. Cell Biol. 24, 341-349. 10.1016/j.ceb.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T., Yang W., Rozamus L. W., Hatada M., Amara J. F., Rollins C. T., Stevenson L. F., Magari S. R., Wood S. A., Courage N. L. et al. (1998). Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA 95, 10437-10442. 10.1073/pnas.95.18.10437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Las Heras J. I., Meinke P., Batrakou D. G., Srsen V., Zuleger N., Kerr A. R. W. and Schirmer E. C. (2013). Tissue specificity in the nuclear envelope supports its functional complexity. Nucleus 4, 460-477. 10.4161/nucl.26872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N., Zhou C., Villen J., Beausoleil S. A., Bakalarski C. E., Elledge S. J. and Gygi S. P. (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 105, 10762-10767. 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droggiti A., Ho C. C.-Y., Stefanis L., Dauer W. T. and Rideout H. J. (2011). Targeted disruption of neuronal 19S proteasome subunits induces the formation of ubiquitinated inclusions in the absence of cell death. J. Neurochem. 119, 630-643. 10.1111/j.1471-4159.2011.07444.x [DOI] [PubMed] [Google Scholar]
- Edbauer D., Winkler E., Haass C. and Steiner H. (2002). Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc. Natl. Acad. Sci. USA 99, 8666-8671. 10.1073/pnas.132277899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles L. M., Chen J., Li L. and Chin L.-S. (2008). Dystonia-associated mutations cause premature degradation of torsinA protein and cell-type-specific mislocalization to the nuclear envelope. Hum. Mol. Genet. 17, 2712-2722. 10.1093/hmg/ddn173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild R. E. and Dauer W. T. (2004). Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. USA 101, 847-852. 10.1073/pnas.0304375101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild R. E. and Dauer W. T. (2005). The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 168, 855-862. 10.1083/jcb.200411026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild R. E., Kim C. E. and Dauer W. T. (2005). Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48, 923-932. 10.1016/j.neuron.2005.11.010 [DOI] [PubMed] [Google Scholar]
- Gupta V. and Bamezai R. N. K. (2010). Human pyruvate kinase M2: a multifunctional protein. Protein Sci. 19, 2031-2044. 10.1002/pro.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokhi V., Ashley J., Nunnari J., Noma A., Ito N., Wakabayashi-Ito N., Moore M. J. and Budnik V. (2013). Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 3, 988-995. 10.1016/j.celrep.2013.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungwirth M., Dear M. L., Brown P., Holbrook K. and Goodchild R. (2010). Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum. Mol. Genet. 19, 888-900. 10.1093/hmg/ddp557 [DOI] [PubMed] [Google Scholar]
- Jungwirth M. T., Kumar D., Jeong D. Y. and Goodchild R. E. (2011). The nuclear envelope localization of DYT1 dystonia torsinA-DeltaE requires the SUN1 LINC complex component. BMC Cell Biol. 12, 24 10.1186/1471-2121-12-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. E., Perez A., Perkins G., Ellisman M. H. and Dauer W. T. (2010). A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. USA 107, 9861-9866. 10.1073/pnas.0912877107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. C., Lusk C. P. and Blobel G. (2006). Karyopherin-mediated import of integral inner nuclear membrane proteins. Nature 442, 1003-1007. 10.1038/nature05075 [DOI] [PubMed] [Google Scholar]
- Kock N., Naismith T. V., Boston H. E., Ozelius L. J., Corey D. P., Breakefield X. O. and Hanson P. I. (2006). Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 15, 1355-1364. 10.1093/hmg/ddl055 [DOI] [PubMed] [Google Scholar]
- Kondo Y., Kondoh J., Hayashi D., Ban T., Takagi M., Kamei Y., Tsuji L., Kim J. and Yoneda Y. (2002). Molecular cloning of one isotype of human lamina-associated polypeptide 1s and a topological analysis using its deletion mutants. Biochem. Biophys. Res. Commun. 294, 770-778. 10.1016/S0006-291X(02)00563-6 [DOI] [PubMed] [Google Scholar]
- Laba J. K., Steen A. and Veenhoff L. M. (2014). Traffic to the inner membrane of the nuclear envelope. Curr. Opin. Cell Biol. 28, 36-45. 10.1016/j.ceb.2014.01.006 [DOI] [PubMed] [Google Scholar]
- Liang C.-C., Tanabe L. M., Jou S., Chi F. and Dauer W. T. (2014). TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Invest. 124, 3080-3092. 10.1172/JCI72830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison C., Pyrpasopoulou A., Theodoropoulos P. A. and Georgatos S. D. (1997). The inner nuclear membrane protein LAP1 forms a native complex with B-type lamins and partitions with spindle-associated mitotic vesicles. EMBO J. 16, 4839-4850. 10.1093/emboj/16.16.4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric M., Shao J., Ryan R. J., Wong C.-S., Gonzalez-Alegre P. and Roller J. J. (2011). A functional role for TorsinA in herpes simplex virus 1 nuclear egress. J. Virol. 85, 9667-9679. 10.1128/JVI.05314-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L., Crimaudo C. and Gerace L. (1995). cDNA cloning and characterization of lamina-associated polypeptide 1C (LAP1C), an integral protein of the inner nuclear membrane. J. Biol. Chem. 270, 8822-8828. 10.1074/jbc.270.15.8822 [DOI] [PubMed] [Google Scholar]
- Muraro N. I. and Moffat K. G. (2006). Down-regulation of torp4a, encoding the Drosophila homologue of torsinA, results in increased neuronal degeneration. J. Neurobiol. 66, 1338-1353. 10.1002/neu.20313 [DOI] [PubMed] [Google Scholar]
- Naismith T. V., Heuser J. E., Breakefield X. O. and Hanson P. I. (2004). TorsinA in the nuclear envelope. Proc. Natl. Acad. Sci. USA 101, 7612-7617. 10.1073/pnas.0308760101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naismith T. V., Dalal S. and Hanson P. I. (2009). Interaction of TorsinA with its major binding partners is impaired by the dystonia-associated ΔGAG deletion. J. Biol. Chem. 284, 27866-27874. 10.1074/jbc.M109.020164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery F. C., Zeng J., Niland B. P., Hewett J., Farley J., Irimia D., Li Y., Wiche G., Sonnenberg A. and Breakefield X. O. (2008). TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J. Cell Sci. 121, 3476-3486. 10.1242/jcs.029454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T., Whiteheart S. W. and Wilkinson A. J. (2004). Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 146, 106-112. 10.1016/j.jsb.2003.11.008 [DOI] [PubMed] [Google Scholar]
- Ohba T., Schirmer E. C., Nishimoto T. and Gerace L. (2004). Energy- and temperature-dependent transport of integral proteins to the inner nuclear membrane via the nuclear pore. J. Cell Biol. 167, 1051-1062. 10.1083/jcb.200409149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius L. J., Hewett J. W., Page C. E., Bressman S. B., Kramer P. L., Shalish C., de Leon D., Brin M. F., Raymond D., Corey D. P. et al. (1997). The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 17, 40-48. 10.1038/ng0997-40 [DOI] [PubMed] [Google Scholar]
- Putyrski M. and Schultz C. (2012). Protein translocation as a tool: the current rapamycin story. FEBS Lett. 586, 2097-2105. 10.1016/j.febslet.2012.04.061 [DOI] [PubMed] [Google Scholar]
- Schaupp A., Sabet O., Dudanova I., Ponserre M., Bastiaens P. and Klein R. (2014). The composition of EphB2 clusters determines the strength in the cellular repulsion response. J. Cell Biol. 204, 409-422. 10.1083/jcb.201305037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer E. C., Florens L., Guan T., Yates J. R. III and Gerace L. (2003). Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 301, 1380-1382. 10.1126/science.1088176 [DOI] [PubMed] [Google Scholar]
- Sinning I., Kleywegt G. J., Cowan S. W., Reinemer P., Dirr H. W., Huber R., Gilliland G. L., Armstrong R. N., Ji X., Board P. G. et al. (1993). Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J. Mol. Biol. 232, 192-212. 10.1006/jmbi.1993.1376 [DOI] [PubMed] [Google Scholar]
- Sosa B. A., Demircioglu F. E., Chen J. Z., Ingram J., Ploegh H. L. and Schwartz T. U. (2014). How lamina-associated polypeptide 1 (LAP1) activates Torsin. Elife 3, e03239 10.7554/eLife.03239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soullam B. and Worman H. J. (1995). Signals and structural features involved in integral membrane protein targeting to the inner nuclear membrane. J. Cell Biol. 130, 15-27. 10.1083/jcb.130.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heyden A. B., Naismith T. V., Snapp E. L., Hodzic D. and Hanson P. I. (2009). LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol. Biol. Cell 20, 2661-2672. 10.1091/mbc.E09-01-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heyden A. B., Naismith T. V., Snapp E. L. and Hanson P. I. (2011). Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum. EMBO J. 30, 3217-3231. 10.1038/emboj.2011.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGompel M. J. W., Nguyen K. C. Q., Hall D. H., Dauer W. T. and Rose L. S. (2015). A novel function for the C. elegans Torsin OOC-5 in nucleoporin localization and nuclear import. Mol. Biol. Cell 26, 1752-1763. 10.1091/mbc.E14-07-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi-Ito N., Doherty O. M., Moriyama H., Breakefield X. O., Gusella J. F., O'Donnell J. M. and Ito N. (2011). dtorsin, the Drosophila ortholog of the early-onset dystonia TOR1A (DYT1), plays a novel role in dopamine metabolism. PLoS ONE 6, e26183 10.1371/journal.pone.0026183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I., Braun H.-P. and Schägger H. (2006). Blue native PAGE. Nat. Protoc. 1, 418-428. 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- Wu W., Lin F. and Worman H. J. (2002). Intracellular trafficking of MAN1, an integral protein of the nuclear envelope inner membrane. J. Cell Sci. 115, 1361-1371. [DOI] [PubMed] [Google Scholar]
- Zhao C., Brown R. S. H., Chase A. R., Eisele M. R. and Schlieker C. (2013). Regulation of Torsin ATPases by LAP1 and LULL1. Proc. Natl. Acad. Sci. USA 110, E1545-E1554. 10.1073/pnas.1300676110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Millen L., Mendoza J. L. and Thomas P. J. (2010). A unique redox-sensing sensor II motif in TorsinA plays a critical role in nucleotide and partner binding. J. Biol. Chem. 285, 37271-37280. 10.1074/jbc.m110.123471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuleger N., Kelly D. A., Richardson A. C., Kerr A. R. W., Goldberg M. W., Goryachev A. B. and Schirmer E. C. (2011). System analysis shows distinct mechanisms and common principles of nuclear envelope protein dynamics. J. Cell Biol. 193, 109-123. 10.1083/jcb.201009068 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.