

Abstract
The study of touch-evoked behavior allows investigation of both the cells and circuits that generate a response to tactile stimulation. We investigate a touch-insensitive zebrafish mutant, macho (maco), previously shown to have reduced sodium current amplitude and lack of action potential firing in sensory neurons. In the genomes of mutant but not wild-type embryos, we identify a mutation in the pigk gene. The encoded protein, PigK, functions in attachment of glycophosphatidylinositol anchors to precursor proteins. In wild-type embryos, pigk mRNA is present at times when mutant embryos display behavioral phenotypes. Consistent with the predicted loss of function induced by the mutation, knock-down of PigK phenocopies maco touch insensitivity and leads to reduced sodium current (INa) amplitudes in sensory neurons. We further test whether the genetic defect in pigk underlies the maco phenotype by overexpressing wild-type pigk in mutant embryos. We find that ubiquitous expression of wild-type pigk rescues the touch response in maco mutants. In addition, for maco mutants, expression of wild-type pigk restricted to sensory neurons rescues sodium current amplitudes and action potential firing in sensory neurons. However, expression of wild-type pigk limited to sensory cells of mutant embryos does not allow rescue of the behavioral touch response. Our results demonstrate an essential role for pigk in generation of the touch response beyond that required for maintenance of proper INa density and action potential firing in sensory neurons.
Keywords: touch response, Nav, glycophosphatidylinositol-anchored protein, Pigk, Rohon-Beard cells
the zebrafish macho (macott261/tt261) mutant was isolated in a large-scale genetic screen for locomotor and behavioral defects (Granato et al. 1996). maco (formerly mao) mutants swim but have a reduced response to touch evident from the time tactile sensitivity first appears (Granato et al. 1996). In addition, maco mutants display aberrant mapping of retinal ganglion cell axons to the tectum, an activity-dependent process (Gneugge et al. 2001; Trowe et al. 1996). Similar to other zebrafish mutations in genes with essential functions, maco is recessive and homozygous mutants survive past embryonic stages and die as larvae. maco mutants also have decreased voltage-gated sodium current (INa) densities and fail to fire action potentials in mechanosensory Rohon-Beard (RB) and retinal ganglion cells (Gnuegge et al. 2001; Ribera and Nüsslein-Volhard 1998). Although RB cells normally exhibit extensive programmed cell death during embryonic and larval stages, these neurons persist longer in maco mutants (Svoboda et al. 2001).
That maco mutants have RBs with reduced INa density and loss of action potential firing suffices, in principle, to explain their behavioral touch sensitivity and prolonged RB survival (Ribera and Nüsslein-Volhard 1998; Svoboda et al. 2001). Similarly, the reduced INa density and lack of action potential firing by retinal ganglion cells in maco mutants can account for their behavioral blindness and mapping errors of retinal ganglion cell axons (Gnuegge et al. 2001; Neuhauss et al. 1999; Trowe et al. 1996).
As INa is reduced in RB and retinal ganglion cells of maco mutants, a reasonable candidate for the affected gene would be one encoding a structural subunit of a voltage-gated sodium (Nav) channel. However, the maco mutation does not map to any of the genomic loci of zebrafish sodium channel α-subunit (scna) or β-subunit (scnb) genes (Chopra et al. 2007; Fein et al. 2008; Novak et al. 2006a,b). In view of this, the maco mutation may reside in a gene that acts to regulate sodium channel function, especially in a developmental context, about which little is known.
In this study, we identify pigk as the gene harboring the maco mutation. The encoded protein, PigK, associates with other subunits to form the transamidase complex that attaches glyocophosphatidylinositol (GPI) residues to immature proteins (Zacks and Garg 2006). pigk is a member of the PIG gene family that encodes over 20 different proteins that reside within the endoplasmic reticulum and are involved either in the synthesis or attachment of GPI anchors to nascent proteins (for review, see Chatterjee and Mayor 2001; Kinoshita 2014; Zacks and Garg 2006). GPI-anchored proteins (GPI-APs) travel through the secretory pathway, and, upon exocytosis, are tethered to the extracellular face of the surface membrane. A previously reported zebrafish mutation, pigu, targets another subunit of the GPI transamidase complex and also results in touch insensitivity and reduced RB INa density (Nakano et al. 2010).
Here, we investigate questions regarding the functional consequences of the maco mutation that is predicted to result in pigk loss-of-function. We also test whether the expression of wild-type (WT) pigk rescues deficits present in maco mutants. Ubiquitous expression of wild-type pigk effectively rescues the touch response in maco mutants, demonstrating that the maco mutation underlies the behavioral phenotype. Further, for maco mutants, expression of wild-type (WT) pigk limited to sensory neurons rescues RB INa amplitudes and the ability to fire an action potential. However, limiting WT pigk expression to RBs does not restore the touch response to maco mutants. Thus we propose that GPI-APs play several essential roles in the touch response.
MATERIALS AND METHODS
Animal care.
All animal care was approved by and performed in accordance with the University of Colorado Institutional Animal Care and Use Committee and animal care guidelines. Zebrafish (Danio rerio) adults were housed in the University of Colorado Anschutz Medical Campus Zebrafish facility. Zebrafish care and breeding were performed as described by Westerfield (1993). Embryos were maintained at 28.5°C in embryo media (EM; in mM: 130 NaCl, 0.5 KCl, 0.02 Na2HPO4, 0.04 KH2PO4, 1.3 CaCl2, 1.0 MgSO4, and 0.4 NaH2CO3, pH 7.2). Developmental staging was done on the basis of external morphological criteria (Kimmel et al. 1995).
Mapping and sequencing.
The macott261/tt261 mutation was mapped to a linkage group and marker using bulked segregant analysis. Fine mapping and sequencing were performed using a Tübingen background at the zebrafish mapping facility at the University of Louisville using previously published methods (Willer et al. 2005).
Reverse transcription and real-time quantitative PCR (RT-QPCR).
RNA was extracted from WT embryos at ∼0.5 (one-cell stage), 22 (25 somite stage), 36, and 48 h postfertilization (hpf) using TRIzol (Ambion, Life Technologies, Grand Island, NY) and Qiagen RNeasy mini kits (Valencia, CA). The extracted RNA was treated with Amplification Grade DNase (Invitrogen, Life Technologies, Carlsbad, CA). RNA was electrophoretically separated on a formaldehyde agarose gel and stained with ethidium bromide. Prominent 16S and 28S stained RNA bands, with 28S band twice as intense as the 16S one, were criteria for adequate RNA integrity.
One Step Super Script kit (Applied Biosystems, Life Technologies) was used for reverse transcription and gene expression on a 7500 Fast Real-Time PCR Instrument (Applied Biosystems, Life Technologies).
The Vector and QPCR Core at the University of Colorado Anschutz Medical Campus performed real-time quantitative polymerase chain reaction (QPCR) assays. All QPCR experiments were performed as biological replicates, that were each analyzed technically in triplicate. Proprietary assays with primers and probes were purchased from Applied Biosystems (Assay ID Dr03082697_m1). The assay spanned the junctions of pigk exons 8 and 9. Cycling conditions were adjusted to those specified in the One Step Super Script kit (Applied Biosystems, Life Technologies). Data were analyzed with ABI 7500 software (version 2.0.6, Applied Biosystems) using all default parameters except that the threshold for the CT standard deviation was decreased from >0.5 to >0.3 to be more stringent. The relative standard curve method was used, and gene expression was normalized to that of the beta actin 2 gene, actb2. The validity of actb2 as the endogenous control was tested by measuring the CT standard deviation of all samples when assayed at equal concentrations. The standard deviation was consistently below 0.5, indicating that actb2 did not vary significantly between samples. Previous work has also validated actb2 as a housekeeping gene appropriate for normalization of zebrafish QPCR results (Casadei et al. 2011).
Morpholino knockdown experiments.
GeneTools (Philomath, OR) designed a translation blocking antisense morpholino (MO) oligonucleotide against a pigk 5′-sequence that included and flanked the start codon (ATG MO; AATAATCCATTATCGTTCGTCACCG). The control MO (CTL MO) was similar to the ATG MO but had 5 mismatched bases (AAaAATCgATTATgGTTCcTCAgCG). MOs were injected into WT embryos at the one-cell stage in volumes of ∼1 nl. The ATG MO was injected at doses ranging between 0.5 and 3.5 ng/nl, while the CTL MO was injected at the highest dose only (3.5 ng/nl).
Touch assay.
Embryos were assayed for touch responsiveness between 30 and 48 hpf, times at which WT embryos respond to touch. The assay consisted of touching the dorsal region of the trunk above the yolk or yolk sac extension 10 times per embryo. Each response was given a score of either one for a robust swimming response, 0.5 for a weak or abnormal response, and zero for no response. Scores were summed for each embryo, with the maximum score being 10 (Pineda et al. 2005). Please note that genotyping (see below) was performed subsequent to behavioral testing, ensuring that responses were scored without knowledge of genotype (blinded).
Genotyping.
Genotyping was performed on embryos produced by a macott261/+ incross, in which 25% were expected to be touch responsive mutants and 75% touch responsive WT and heterozygous embryos (siblings). The maco mutation does not introduce a change in restriction enzyme sequence, which is normally the basis for an easy in-house genotype assay. Consequently, a commercial vendor (Transnetyx, Cordova, TN) performed the genotyping using a specific proprietary probe for pigk.
Transgenic expression of pigk.
Transgenic expression of WT pigk and mutant pigk (pigk_M1T) was achieved using Tol2 constructs (Kwan et al. 2007). Tol2 plasmids were constructed with either the ubiquitin B (ubb) promoter (Mosimann et al. 2011) for ubiquitous expression, or the CREST3 enhancer (kindly provided by Dr. Alvaro Sagasti, UCLA; Higashijima et al. 2000; Palanca et al. 2013; Uemura et al. 2005) for selective expression in sensory neurons. The 3′-element of the transgene contained an IRES-EGFP (Kwan et al. 2007). On the basis of GFP fluorescence, CREST3, but not ubb, allowed for successful translation of the IRES-EGFP reporter. The final Tol2 construct also included a transgene with the cardiac myosin light chain promoter driving expression of GFP, to allow rapid screening of transgenesis by examination of GFP expression in hearts.
Transposase RNA was synthesized in vitro using the pCS2FA-transposase plasmid (Kawakami 2004). The plasmid was linearized using NotI-HF (New England Biolabs, Ipswich, MA), purified (Qiagen PCR Purification Kit, Valencia, CA), and RNA was transcribed in vitro (mMessage mMachine SP6 kit, Ambion, Life Technologies, Grand Island, NY). The RNA was then purified using the Qiagen RNAeasy mini kit (Valencia, CA).
Approximately 25 pg of the Tol2 construct and 25 pg of transposase RNA were coinjected into embryos of WT clutches or macott261/+ incrosses at the one-cell stage. Embryos with GFP+ hearts were assayed for touch responsiveness at 48 hpf, as described above.
Confocal microscopy.
Embryos were mounted laterally and imaged using a 3i Marianas spinning disc confocal microscope [Intelligent Imaging Innovations (3i), Denver, CO]. For all embryos at a given stage, the same settings were used to acquire sense and antisense images. Images were subsequently processed and analyzed using ImageJ (Schneider et al. 2012).
Embryo preparation and electrophysiology.
For electrophysiological study, we mounted embryos on glass slides in the presence of Tricaine (MS-222, Sigma-Aldrich, St. Louis, MO) using Vetbond (3M, St. Paul, MN) as described previously (Ribera and Nüsslein-Volhard 1998). Embryos were then killed by transection of the hindbrain and the skin was removed with glass dissecting needles. Tricaine was then washed out with at least three rinses of the recording solution over a period of 5 min. Using a glass dissecting needle, the meninges covering the dorsal side of the spinal cord was cut to reveal RB cells.
All recordings were performed at room temperature using an Axopath 200B amplifier (Axon Instruments/Molecular Devices, Sunnyvale, CA). Data were acquired with pClamp10 and analyzed with Clampfit10 (Molecular Devices, Sunnyvale, CA). INa and action potential recordings were performed on RB cells in either uninjected embryos or ones injected with a transgenic construct that allowed for CREST3-driven RB-selective expression of WT or mutant pigk (pigk-IRES-EGFP or pigk_M1T-IRES-EGFP, respectively). In injected embryos, recordings were obtained from RB cells expressing GFP. Genetic identity (maco+/+, macott261/+, or macott261/tt261) was determined after electrophysiology as described above. As genotyping was done subsequent to electrophysiology, recordings were obtained blinded to knowledge of genotype.
Voltage-clamp recordings of RB INa of 48 hpf maco sibling (maco+/+, macott261/+) and mutant (macott261/tt261) embryos were performed as previously described (Ribera and Nüsslein-Volhard 1998). Cells were held at −80 mV, and INa was elicited by a series of 20-ms depolarizing voltage steps in 10-mV increments. The bath solution consisted of (in mM) 135 NaCl, 3 KCl, 20 TEA-Cl, 5 MnCl2 (4 H2O), 5 HEPES (pH 7.4; 310 mOsm). The pipette solution contained (in mM) 120 N-methyl-d-glucamine, 20 TEA-Cl, 11 EGTA, 1 CaCl2 (2H2O), 5 NaCl, 10 HEPES (pH 7.2; 300 mOsm). Glass electrodes with resistances varying between 1.1 and 7.1 MΩ (average = 3.8 ± 1.5 MΩ) were used.
Current-clamp methods were used to elicit and record RB active membrane responses to current injection, as previously described (Moreno and Ribera 2009). The intracellular pipette solution contained (in mM) 135 KCl, 10 EGTA, and 10 HEPES, pH 7.4. The extracellular bath solution consisted of (in mM): 125 NaCl, 2 KCl, 10 CaCl2, and 5 HEPES, pH 7.4. α-Bungarotoxin (1 μM) was included in the bath to immobilize embryos during recordings. To evoke RB active responses (e.g., action potentials), we injected depolarizing current, 1 ms in duration, that increased by 0.05 nA in each subsequent trial. The interstimulus interval was 1 s to avoid possible inactivation of sodium channels.
To compare the properties of the different types of responses evoked by current injection, we measured their amplitudes and rates of rise (dV/dt). For each cell, we analyzed the response that occurred in the trial after the first one that had an active response. In independent experiments, we verified that the small-amplitude responses were active by confirming their sensitivity to 1 μM tetrodotoxin. The amplitude was calculated as the difference between the resting membrane potential and the active response peak. The rate of rise was calculated for the period of time between the end of current injection and peak of the active response. To be scored as an action potential, an active response had to meet two criteria: 1) amplitude ≥ 20 mV; 2) dV/dt > 40 mv/ms. We also measured the minimum amount of current required to generate an active response (rheobase).
Statistical analysis.
Statistical significance was assessed using nonparametic tests (Mann-Whitney, Kruskal-Wallis) with post hoc correction (Bonferroni Dunn) for multiple comparisons using InStat software (GraphPad, La Jolla, CA). P values < 0.05 were indicative of statistical significance and are provided in the figure legends. As indicated in the figure legends, data are presented either as means ± SD, means ± SE, or box plots that indicate the median, 25th, 75th, and 90th percentiles.
RESULTS
Maco mutants carry a mutation within the pigk gene.
The maco mutation was first mapped to the second chromosome using bulked segregant analysis and closely linked to Simple Sequence Length Polymorphism (SSLP) marker zC199A5 (Fig. 1A). Sequencing and comparison of WT, mutant, and sibling DNA identified the pigk gene as the locus of a mutation present in homozygous mutants but not WT embryos.
Fig. 1.
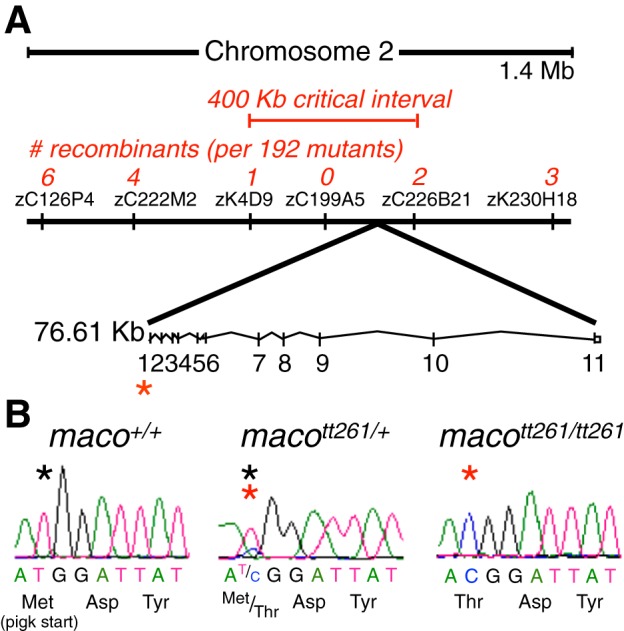
macott261/tt261 embryos have a mutation in the pigk start codon. A: bulked segregant analysis was first used to map the macott261 mutation to a region of Chromosome 2. Subsequent fine mapping with Simple Sequence Length Polymorphism (SSLP) markers identified a region with no recombinants. This area was sequenced, revealing the macott261 locus in the pigk start codon. Asterisks designate the locus of the maco mutation. B: sequence chromatograms revealed a thymidine to cytosine substitution in the pigk start codon. Asterisks denote the residue of interest (black asterisk, wild-type sequence; red asterisk, mutant sequence).
The mutation consists of a thymidine to cytosine substitution in the pigk start codon, corresponding to an amino acid change of methionine to threonine (Fig. 1B). The next potential in-frame start codon occurs downstream of the signal sequence at amino acid 82. These results suggest that the identified mutation would result in loss of the signal sequence peptide that is required for translocation to the endoplasmic reticulum lumen, the normal site of PigK transamidase activity.
For understanding the consequences of gene mutation in zebrafish, it is important to consider the possibility of a fully or partially redundant gene due to the genomewide duplication event that occurred during teleost evolution (Amores et al. 1998; Holland and Williams 1990; Lopreato et al. 2001; Stock et al. 1996; Vandepoele et al. 2004). Given that maco mutants do have specific deficits and ultimately die as larvae, if a duplicated pigk gene were to exist, it only partially compensates for loss of function of the one studied here. Further, search of the most recent Genome Resource Consortium GRCz10 reference assembly makes this possibility unlikely since we found no potential candidate for a duplicated pigk gene.
pigk mRNA is present at the one-cell stage and embryonic stages.
GPI-APs are present in most cells, have diverse functions, and comprise ∼0.5% of proteins in a wide range of eukaryotes (Eisenhaber at al. 2001; for review, see Kinoshita et al. 2008; Kinoshita 2014). On this basis, we expected pigk to be expressed during embryogenesis. To investigate the temporal expression pattern of pigk mRNA, we performed reverse transcription on RNA extracted from WT and maco mutant embryos at several stages (one-cell; 24, 36, and 48 hpf), that was then followed by real-time quantitative PCR. We detect the presence of pigk mRNA at the one-cell stage (Fig. 2A), prior to the onset of zygotic transcription, indicating that the mRNA is maternally provided (Kane and Kimmel 1993). In WT embryos, pigk mRNA expression occurs throughout embryogenesis (Fig. 2B). Further, between 22 and 48 hpf, normalized pigk mRNA levels increase (P < 0.05; Fig. 2B).
Fig. 2.
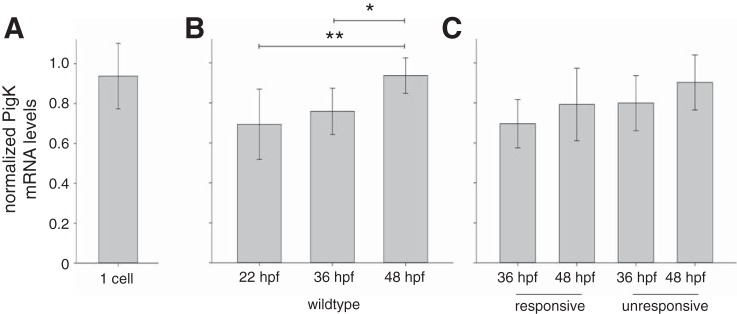
pigk mRNA is maternally expressed and detected during embryonic stages of development. To test for the presence of pigk mRNA during embryonic development, we performed qPCR using RNA extracted from wild-type (WT) embryos, touch-unresponsive maco embryos, and their responsive siblings. Data are presented as means ± SD, with pigk mRNA levels normalized to those of actb2 mRNA (see materials and methods). A: qPCR detected pigk mRNA at the one-cell stage, indicating that the transcripts are maternally provided. B: in WT embryos, pigk expression levels increased between 22 and 48 h postfertilization (hpf). *P < 0.05, **P < 0.01. C: both responsive (maco+/+ and macott261/+, pooled) and unresponsive (macott261/tt261) sibling embryos expressed pigk mRNA at 36 and 48 hpf. At both time points, unresponsive and responsive siblings had similar pigk expression levels.
We also compared the temporal expression of pigk RNA in maco touch unresponsive mutants and their responsive siblings. We detect pigk mRNA in both touch-responsive sibling and touch-unresponsive maco embryos. Moreover, the normalized levels of pigk mRNA are not decreased in unresponsive maco embryos (Fig. 2C). These results indicate that the maco mutation does not lead to nonsense-mediated decay of pigk transcripts.
PigK knockdown phenocopies maco embryonic touch insensitivity and reduced RB INa amplitudes.
As mentioned, the identified mutation in the pigk gene will eliminate the encoded protein's signal sequence, required for translocation to the endoplasmic reticulum lumen, the site of PigK's transamidase function (Fig. 1). Accordingly, we would predict that the maco mutation results in loss of PigK function, an effect that would be phenocopied by knockdown of PigK protein. To test this, we used antisense MO methods to knockdown PigK in WT embryos and assayed effects on two known maco phenotypes: decreased touch response and reduced RB INa density.
maco mutants have touch scores of ∼0, while WT siblings show scores near 10 (Fig. 3A). At all doses tested (0.5–3.5 ng/nl), PigK MO injection results in a significant decrease in the touch response score vs. that resulting from injection of CTL MO (3.5 ng/nl; Fig. 3B).
Fig. 3.
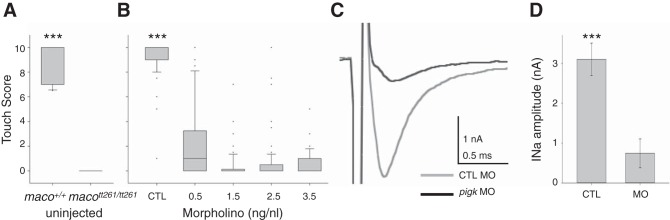
PigK knockdown phenocopies maco behavior. A: maco+/+ embryos had significantly higher touch response scores than did macott261/tt261 embryos. (n: 10, maco+/+; 12, macott261/tt261). B: PigK morpholino (MO) led to a dose-dependent decrease in touch scores. In contrast, injection of CTL MO had no effect on touch sensitivity. (n = 73, 3.5 ng/nl CTL MO; 76, 0.5 ng/nl PigK MO; 61, 1.5 ng/nl PigK MO; 71, 2.5 ng/nl PigK MO; 72, 3.5 ng/nl PigK MO). For the graphs of A and B, solid horizontal lines designate the median, boxes indicate the 25th–75th percentiles, and whiskers show 90th percentile. C: injection of PigK MO also led to reduction of RB INa amplitudes. D: INa amplitudes were decreased significantly in embryos injected with PigK MO. Data are presented as means ± SE. ***P < 0.001 (n = 3, 3.5 ng/nl MO CTL; 5, 3.5 ng/nl PigK MO).
Another phenotype of maco mutants is reduction of INa amplitudes in RB cells (Ribera and Nüsslein-Volhard 1998). The peak INa amplitudes recorded from RB cells in PigK MO injected embryos are significantly reduced compared with those in embryos injected with control MO (Fig. 3, C and D). Overall, these data support the view that the mutation identified in maco mutants (Fig. 1) leads to loss of function of the pigk gene.
WT pigk rescues the maco phenotype.
If the mutation that we have identified in the pigk gene underlies the maco phenotype, then overexpression of WT pigk in mutant embryos should rescue the deficits. Our first attempt to test this prediction consisted of injecting WT pigk mRNA into maco mutants, as this approach successfully restored the touch response in pigumi310/mi310 mutants (Nakano et al. 2010). However, pigk RNA injections did not lead to rescue of the maco phenotype, even at RNA concentrations that produced significant embryonic defects and lethality (data not shown).
As an alternative approach, we drove expression of WT pigk transgenically in maco mutants. First, we injected a Tol2 construct containing the ubb promoter, ubb:pigk-IRES-EGFP, to drive expression of WT pigk in all cells (Mosimann et al. 2011). The DNA was injected into the progeny of a macott261/+ incross. Each embryo was assayed for touch responsiveness at 48 hpf and subsequently genotyped.
For our first comparisons, we focus on the touch scores of uninjected maco+/+, macott261/+, and macott261/tt261 embryos. There is a significant difference between the touch scores of maco+/+ vs. macott261/tt261 (Fig. 4A, column 1 vs. column 3), and macott261/+ vs. macott261/tt261 (Fig. 4A, column 2 vs. column 3) embryos. In contrast, the touch scores of maco+/+ vs. macott261/+ embryos (Fig. 4A, column 1 vs. column 2) do not differ. These results are consistent with maco being a recessive mutation (Granato et al. 1996). Further, maco+/+ and macott261/+ embryos injected with the ubb:pigk-IRES-EGFP transgene do not have significantly different touch scores vs. those of uninjected maco+/+ or macott261/+ embryos (Fig. 4B, column 2 vs. Fig. 4A, columns 1 and 2). The latter result suggests that injection of the transgenic construct per se does not affect touch sensitivity (but see below).
Fig. 4.
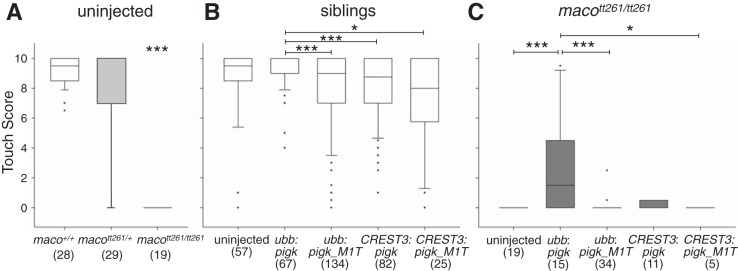
Ubiquitous but not RB-restricted expression of WT pigk rescues touch responsiveness in macott261/tt261 embryos. One-cell-stage embryos were either not injected or injected with the indicated transgenic constructs. Touch responsiveness was assayed at 48 hpf, and genotyping was performed after behavioral testing. In the graphs, the solid horizontal lines designate the median score, boxes indicate the 25th–75th percentile range, and whiskers show the 90th percentile. Sample size (n) is indicated within the figure. The data presented in A for uninjected embryos are reshown in B and C as the uninjected conditions. A: uninjected WT (maco+/+; column 1) and heterozygote (macott261/+; column 2) embryos have significantly higher touch scores than do sibling macott261/tt261 mutants (column 3). ***P < 0.001. B: data were pooled for maco+/+ and macott261/+ embryos injected with the same transgene. Regardless of transgene injection, the different sibling groups all had touch scores that were closer to 10 than 0. However, sibling embryos injected with ubb:pigk-IRES-EGFP (column 2) had slightly higher median scores than those injected with ubb:pigk_M1T-IRES-eGFP (column 3) or either of the CREST3 driven transgenes (columns 4 and 5). *P < 0.05, ***P < 0.001. C: data for embryos that were genotypically homozygous for the maco mutation (macott261/tt261). macott261/tt261 embryos injected with ubb:pigk-IRES-EGFP (column 2) had significantly greater touch scores than did uninjected macott261/tt261 embyros (column 1) or those injected with ubb:pigk_M1T-IRES-EGFP (column 3), CREST3:pigk-IRES-EGFP (column 4), or CREST3:pigk_M1T-IRES-EGFP (column 5). *P < 0.05, ***P < 0.001.
For subsequent comparisons, we pooled the touch scores of maco+/+ and macott261/+ groups (“siblings”) as they did not differ significantly. First, we compare the touch scores of uninjected siblings (Fig. 4B, column 1) to those injected with ubb:pigk-IRES-EGFP or ubb:pigk_M1T-IRES-EGFP (Fig. 4B, columns 2 and 3). For these comparisons, there are no significant differences, consistent again with the view that injection of a transgenic construct does not affect the touch response of sibling embryos. However, siblings injected with ubb:pigk-IRES-EGFP (Fig. 4B, column 2) do have significantly higher touch scores than those injected with ubb:pigk_M1T-IRES-EGFP (Fig. 4B, column 3), raising the possibility that overexpression of mutant PigK might have a mild negative effect on touch sensitivity.
We next assessed whether transgene injection could restore touch sensitivity to mutant embryos by comparing touch scores of uninjected macott261/tt261 embryos and macott261/tt261 embryos expressing a transgene (Fig. 4C). macott261/tt261 embryos injected with ubb:pigk-IRES-EGFP (Fig. 4C, column 2) have significantly higher touch scores than do uninjected macott261/tt261 embryos (Fig. 4C, column 1) or macott261/tt261 embryos injected with the mutant pigk transgene (Fig. 4C, column 3). Importantly, ubiquitous expression of WT pigk, but not pigk_M1T, partially restores the touch response of mutant embryos, supporting the view that the pigk mutation present in maco mutants (Fig. 1) underlies their touch-insensitive behavioral phenotype.
We next tested whether transgenic overexpression of WT pigk selectively in RB cells of macott261/tt261 embryos could also rescue the touch response of maco mutants. For these experiments, we used Tol2 constructs containing the CREST3 enhancer, rather than the ubb promoter, to limit expression to sensory neurons.
Control experiments, performed on sibling embryos (maco+/+, macott261/+), show that CREST3 driven expression of either WT pigk or pigk_M1T does not lead to significantly different touch scores compared with those of uninjected sibling embryos (Fig. 4B, columns 4 and 5 vs. column 1). However, as for injection of the ubb:pigk_M1T-IRES-EGFP construct, injection of either CREST3:pigk-IRES-EGFP or CREST3:pigk_M1T-IRES-EGFP (Fig. 4B, columns 4 and 5) has a small but significant effect on touch sensitivity, compared with that of embryos injected with the ubb:pigk-IRES-EGFP construct. The bases for the these small reductions in touch sensitivity are not clear. Overall, the median touch scores of siblings injected with either CREST3:pigk-IRES-EGFP or CREST3:pigk_M1T-IRES-EGFP (∼8; Fig. 4B, columns 4 and 5) are close to those of uninjected touch-sensitive siblings (∼9; Fig. 4B, column 1).
We next considered the effects of RB-limited pigk expression on touch sensitivity of mutant embryos. maco mutants injected with CREST3:pigk_M1T-IRES-EGFP (Fig. 4C, column 5) have touch scores that are similar to those of uninjected mutants (Fig. 4C, column 1). However, in contrast to the effects of ubb-driven ubiquitous expression of WT pigk (Fig. 4C, column 2), genotypically macott261/tt261 embryos injected with CREST3:pigk-IRES-EGFP (Fig. 4C, column 4) have touch scores that are ∼0 and not significantly different from those of the uninjected macott261/tt261 group (Fig. 4C, column 1).
Overall, these results indicate that ubiquitous expression of WT pigk, but not pigk_M1T, restores the touch response to mutant embryos. In contrast, CREST3-driven RB-limited expression of WT pigk does not rescue the touch response.
Expression of WT pigk in RB cells of maco mutants restores INa amplitudes.
Possible explanations for the lack of rescue of the touch response by the CREST3:pigk-IRES-EGFP transgene include 1) an insufficient number of RB cells expressing WT pigk, or 2) the transgene led to low levels of WT pigk expression in RB cells, below that required to maintain normal INa amplitudes. However, on the basis of GFP fluorescence, multiple RB cells express the transgene in injected embryos (Fig. 5, A, B, and D). Moreover, Douglass et al. (2008) demonstrated that activation of a single RB cell sufficed to elicit a behavioral response.
Fig. 5.

Overexpression of WT pigk in RB cells partially rescues RB INa. A: in a single embryo injected with CREST3:pigk-IRES-EGFP, several GFP+ RB cells are present (left, bright field; center, fluorescent image; right, merge). Arrowheads point to GFP+ RB cells. Scale Bar, 250 μm. B: the GFP+ RB cells in the box in the middle panel of A are shown at higher magnification. This region corresponds to that assayed for touch. Scale bar, 50 μm. C: a GFP+ RB cell in an embryo injected with CREST3:pigk-IRES-EGFP has a relatively normal extensive peripheral arbor. Scale Bar, 50 μm. D: the spinal cord of a CREST3:pigk-IRES-EGFP injected embryo is mounted and minimally dissected in preparation for electrophysiology. The left panel is a bright field image, with arrows indicating RB cells that are identified on the basis of position and size of soma (Ribera and Nüsslein-Volhard 1998). The middle panel shows the GFP fluorescence of several RB cells. The right panel is a merged image of the brightfield and fluorescent images. Scale bar, 20 μm. E–G: INa was recorded from RB cells in embryos that were either uninjected WT (E), uninjected macott261/tt261 (F), or macott261/tt261 injected (G) with the CREST3:pigk-IRES-EGFP transgene. For injected embryos, currents were recorded from GFP+ RB cells. H–J: the graphs of H–J present the absolute peak INa amplitude values as means ± SE. Samples size (n) information is provided within the figure. H: RB cells of uninjected maco+/+ (WT) and macott261/+ sibling embryos had INa amplitudes that were significantly larger than those recorded from RB cells in macott261/tt261 uninjected embryos. ***P < 0.001. I: for the comparisons of this graph, data obtained from RBs in maco+/+ (WT) and macott261/+ siblings were pooled. Peak INa amplitudes recorded from RBs in uninjected siblings (column 1) or those injected with either the CREST3:pigk-IRES-EGFP (column 2) or CREST3:pigk_M1T-IRES-EGFP (column 3) transgenes were compared. Transgene injection did not have a significant effect on peak INa amplitude. J: peak INa amplitudes recorded from RBs in uninjected macott261/tt261 embryos (column 1) or those in macott261/tt261 embryos injected with either the CREST3:pigk-IRES-EGFP (column 2) or the CREST3:pigk_M1T-IRESeGFP (column 3) transgenes are shown. Injection of the CREST3:pigk-IRES-EGFP, but not CREST3:pigk_M1T-IRESeGFP, transgene led to a significant increase in RB peak INa amplitude. **P < 0.01, ***P < 0.001.
As noted, GFP expression identified RB cells that successfully expressed the transgene (Fig. 5, A–D). This allowed us to test directly whether transgenic expression of WT pigk in a RB cell of a maco mutant could rescue INa amplitude and action potential firing. RB INa amplitude is significantly reduced in macott261/tt261 compared with maco+/+ or macott261/+ embryos (Fig. 5, E–H), as shown previously (Ribera and Nüsslein-Volhard 1998). For control purposes, we then determined the effects of CREST3-driven expression of WT pigk or pigk_M1T in the context of a responsive sibling (maco+/+, macott261/+) (Fig. 5I). RB INa amplitudes recorded from injected genotypically maco+/+ and macott261/+ embryos (siblings) do not differ and were pooled for subsequent comparisons. In RBs of siblings that were either uninjected (Fig. 5I, column 1) or injected with CREST3:pigk-IRES-EGFP (Fig. 5I, column 2) or CREST3:pigk_M1T-IRES-EGFP (Fig. 5I, column 3), INa amplitudes do not significantly differ. This finding suggests that expression of WT pigk or pigk_M1T does not affect INa amplitudes in the context of the sibling genotype (maco+/+, macott261/+).
We then compared data obtained from genotypically macott261/tt261 embryos. For RBs in mutant embryos injected with CREST3:pigk-IRES-EGFP, INa amplitudes are significantly larger than those recorded from RBs of uninjected embryos (Fig. 5J, column 2 vs. column 1) or embryos expressing pigk_M1T in RB cells (Fig. 5J, column 2 vs. column 3). This finding indicates that expression of WT pigk in RB cells suffices to rescue INa amplitude in macott261/tt261 embryos.
WT pigk rescues action potential firing in RB cells of maco mutants.
RB cells of macott261/tt261 embryos not only have reduced INa amplitudes but they also fail to fire action potentials (Ribera and Nüsslein-Volhard 1998). We next tested if the rescue of RB INa amplitude in macott261/tt261 embryos by transgenic expression of WT pigk also allows for recovery of not only RB INa amplitude (Fig. 5) but also firing of action potentials. We performed current-clamp recordings from RBs in uninjected WT and macott261/tt261 embryos, and macott261/tt261 embryos injected with the CREST3:pigk-IRES-EGFP.
In WT uninjected embryos, 5/5 RB cells fire large amplitude, rapidly rising active responses, characteristic of action potentials (e.g., Fig. 6A; see materials and methods; Table 1). In contrast, only 3/16 RBs from uninjected macott261/tt261 embryos display an active response (Table 1; e.g., Fig. 6A). Further, these active responses rise slowly, are of small amplitude, and do not meet the criteria to be considered action potentials (Table 1). In contrast, for 8/8 RBs of macott261/tt261 embryos injected with the CREST3:pigk-IRES-EGFP construct, current injection elicits active responses and 50% of these responses were of sufficient amplitude and rate of rise to be considered action potentials (e.g., Fig. 6A; Table 1). There is no significant difference in the active response amplitudes or dV/dt's recorded from RBs of WT vs. macott261/tt261 embryos injected with CREST3:pigk-IRES-EGFP (Fig. 6, B and C).
Fig. 6.
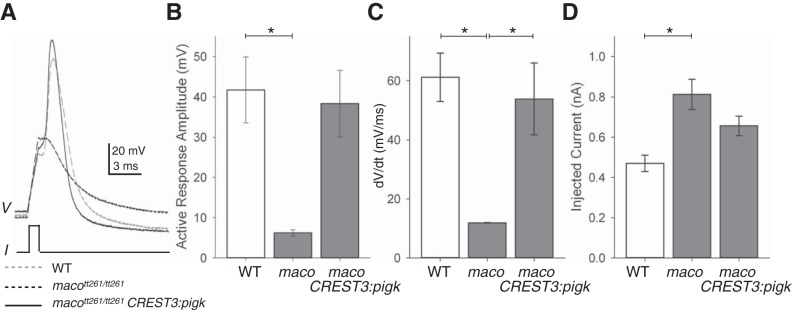
Overexpression of WT pigk in macott261/tt261 RB cells restores the ability to fire an action potential. A: action potentials were elicited by current injection (I), and the membrane potential responses (V) of RB cells in 48 hpf embryos were recorded. Exemplar responses are shown for RBs from embryos that were either uninjected WT (dashed gray line), or macott261/tt261 (dashed black line) or macott261/tt261 injected with the CREST3:WTPigk-IRES-EGFP transgene (solid black line). B: the amplitudes of the active membrane responses recorded from RBs in WT (column 1) were significantly larger than those recorded from RBs in uninjected macott261/tt261 embryos (column 2). *P < 0.05. C: the rates of rise (dV/dt) of the active membrane responses recorded from RBs in either WT (column 1) or macott261/tt261 injected with CREST3:pigk-IRES-EGFP (column 3) embryos were significantly faster than those of RBs in uninjected macott261/tt261 embryos (column 2). *P < 0.05. D: to elicit an active response, significantly more current was required for RBs of macott261/tt261 (column 2) vs. than that required for RBs in WT embryos (column 1). In addition, as shown in B and C and Table 1, the active membrane responses of RB cells in maco mutants did not meet the criteria for being considered an action potential (see materials and methods). *P < 0.05.
Table 1.
RB active responses in control, mutant, and “rescued” embryos
| Genotype | maco+/+ | macott261/tt261 | macott261/tt261 |
|---|---|---|---|
| Transgene | none | none | CREST3:pigk |
| Number of RBs tested | 5 | 16 | 8 |
| Number with active responses | 5 (100%) | 3 (19%) | 8 (100%) |
| Number of firing action potentials* | 5 (100%) | 0 (0%) | 4 (50%) |
Action potentials are active responses with amplitudes >20 mV and dV/dt >40 mV/ms (see materials and methods).
We also compared the amplitude of the minimum amount of current required to elicit any type of active response (rheobase). RB cells in WT embryos or macott261/tt261 embryos injected with CREST3:pigk-IRES-EGFP do not differ in the minimum amount of injected current required to elicit an action potential (Fig. 6D). Further, RB cells in WT embryos fire action potentials, in response to significantly less current than that required to elicit even the slow, small-amplitude active responses of a subset of RBs in macott261/tt261 embryos (Fig. 6D).
Taken together, these data indicate that CREST3-driven expression of WT pigk suffices to rescue INa current amplitude and action potential firing in RB cells of mutant embryos (Figs. 5 and 6). However, despite this functional rescue of two PigK-dependent RB properties, transgenic expression of WT pigk in RB cells of maco mutants does not restore the touch response to mutant embryos (Fig. 4C).
DISCUSSION
We report that the maco phenotype is due to a single nucleotide substitution in the pigk gene (Fig. 1). The encoded protein, PigK, functions as an essential subunit of the GPI transamidase complex and thus is required for biosynthesis of GPI-APs. PigK's function in GPI attachment does not provide obvious insights into the molecular basis of the decreased touch response of maco mutants. Consequently, the possibility of additional, presently unknown functions for PigK emerges. However, the finding that another zebrafish PIG mutant, pigumi310/mi310, also has a reduced touch response and decreased RB INa density (Nakano et al. 2010) supports the view that defective GPI-AP formation underlies the maco and pigu touch-insensitive phenotypes and reduced RB INa density.
Our studies also provide new insights regarding the requirements for GPI transamidase function in the touch response. While ubiquitous expression of WT pigk in maco mutants rescues the touch response, selective expression of pigk in RB cells does not, even though RB INa density and action potential firing recover (Figs. 4–6). Previous work has demonstrated that the firing of one RB cell suffices to generate the behavior associated with the touch response (Douglass et al. 2008). Taken together, the results reveal that the touch response has additional requirements for GPI transamidase function beyond maintenance of normal INa density and action potential firing in RB cells. This conclusion was not expected, given that the reductions in RB INa density and the resultant loss of action potential firing should suffice to explain the behavioral touch insensitivity.
Identification of a single base change in pigk provides the long-sought answer for the identity of the gene responsible for the maco phenotype. As is often true, the answer to one question leads to several new ones. In particular, given the large number of GPI-APs (Kinoshita 2014), one might expect that the maco mutation would lead to pleiotropic rather than the observed specific phenotypes (Gneugge et al. 2001; Granato et al. 1996; Neuhauss et al. 1999; Ribera and Nüsslein-Volhard 1998; Svoboda et al. 2001; Trowe et al. 1996). Moreover, given that GPI-APs play diverse essential functions during embryogenesis, loss of transamidase function would be expected to result in embryonic lethality. In mice, deficiency of PigA function does lead to embryonic lethality (Kawagoe et al. 1996; Keller et al. 1999; Tremml et al. 1999). However, maco mutants survive to larval stages (Granato et al. 1996).
Partial answers to these questions emerge from study of inherited human diseases resulting from mutations in genes involved in GPI-AP biosynthesis (for review, see Kinoshita 2014). Twenty-two human PIG genes exist as well as four PGAP (Post GPI Attachment to Proteins) genes (Almeida et al. 2006; Bosch et al. 2015; Fujiwara et al. 2015; Hansen et al. 2013; Horn et al. 2014; Howard et al. 2014; Krawitz et al. 2013; Murakami et al. 2012; Murakami et al. 2014; Nakamura et al. 2014; Ng et al. 2012; Ohba et al. 2014; Thompson et al. 2012). PIG genes are involved in GPI synthesis and attachment to proproteins, while PGAP genes are required for final remodeling of the GPI residue after attachment to a protein (Kinoshita 2014). Similar to maco mutants, humans carrying PIG or PGAP mutations typically survive well beyond embryogenesis. The notable exception are males that carry mutations in the X-linked PIGA gene and die at delivery or soon afterwards (Johnston et al. 2012).
Reduction rather than complete loss of surface GPI-AP expression might account for the more typical extended survival of individuals carrying PIG or PGAP mutations (Kinoshita 2014). For example, in the case of PIGM, the mutations target the promoter region and result in decreased but not total loss of pigm transcription (Almeida et al. 2006). Similarly, for coding region mutations in the human PIGA, PIGL, and PIGN genes, surface GPI-AP expression persists albeit at severely reduced levels (Johnston et al. 2012; Ng et al. 2012; Ohba et al. 2014). An additional possible explanation for the extended survival may be partial compensation provided by secretion of proteins that would normally have been GPI-APs and tethered to the cell surface (e.g., McKean and Niswander 2012). In support of this possibility, mutations in PIGO and PIGV lead to increased serum levels of alkaline phosphatase, a protein that is normally tethered to the cell surface via a GPI anchor (Fujiwara et al. 2015; Horn et al. 2014; Murakami et al. 2012; Nakamura et al. 2014).
Similarly, maco mutants survive to larval stages. It is not known how many GPI-APs exist in zebrafish nor what effect the maco mutation has on their surface expression. A possible mitigating factor may be maternally provided pigk mRNA (Fig. 2), that could allow for sufficient biosynthesis of GPI-APs to meet the needs of early embryogenesis. However, injection of the PigK MO, which would block translation of maternally provided pigk mRNA, does not lead to increased lethality compared with that observed for WT or maco mutant embryos (data not shown). In contrast, injection of MO targeting another member of the GPI transamidase complex, pigt, does result in early embryonic defects (Kvarnung et al. 2013). While the basis for the different effects of MOs targeting pigk vs. pigt is not clear, it is possible that maternal stores provide not only pigk mRNA but also PigK protein. Alternatively, the effects detected by injection of PigT MO may reflect the notorious nonspecific effects of this antisense method, even though control injections and rescue experiments suggest accurate targeting of the PigT MO (Kvarnung et al. 2013; but see, Kok et al. 2015).
The human conditions do not provide good answers to questions concerning the basis for the specific rather than pleiotropic effects of PIG or PGAP mutations. The total repertoire of symptoms observed collectively in humans carrying PIG or PGAP mutations are remarkably diverse, consistent with the large number of GPI-APs (> 150 in humans) and their known essential roles (Kinoshita 2014). However, any one individual carrying a PIG or PGAP mutation presents with a limited range of deficits (e.g., Almeida et al. 2006; Hansen et al. 2013; Horn et al. 2011; Johnston et al. 2012; Krawitz et al. 2012; Kvarnung et al. 2013; Maydan et al. 2011; Murakami et al. 2012; 2014; Nakashima et al. 2014; Ohba et al. 2014). Similar to zebrafish maco (pigk) and pigu mutants, human PIG or PGAP mutations typically result in impaired neuronal development and/or function.
How might GPI-APs be involved in the touch response?
The touch response initiates in the free mechanosensitive nerve endings of RB processes that innervate the skin (Clarke et al. 1984). RBs then contact the Mauthner neuron in the hindbrain as well as a local interneuron, the commissural primary ascending interneuron (CoPA). CoPAs in turn excite other interneurons (see below), ultimately leading to firing of motor neurons and their innervated muscles (Bernhardt et al. 1990; Downes and Granato 2006; Gleason et al. 2003; Hale et al. 2001; Low et al. 2012; Metcalfe et al. 1990; Pietri et al. 2009). Thus, disruption of any step ranging from the initial mechanotransduction, to generation and conduction of an action potential in any of the involved neurons, to synaptic transmission both centrally as well in the periphery at the neuromuscular junction and finally muscle contraction, could potentially be affected by loss of PigK function. That maco mutant embryos swim spontaneously, however, points to requirements for PigK prior to muscle.
Study of the touch-insensitive mutant, fakir (far), raises the possibility that a potential site for GPI-AP-dependent function in the touch response is the RB presynaptic terminal (Low et al. 2012). far mutants carry a mutation in the cacna1ab gene that encodes the Cav2.1b pore-forming α-subunit of P/Q type calcium channel complexes. P/Q-type calcium channel complexes also contain α2δ subunits, which are GPI-APs (Davies et al. 2010). Further, P/Q calcium channels play important roles in the release of neurotransmitter from presynaptic terminals in vertebrate species including zebrafish (e.g., Wen et al. 2013). In zebrafish, RB neurons show abundant expression of cacna1ab (Low et al. 2012). Moreover, far mutants appear to have normal motor network and muscle function but defective touch-dependent activation of motor function, similar to maco mutants. Taken together, these results suggest that RB presynaptic terminals might require α2δ calcium channel subunits for neurotransmitter release and thus are a candidate for a site within the touch circuit requiring GPI-AP function.
Using a combination of lesion, behavioral, electrophysiological, and morphological approaches, Pietri et al. (2009) obtained evidence for an interneuron microcircuit, the rostral loop, that is essential for the touch response. This microcircuit is located in the rostral spinal cord and transforms ascending sensory information emanating from RB activation of CoPAs into descending motor commands. Candidates for interneurons with appropriate soma locations and descending axonal projections include CiD (Circumferential Descending) and IC (Ipsilateral Caudal projecting) cells (Bernhardt et al. 1990; Hale et al. 2001; Mendelson, 1986). In combination with our results, the findings of Pietri et al. (2009) point to CiD and IC interneurons as other candidates for touch circuit sites with GPI-AP-dependent function.
Additional insights into possible sites of GPI-AP-dependent functions emerge by considering the large reduction of INa density in RB cells of maco mutants (Ribera and Nüsslein-Volhard 1998). The scn8aa gene underlies the majority of RB INa, thus making it a potential target of GPI-AP-dependent regulation (Low et al. 2010; Novak et al. 2006b; Pineda et al. 2005). While dorsal interneurons in the vicinity of RBs appear to have normal INa densities, scn8aa expression is also detected in as yet unidentified interneurons, with somas located in positions ventral to those of RBs and similar to those of CoPA, CiD, and IC interneurons (Novak et al. 2006b; Ribera and Nüsslein-Volhard 1998). In addition, subsets of motor neurons express scn8aa (Novak et al. 2006b; Yonkers and Ribera 2009). These findings point to these unidentified scn8aa-expressing interneurons and motor neurons as additional candidate sites for GPI-AP-dependent functions.
Study of touch-insensitive zebrafish scn8aa mutants, however, imposes some caveats for use of the scn8aa expression pattern as the basis to identify potential sites of GPI-AP function in the touch circuit (Low et al. 2010). In touch-insensitive scn8aa mutants, RB cells maintain the ability to fire action potentials despite having reduced INa density (Low et al. 2010). This caveat suggests that mutation of pigk may affect more than one type of sodium channel complex. Potential candidates are complexes containing α-subunits encoded by other scna genes expressed in RB neurons, i.e., scn1lab and scn8ab (Novak et al. 2006b). Loss of pigk function either spares some sodium channel complexes or reduces but does not abolish their function, because a small-amplitude INa persists in RBs of maco mutants (Ribera and Nüsslein-Volhard 1998).
Nakano et al. (2010) found that surface expression of a sodium channel β-subunit was reduced in isolated, dissociated RB cells of maco mutants compared with those of control embryos. On this basis they hypothesized that embryos require GPI-AP(s) for proper surface membrane trafficking of Nav complexes and normal INa amplitude in RB cells, and consequently for touch sensitivity. Using MOs, they undertook a candidate approach to test whether knockdown of one or a combination of GPI-APs could lead to touch sensitivity. On the basis of known interactions with Nav complexes, a promising candidate is Contactin1 (McEwen and Isom 2004; Rush et al. 2005). However, contactin1a MO injection did not result in touch-insensitive embryos (Nakano et al. 2010). Similarly, we found that knockdown of Contactin1 does not phenocopy maco touch insensitivity (data not shown). These results are consistent with the demonstration that, when studied in vivo, mammalian Contactin1 associates with Nav complexes containing Nav1.8 and Nav1.9, but not Nav1.6 or Nav1.7, α-subunits (Rush et al. 2005). Nav1.8 and Nav1.9 are mammalian orthologs of zebrafish nav12aa and nav12ab, while Nav1.6 and Nav1.7 are orthologous to nav1.6 (nav1.6a, nav1.6b) and nav1.1L (nav1.1La, nav1.1Lb) duplicates, respectively (Novak et al. 2006a).
Using the MO approach, Nakano et al. (2010) tested additional candidates including Tag-1, Contactin4a, Contactin4b, and Contactin5, either individually or in combination with each other. However, in no case was touch insensitivity observed. In view of our findings, however, the touch response is not a reliable surrogate for RB INa function. If the goal is to identify the GPI-AP(s) involved in regulation of RB INa amplitude, the results of candidate gene experiments warrant direct assay of RB INa.
The study of zebrafish touch-insensitive mutants began with the isolation of several mutations that resulted in embryonic touch sensitivity (Granato et al. 1996). The next advance consisted of the demonstration that, in four of six touch-insensitive mutants studied, RB cells have reduced INa density (Ribera and Nüsslein-Volhard 1998). The latter finding indicates that screening for touch sensitivity serves as an unexpected but efficient method for isolation of genes that affect RB INa. Recently, alligator (ali), one of the four mutants with reduced RB INa density (Ribera and Nüsslein-Volhard 1998), was shown to carry a mutation in rnf121 [Really Interesting New Gene (RING) finger protein 121] encoding an E3-ubiquitin ligase (Ogino et al. 2015). RNF121 ensures proper localization of correctly folded RB sodium channel complexes by regulating two alternative Nav channel fates, either degradation by the proteasome or surface membrane insertion. A candidate approach would have been unlikely to uncover pigk, pigu or rnf121 as genes involved in regulation of sensory neuron INa, given their previously (rnf121) and still (pigk, pigu) unknown roles in sodium channel cell biology. Taken together, the study of zebrafish touch-insensitive mutants demonstrates the ability of forward genetics to uncover genes that are novel or have unexpected functions.
At present, two touch-insensitive mutants, crocodile (cro) and steifftier (ste), with reduced RB INa amplitude (Ribera and Nüsslein-Volhard, 1998) still await identification of their lesioned genes. Thus the 1996 screen for locomotion mutants (Granato et al. 1996) may provide yet more insights into the novel genes and mechanisms that regulate voltage-gated sodium channel function in a developmental context.
GRANTS
National Institutes of Health (NIH) Grants R01-NS-025217 and R01-NS-038937 (A. B. Ribera) supported this work. Contents are the authors' sole responsibility and do not necessarily represent official NIH views. R. Geisler acknowledges funding from the German Human Genome Project (DHGP 01 KW 9627) and the European Commission's 7th Framework Programme (ZF-HEALTH project). QPCR and confocal imaging were performed using services and equipment of the University of Colorado Anschutz Medical Campus Vector and QPCR Core and Advance Light Microscopy Core, respectively. These cores are supported in part by NIH Awards P30-NS-048154 and UL1-RR-025780.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.C., M.A.Y., J.R.W., R.G.G., R.G., S.C.N., and A.B.R. conception and design of research; V.C., M.A.Y., M.B.T., J.R.W., G.B.W., R.G., S.C.N., and A.B.R. performed experiments; V.C., M.A.Y., J.R.W., G.B.W., R.G.G., R.G., S.C.N., and A.B.R. analyzed data; V.C., M.A.Y., J.R.W., R.G.G., R.G., S.C.N., and A.B.R. interpreted results of experiments; V.C., M.A.Y., J.R.W., and A.B.R. prepared figures; V.C. and A.B.R. drafted manuscript; V.C., R.G., and A.B.R. edited and revised manuscript; V.C., M.A.Y., M.B.T., J.R.W., G.B.W., R.G.G., R.G., S.C.N., and A.B.R. approved final version of manuscript.
ACKNOWLEDGMENTS
V. Carmean and A. B. Ribera thank members of the Ribera lab for helpful discussion, K. Williams and the University of Colorado Anschutz Medical Campus Vector Core for performing the QPCR experiments, and R. Moldovan for expert assistance with confocal microscopy work.
Present address of M. A. Yonkers: Herbert Eye Institute, University of California, Irvine, CA.
Present address of J. R. Willer: Center for Human Disease Modeling, Duke University, Durham, NC.
Present address of R. G. Gregg; Institute of Toxicology and Genetics, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen, Germany.
Present address of S. C. Neuhauss: Institute of Molecular Life Sciences, Neuroscience Center Zurich and Center for Integrative Human Physiology, University of Zurich, Zurich, Switzerland.
REFERENCES
- Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, Richards S, Patterson S, Kotsianidis I, Mollica L, Crawford DH, Baker A, Ferguson M, Roberts I, Houlston R, Kinoshita T, Karadimitris A. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 12: 846–851, 2006. [DOI] [PubMed] [Google Scholar]
- Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, Westerfield M, Ekker M, Postlethwait JH. Zebrafish hox clusters and vertebrate genome evolution. Science 282: 1711–1714, 1998. [DOI] [PubMed] [Google Scholar]
- Bernhardt RR, Chitnis AB, Lindamer L, Kuwada JY. Identification of spinal neurons in the embryonic and larval zebrafish. J Comp Neurol 302: 603–616, 1990. [DOI] [PubMed] [Google Scholar]
- Bosch DG, Boonstra FN, Kinoshita T, Jhangiani S, de Ligt J, Cremers FP, Lupski JR, Murakami Y, de Vries BB. Cerebral visual impairment and intellectual disability caused by PGAP1 variants. Eur J Hum Genet 2015. March 25. doi: 10.1038/ejhg.2015.42 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadei R, Pelleri MC, Vitale L, Facchin F, Lenzi L, Canaider S, Strippoli P, Frabetti F. Identification of housekeeping genes suitable for gene expression analysis in the zebrafish. Gene Express Patterns 11: 271–276, 2011. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Mayor S. The GPI-anchor and protein sorting. Cell Mol Life Sci 58: 1969–1987, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra SS, Watanabe H, Zhong TP, Roden DM. Molecular cloning and analysis of zebrafish voltage-gated sodium channel beta subunit genes: implications for the evolution of electrical signaling in vertebrates. BMC Evol Biol 7: 113, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JD, Hayes BP, Hunt SP, Roberts A. Sensory physiology, anatomy and immunohistochemistry of Rohon-Beard neurones in embryos of Xenopus laevis. J Physiol 348: 511–525, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS, Pratt WS, Dolphin AC. The α2δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a post-translational modification essential for function. Proc Natl Acad Sci USA 107: 1654–1659, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass AD, Kraves S, Deisseroth K, Schier AF, Engert F. Escape behavior elicited by single, channelrhodopsin-2-evoked spikes in zebrafish somatosensory neurons. Curr Biol 18: 1133–1137, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes GB, Granato M. Supraspinal input is dispensable to generate glycine-mediated locomotive behaviors in the zebrafish embryo. J Neurobiol 66: 437–451, 2006. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: analysis of protein sequence data from complete genomes. Prot Eng 14: 17–25, 2001. [DOI] [PubMed] [Google Scholar]
- Fein AJ, Wright MA, Slat EA, Ribera AB, Isom LL. scn1bb, a zebrafish ortholog of SCN1B expressed in excitable and nonexcitable cells, affects motor neuron axon morphology and touch sensitivity. J Neurosci 28: 12510–12522, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Murakami Y, Niihori T, Kanno J, Hakoda A, Sakamoto O, Okamoto N, Funayama R, Nagashima T, Nakayama K, Kinoshita T, Kure S, Matsubara Y, Aoki Y. Mutations in PIGL in a patient with Mabry syndrome. Am J Med Genet A 167: 777–785, 2015. [DOI] [PubMed] [Google Scholar]
- Gleason MR, Higashijima S, Dallman J, Liu K, Mandel G, Fetcho JR. Translocation of CaM kinase II to synaptic sites in vivo. Nat Neurosci 6: 217–218, 2003. [DOI] [PubMed] [Google Scholar]
- Gnuegge L, Schmid S, Neuhauss SC. Analysis of the activity-deprived zebrafish mutant macho reveals an essential requirement of neuronal activity for the development of a fine-grained visuotopic map. J Neurosci 21: 3542–3548, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato M, van Eeden FJ, Schach U, Trowe T, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123: 399–413, 1996. [DOI] [PubMed] [Google Scholar]
- Hale ME, Ritter DA, Fetcho JR. A confocal study of spinal interneurons in living larval zebrafish. J Comp Neurol 437: 1–16, 2001. [DOI] [PubMed] [Google Scholar]
- Hansen L, Tawamie H, Murakami Y, Mang Y, ur Rehman S, Buchert R, Schaffer S, Muhammad S, Bak M, Nöthen MM, Bennett EP, Maeda Y, Aigner M, Reis A, Kinoshita T, Tommerup N, Baig SM, Abou Jamra R. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am J Hum Genet 92: 575–583, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Hotta Y, Okamoto H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J Neurosci 20: 206–218, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PW, Williams NA. Conservation of engrailed-like homeobox sequences during vertebrate evolution. FEBS Lett 277: 250–252, 1990. [DOI] [PubMed] [Google Scholar]
- Horn D, Wieczorek D, Metcalfe K, Baric I, Palezac L, Cuk M, Petkovic Ramadza D, Kruger U, Demuth S, Heinritz W, Linden T, Koenig J, Robinson PN, Krawitz P. Delineation of PIGV mutation spectrum and associated phenotypes in hyperphosphatasia with mental retardation syndrome. EJHG 22: 762–767, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MF, Murakami Y, Pagnamenta AT, Daumer-Haas C, Fischer B, Hecht J, Keays DA, Knight SJ, Kölsch U, Krüger U, Leiz S, Maeda Y, Mitchell D, Mundlos S, Phillips JA 3rd, Robinson PN, Kini U, Taylor JC, Horn D, Kinoshita T, Krawitz PM. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am J Hum Genet 94: 278–287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Gropman AL, Sapp JC, Teer JK, Martin JM, Liu CF, Yuan X, Ye Z, Cheng L, Brodsky RA, Biesecker LG. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 90: 295–300, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane DA, Kimmel CB. The zebrafish midblastula transition. Development 119: 447–456, 1993. [DOI] [PubMed] [Google Scholar]
- Kawagoe K, Kitamura D, Okabe M, Taniuchi I, Ikawa M, Watanabe T, Kinoshita T, Takeda J. Glycosylphosphatidylinositol-anchor-deficient mice: implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood 87: 3600–3606, 1996. [PubMed] [Google Scholar]
- Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol 77: 201–222, 2004. [DOI] [PubMed] [Google Scholar]
- Keller P, Tremml G, Rosti V, Bessler M. X inactivation and somatic cell selection rescue female mice carrying a Piga-null mutation. Proc Natl Acad Sci USA 96: 7479–7483, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310, 1995. [DOI] [PubMed] [Google Scholar]
- Kinoshita T. Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proc Jpn Acad Ser B Phys Biol Sci 90: 130–143, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T, Fujita M, Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J Biochem 144: 287–294, 2008. [DOI] [PubMed] [Google Scholar]
- Kok FO, Shin M, Ni CW, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I, DeSantis DF, Sheppard-Tindell S, Ebarasi L, Betsholtz C, Schulte-Merker S, Wolfe SA, Lawson ND. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev Cell 32: 97–108, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SC, Blennow E, Bergmann M, Stodberg T, Makitie O, Anderlid BM, Bryceson YT, Nordenskjold M, Nordgren A. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 50: 521–528, 2013. [DOI] [PubMed] [Google Scholar]
- Krawitz PM, Murakami Y, Riess A, Hietala M, Krüger U, Zhu N, Kinoshita T, Mundlos S, Hecht J, Robinson PN, Horn D. PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet 92: 584–589 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236: 3088–3099, 2007. [DOI] [PubMed] [Google Scholar]
- Lopreato GF, Lu Y, Southwell A, Atkinson NS, Hillis DM, Wilcox TP, Zakon HH. Evolution and divergence of sodium channel genes in vertebrates. Proc Natl Acad Sci USA 98: 7588–7592, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SE, Zhou W, Choong I, Saint-Amant L, Sprague SM, Hirata H, Cui WW, Hume RI, Kuwada JY. Na(v)1.6a is required for normal activation of motor circuits normally excited by tactile stimulation. Dev Neurobiol 70: 508–522, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SE, Woods IG, Lachance M, Ryan J, Schier AF, Saint-Amant L. Touch responsiveness in zebrafish requires voltage-gated calcium channel 21b. J Neurophysiol 108: 148–159, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydan G, Noyman I, Har-Zahav A, Neriah ZB, Pasmanik-Chor M, Yeheskel A, Albin-Kaplanski A, Maya I, Magal N, Birk E, Simon AJ, Halevy A, Rechavi G, Shohat M, Straussberg R, Basel-Vanagaite L. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 48: 383–389, 2011. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Isom LL. Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem 279: 52744–52752, 2004. [DOI] [PubMed] [Google Scholar]
- McKean DM, Niswander L. Defects in GPI biosynthesis perturb Cripto signaling during forebrain development in two new mouse models of holoprosencephaly. Biol Open 1: 874–883, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson B. Development of reticulospinal neurons of the zebrafish. II. Early axonal outgrowth and cell body position. J Comp Neurol 251: 172–184, 1986. [DOI] [PubMed] [Google Scholar]
- Metcalfe WK, Myers PZ, Trevarrow B, Bass MB, Kimmel CB. Primary neurons that express the L2/HNK-1 carbohydrate during early development in the zebrafish. Development 110: 491–504, 1990. [DOI] [PubMed] [Google Scholar]
- Moreno RL, Ribera AB. Zebrafish motor neuron subtypes differ electrically prior to axonal outgrowth. J Neurophysiol 102: 2477–2484, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 138: 169–177, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Kanzawa N, Saito K, Krawitz PM, Mundlos S, Robinson PN, Karadimitris A, Maeda Y, Kinoshita T. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem 287: 6318–6325, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Tawamie H, Maeda Y, Büttner C, Buchert R, Radwan F, Schaffer S, Sticht H, Aigner M, Reis A, Kinoshita T, Jamra RA. Null mutation in PGAP1 impairing Gpi-anchor maturation in patients with intellectual disability and encephalopathy. Plos Gen 10: e1004320, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Osaka H, Murakami Y, Anzai R, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Kinoshita T, Matsumoto N, Saitsu H. PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia 55: e13–e17, 2014. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Fujita M, Ogino K, Saint-Amant L, Kinoshita T, Oda Y, Hirata H. Biogenesis of GPI-anchored proteins is essential for surface expression of sodium channels in zebrafish Rohon-Beard neurons to respond to mechanosensory stimulation. Development 137: 1689–1698, 2010. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Kashii H, Murakami Y, Kato M, Tsurusaki Y, Miyake N, Kubota M, Kinoshita T, Saitsu H, Matsumoto N. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogen 15: 193–200, 2014. [DOI] [PubMed] [Google Scholar]
- Neuhauss SC, Biehlmaier O, Seeliger MW, Das T, Kohler K, Harris WA, Baier H. Genetic disorders of vision revealed by a behavioral screen of 400 essential loci in zebrafish. J Neurosci 19: 8603–8615, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng BG, Hackmann K, Jones MA, Eroshkin AM, He P, Wiliams R, Bhide S, Cantagrel V, Gleeson JG, Paller AS, Schnur RE, Tinschert S, Zunich J, Hegde MR, Freeze HH. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 90: 685–688, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak AE, Jost MC, Lu Y, Taylor AD, Zakon HH, Ribera AB. Gene duplications and evolution of vertebrate voltage-gated sodium channels. J Mol Evol 63: 208–221, 2006a. [DOI] [PubMed] [Google Scholar]
- Novak AE, Taylor AD, Pineda RH, Lasda EL, Wright MA, Ribera AB. Embryonic and larval expression of zebrafish voltage-gated sodium channel alpha-subunit genes. Dev Dyn 235: 1962–1973, 2006b. [DOI] [PubMed] [Google Scholar]
- Ogino K, Low SE, Yamada K, Saint-Amant L, Zhou W, Muto A, Asakawa K, Nakai J, Kawakami K, Kuwada JY, Hirata H. RING finger protein 121 facilitates the degradation and membrane localization of voltage-gated sodium channels. Proc Natl Acad Sci USA 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Okamoto N, Murakami Y, Suzuki Y, Tsurusaki Y, Nakashima M, Miyake N, Tanaka F, Kinoshita T, Matsumoto N, Saitsu H. PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics 15: 85–92, 2014. [DOI] [PubMed] [Google Scholar]
- Palanca AM, Lee SL, Yee LE, Joe-Wong C, Trinh le A, Hiroyasu E, Husain M, Fraser SE, Pellegrini M, Sagasti A. New transgenic reporters identify somatosensory neuron subtypes in larval zebrafish. Dev Neurobiol 73: 152–167, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietri T, Manalo E, Ryan J, Saint-Amant L, Washbourne P. Glutamate drives the touch response through a rostral loop in the spinal cord of zebrafish embryos. Dev Neurobiol 69: 780–795, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda RH, Heiser RA, Ribera AB. Developmental, molecular, and genetic dissection of INa in vivo in embryonic zebrafish sensory neurons. J Neurophysiol 93: 3582–3593, 2005. [DOI] [PubMed] [Google Scholar]
- Ribera AB, Nüsslein-Volhard C. Zebrafish touch-insensitive mutants reveal an essential role for the developmental regulation of sodium current. J Neurosci 18: 9181–9191, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Craner MJ, Kageyama T, Dib-Hajj SD, Waxman SG, Ranscht B. Contactin regulates the current density and axonal expression of tetrodotoxin-resistant but not tetrodotoxin-sensitive channel in DRG neurons. Eur J Neurosci 22: 39–49, 2005. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock DW, Ellies DL, Zhao Z, Ekker M, Ruddle FH, Weiss KM. The evolution of the vertebrate Dlx gene family. Proc Natl Acad Sci USA 93: 10858–10863, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda KR, Linares AE, Ribera AB. Activity regulates programmed cell death of zebrafish Rohon-Beard neurons. Development 128: 3511–3520, 2001. [DOI] [PubMed] [Google Scholar]
- Thompson MD, Roscioli T, Marcelis C, Nezarati MM, Stolte-Dijkstra I, Sharom FJ, Lu P, Phillips JA, Sweeney E, Robinson PN, Krawitz P, Yntema HG, Andrade DM, Brunner HG, Cole DE. Phenotypic variability in hyperphosphatasia with seizures and neurologic deficit (Mabry syndrome). Am J Med Genet A 158A: 553–558, 2012. [DOI] [PubMed] [Google Scholar]
- Tremml G, Dominguez C, Rosti V, Zhang Z, Pandolfi PP, Keller P, Bessler M. Increased sensitivity to complement, and a decreased red blood cell life span in mice mosaic for a nonfunctional Piga gene. Blood 94: 2945, 1999. [PubMed] [Google Scholar]
- Trowe T, Klostermann S, Baier H, Granato M, Crawford AD, Grunewald B, Hoffmann H, Karlstrom RO, Meyer SU, Muller B, Richter S, Nusslein-Volhard C, Bonhoeffer F. Mutations disrupting the ordering and topographic mapping of axons in the retinotectal projection of the zebrafish, Danio rerio. Development 123: 439–450, 1996. [DOI] [PubMed] [Google Scholar]
- Uemura O, Okada Y, Ando H, Guedj M, Higashijima S, Shimazaki T, Chino N, Okano H, Okamoto H. Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev Biol 278: 587–606, 2005. [DOI] [PubMed] [Google Scholar]
- Vandepoele K, De Vos W, Taylor JS, Meyer A, Van de Peer Y. Major events in the genome evolution of vertebrates: paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc Natl Acad Sci USA 101: 1638–1643, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Linhoff MW, Hubbard JM, Nelson NR, Stensland D, Dallman J, Mandel G, Brehm P. Zebrafish calls for reinterpretation for the roles of P/Q calcium channels in neuromuscular transmission. J Neurosci 33: 7384–7392, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio). Eugene, OR: Westerfield, 1993. [Google Scholar]
- Willer GB, Lee VM, Gregg RG, Link BA. Analysis of the zebrafish perplexed mutation reveals tissue-specific roles for de novo pyrimidine synthesis during development. Genetics 170: 1827–1837, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonkers MA, Ribera AB. Molecular components underlying nongenomic thyroid hormone signaling in embryonic zebrafish neurons. Neural Dev 4: 20, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacks MA, Garg N. Recent developments in the molecular, biochemical and functional characterization of GPI8 and the GPI-anchoring mechanism. Mol Membr Biol 23: 209–225, 2006. [DOI] [PubMed] [Google Scholar]