

Introduction: Alzheimer's disease (AD) begins to develop decades prior to its clinical manifestation (Sperling et al., 2011), and while it is the most common form of dementia, as of yet there is no cure. Two of the most researched pathological features contributing to disease development are the extracellular amyloid plaques composed of amyloid-beta proteins (Aβ) and neurofibrillary tangles of tau proteins. Another feature of AD is the progression of early neuronal excitability/hyperactivity to silencing/hypoactivity (Palop and Mucke, 2010), with hypoactivity explained by the synaptic failure hypothesis (Selkoe, 2002). In vitro and animal model studies have demonstrated that Aβ pathology associates with increased excitability of hippocampal neurons. Furthermore, hippocampal hyperactivity has been reported in human subjects at high risk for developing AD. In an effort to identify possible factors involved in this progression of neuronal activity in AD, we reviewed the literature and identified a novel interaction between Aβ, glycogen, neurons, and astrocytes with a focus on events in the synapse.
Neuronal activity: Within the brain, Aβ is released at the presynaptic sites, and increased neuronal activity leads to higher levels of Aβ within the interstitium (Palop and Mucke, 2010). Normal levels of Aβ (pM) augment synaptic activity (Palop and Mucke, 2010; Puzzo and Arancio, 2013; Fogel et al., 2014), but higher levels of Aβ (nM) act as a negative feedback mechanism and inhibit synaptic activity (Palop and Mucke, 2010; Puzzo and Arancio, 2013). Thus, under normal conditions, elevated levels of Aβ would be expected to homeostatically inhibit neuronal activity. However, in AD there is chronic elevation of interstitial Aβ levels because of oversecretion due to neuronal hyperactivity (Cirrito et al., 2005) and reduced clearance at the blood-brain barrier (Bateman et al., 2006). This elevation of Aβ then results in misfolding and accumulation into pathological amyloid plaques (Selkoe, 2002; Palop and Mucke, 2010). These plaques then serve as a reservoir of toxic oligomeric Aβ species (Selkoe, 2002; Sheng et al., 2012; Klein, 2013) that may lead to neuronal and synaptic loss. This synaptic loss has been directly correlated to decreased cognition in rigorous studies by Scheff (Scheff and Price, 2006). Chronic elevation of Aβ leads to negative feedback (Puzzo and Arancio, 2013) and neuronal hypoactivity (Sheng et al., 2012).
Synaptic energy: Metabolic dysfunction and neuronal excitability are linked, as synaptic activity is coupled to energy supply, with 97% of the energy for a resting neuron being supplied by the mitochondria. The increased energy demands of active neurons are met with an increased reliance on glycolysis (Bélanger 2011) with astrocytes increasing additional nutrient supply via the glycogen-lactate shunt (Bélanger et al., 2011; Stobart and Anderson, 2013). Stimulation of astrocytes results in increased intracellular calcium levels, which then result in the following: glucose uptake, vasodilation, astrocyte glycogenolysis, and release of adenosine and adenosine triphosphate (ATP) (Maragakis and Rothstein, 2006; Hertz et al., 2007; Bélanger et al., 2011; Obel et al., 2012). Thus, increased synaptic activity results in increased nutrient delivery in the brain (Bélanger et al., 2011). Pathologically, astrocyte dysfunction with reduction of nutrient delivery to the neurons could in part contribute to the metabolic dysfunction (decreased nutrient delivery and glucose utilization), leading to AD pathology.
Glycogen: The main energy supplies in the brain are glucose and glycogen. Glycogen is primarily located within astrocytes, stimulated by insulin (Brown, 2004), and serving as the largest energy reserve in the brain (Bélanger et al., 2011). Astrocytes convert glycogen stores to lactate, which is then supplied to the neurons (Bélanger et al., 2011). There is a tight coupling between neuronal activity and glycogen mobilization (Bélanger et al., 2011), and as such, glycogen stores correlate with cognition (Bélanger et al., 2011). Importantly, astrocyte glycogen is concentrated around synapses and is known to be important for synaptic activity, and associated with greater synapse protection (Brown, 2004; Bélanger et al., 2011). While glycogenolysis satisfies increased synaptic energy demands in the short term, it also occurs when glucose levels are normal (Brown, 2004; Hertz et al., 2007; Bélanger et al., 2011). This is likely because the energy yield is 50% greater when using glycogen instead of glucose as a substrate for glycolysis (Hertz et al., 2007; Obel et al., 2012). Thus decreased glycogen levels correlate with decreased cognition and synaptic loss.
Measurement of glycogen levels throughout the course of AD is not currently documented in the literature, but would be warranted to further understand any possible correlation between glycogen levels and cognition in AD. We propose that glycogen levels may be reduced in AD, with decreased glycogen synthesis and increased glycogenolysis occurring at the same time (Figure 1). First, glycogen synthase kinase 3β (GSK-3β) activity is upregulated in AD (Llorens-Martín et al., 2014), and because GSK-3β inhibits glycogen synthase (Brown, 2004), this will result in diminished glycogen synthesis. Tangentially, GSK-3β is also associated with tau pathology in AD (Llorens-Martín et al., 2014). Second, adenosine binding to adenosine A2 receptors (A2A) increases the rate of glycogenolysis, at least in cultured astrocytes and brain slices (Xu et al., 2014). The A2A are located on synapses where they alter metabolism, neurotransmitter release (acetylcholine and glutamate), and modulate cognitive function (Gomes et al., 2011). Of note, the pathological upregulation of A2A receptors in AD (Gomes et al., 2011) may result in depletion of brain glycogen levels. Thus the loss of glycogen is linked to decreased cognition (Stobart and Anderson, 2013) and synaptic dysfunction (Sheng et al., 2012).
Figure 1.
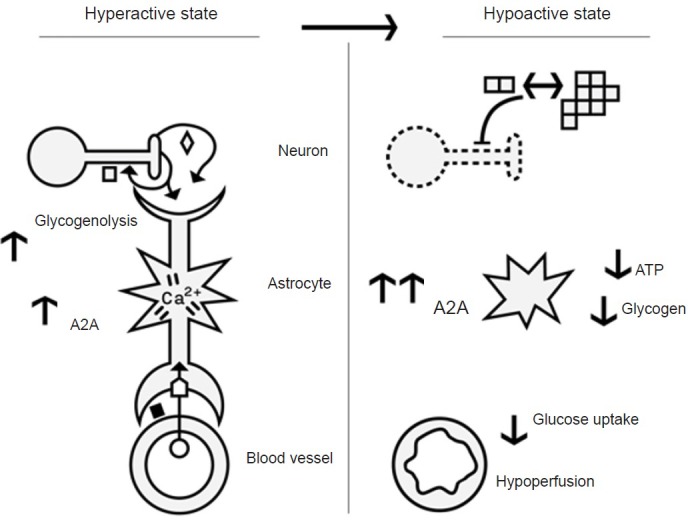
Depiction of changes in astrocytes, neurons and blood vessels in the hyper- and hypoactive states in Alzheimer's disease (AD).
Hyperactive state: Increased neuronal activity results in the synaptic release of amyloid-beta (Aβ) and glutamate, with Aβ (pM levels) feedback to further increase synaptic activity. Neuronal activity (e.g., glutamate ◇) stimulates the astrocytes and results in several changes in the astrocyte and their glycogen concentration. For example, increased levels of calcium lead to increased glycogenolysis, glucose uptake and vasoactive substance release by the astrocyte end-feet surrounding blood vessels. Homeostatic mechanisms, such as elevated levels of Aβ (nM) and adenosine A2A re-ceptors (A2A) exist to reduce neuronal hyperactivity. A pathological hyperactive state occurs early on in AD progression, during the stage of mild cognitive impairment (MCI). It is likely that dysregulation of the homeostatic mechanisms of Aβ and A2A, along with depleted glycogen reserves lead to subsequent hypoactivity. Hypoactive state: This state occurs later in AD, and we speculate it to be the result of chronic hyperactivity. For example, it is known that the combination of increased synaptic secretion and reduced clearance of Aβ result in the misfolding and polymerization of Aβ, which accumulate as amyloid plaques (multiple □). These plaques serve as a reservoir of oligomeric Aβ (dimer □). The elevated levels of Aβ (nM) inhibit synaptic activity and result in neuronal death (death depicted by dotted lines). Astrocytes also become activated (depicted by star shape), but this is not discussed in the text. In AD, cerebrospinal fluid (CSF) Adenosine triphosphate (ATP) levels (Coskuner and Murray, 2014), brain glucose utilization, and blood flow are all decreased (reviewed by Murray et al., 2011), while A2A levels are upregulated (Gomes et al., 2011). Although glycogen levels have not been measured in AD, based on reasons given in the text, we predict that glycogen levels will be low. Thus we have linked Aβ, glycogen, and A2A levels to the progression of hyperactivity to hypoactivity. ◇ Glutamate; ⌂ Glucose transporter; ◼ vasoactive sub-stances; □ amyloid beta (Aβ) - monomer, oligomer and fibril (shown as dimers and multimers respectively); ○ glucose.
Speculation as to the cause of initial neuronal hyperactivity: We examined possible factors involved in the progression of neuronal hyperactivity to hypoactivity in AD (Figure 1), but have not addressed the initial causation of hyperactivity. One hypothesis is that the hyperactivity compensates (Mormino et al., 2012; Elman et al., 2014) for neuronal and synaptic loss during early AD. The studies by Scheff indicate that synaptic loss in early AD, directly correlated to cognitive loss (Scheff and Price, 2006). These studies also found that although there was a loss of synapse numbers, in the remaining synapses there was an increase in the size of the total contact areas, which the authors suggested to be a synaptic compensatory mechanism in response to AD pathology (Scheff and Price, 2006). The initial neuronal loss may be due to metabolic dysfunction via reduced glucose utilization (e.g., insulin resistance in the brain, also termed Type 3 diabetes) or hypoperfusion (Beach et al., 2007; De La Monte, 2012). Metabolic dysfunction increases Aβ generation (reviewed by Murray et al., 2011), and reduced ATP levels increase formation of Aβ oligomers, at least in vitro (Coskuner and Murray, 2014). Thus the elevation of Aβ will initially lead to neuronal hyperactivity (Puzzo and Arancio, 2013), and any generated toxic Aβ oligomers will cause cell death (Klein, 2013) (Figure 1). Thus, it is quite possible that metabolic dysfunction underlies the early changes in AD, including neuronal hyperactivity and death. This suggestion is bolstered by epidemiological findings where metabolic diseases, such as obesity in middle age and type 2 diabetes, are associated with increased risk of AD (Barnes and Yaffe, 2011).
Conclusion: Neuronal hyperactivity may compensate for cognitive declines in AD. Such hyperactivity results in elevated Aβ, A2A and reduced glycogen stores, which would normally feedback to reduce hyperactivity (Figure 1). Pathologically, chronic activation of such feedback would result in hypoactivity and cell death. We have used the current literature to delineate these linkages between Aβ, glycogen and neuronal activity in AD. Thus, excessive Aβ generation, astrocyte stimulation during neuronal hyperactivity, along with reduced glycogen storage, may lead to neuronal hypoactivity. We look forward to direct experimental verification of these links, which we have identified within existing publications.
We thank Vern Giammartino (http://www.verngiammartino.com/) for the graphical depiction of this process. We also acknowledge and apologize that due to a limitation of number of references not all relevant publications were cited.
References
- Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol. 2011;10:819–828. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE. Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Brown AM. Brain glycogen re-awakened. J Neurochem. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Coskuner O, Murray IV. Adenosine triphosphate (ATP) reduces amyloid-β protein misfolding in vitro. J Alzheimers Dis. 2014;41:561–574. doi: 10.3233/JAD-132300. [DOI] [PubMed] [Google Scholar]
- de la Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res. 2012;9:35–66. doi: 10.2174/156720512799015037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elman JA, Oh H, Madison CM, Baker SL, Vogel JW, Marks SM, Crowley S, O’Neil JP, Jagust WJ. Neural compensation in older people with brain amyloid-β deposition. Nat Neurosci. 2014;17:1316–1318. doi: 10.1038/nn.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O’Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, Isacoff EY, Hirsch JA, Slutsky I. APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep. 2014;7:1560–1576. doi: 10.1016/j.celrep.2014.04.024. [DOI] [PubMed] [Google Scholar]
- Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta. 2011;1808:1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Klein WL. Synaptotoxic amyloid-β oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J Alzheimers Dis. 2013;33(Suppl 1):S49–65. doi: 10.3233/JAD-2012-129039. [DOI] [PubMed] [Google Scholar]
- Llorens-Martín M, Jurado J, Hernández F, Avila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Mormino EC, Brandel MG, Madison CM, Marks S, Baker SL, Jagust WJ. Aβ Deposition in aging is associated with increases in brain activation during successful memory encoding. Cereb Cortex. 2012;22:1813–1823. doi: 10.1093/cercor/bhr255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IV, Proza JF, Sohrabji F, Lawler JM. Vascular and metabolic dysfunction in Alzheimer's disease: a review. Exp Biol Med (Maywood) 2011;236:772–782. doi: 10.1258/ebm.2011.010355. [DOI] [PubMed] [Google Scholar]
- Obel LF, Müller MS, Walls AB, Sickmann HM, Bak LK, Waagepetersen HS, Schousboe A. Brain glycogen-new perspectives on its metabolic function and regulation at the subcellular level. Front Neuroenergetics. 2012;4:3. doi: 10.3389/fnene.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Arancio O. Amyloid-β peptide: Dr. Jekyll or Mr. Hyde? J Alzheimers Dis. 33(Suppl 1):S111–120. doi: 10.3233/JAD-2012-129033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol 4. 2012 doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobart JL, Anderson CM. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front Cell Neurosci. 2013;7:38. doi: 10.3389/fncel.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Song D, Bai Q, Zhou L, Cai L, Hertz L, Peng L. Role of glycogenolysis in stimulation of ATP release from cultured mouse astrocytes by transmitters and high K + concentrations. ASN Neuro. 2014;6:e00132. doi: 10.1042/AN20130040. [DOI] [PMC free article] [PubMed] [Google Scholar]