

Alzheimer's disease (AD) is a neurodegenerative disorder which is remarkably characterized by pathological hallmarks that include neurofibrillary tangles, neuronal loss extracellular senile plaques containing aggregated amyloid beta (Aβ), and neurofibrillary tangles composed of the hyper- phosphorylated form of the microtubule protein tau. It is the most common form of dementia which is characterized by severe neurodegenerative changes such as loss of neurons and synapses in brain (Kamat et al., 2014). Several well-known mutations that lead to early-onset familial AD exist. However, these circumstances only reason for a few percentages of total AD cases.
Streptozotocin (STZ) is a glucosamine-nitrosourea compound which was originally identified as an antibiotic. It is toxic to beta cells of pancreas and usually transported through glucose transporter 2 and commonly used to induce experimental diabetes in animals. STZ administration through route such as intracebroventricular or intraperitoneal injection produces reduced cognition and increased cerebral aggregated Aβ fragments, total tau protein, and Aβ deposits. These changes were accompanied with decreased glycogen synthase kinase (GSK-3) alpha/beta ratio (phosphorylated/total) in the brain. Administration of STZ in a rodent's brain has been shown to produce neuroinflammation, oxidative stress and biochemical alterations, which is considered to be a valid experimental model of the early pathophysiological changes in neurodegenerative disease. STZ-induced spatial learning deficit in Morris water maze test and tau phosphorylation in rodent's brain produces sporadic AD (sAD) like pathology (Gao et al., 2014). Tau protein and Aβ are regarded as the main pathological features which are responsible for the pathological cascade which results in dementia i.e., loss of cognitive function, neuropsychiatric changes and finally causes death of neurons. On the other hand, in line with the recent findings of sAD being recognized as an insulin resistant brains state, a new non-transgenic animal model has been proposed as a representative model of sAD. The STZ treated animals develop insulin resistant brains state which is associated with memory impairment, progressive cholinergic deficits, glucose hypometabolism, oxidative stress and neurodegeneration that share many features in common with sAD in humans. Reports suggested that STZ exacerbates AD-like changes which were similar to the triple transgenic mice and other pathophysiology such as processing of amyloid precursor protein (APP), glucose metabolism, insulin signaling, synaptic function, protein kinases, and apoptosis (Chen et al., 2014). Triple transgenic mice consists of 3 rare familial mutant genes that in humans independently produce devastating Aβ protein precursor (AβPP), presenilin-1, and frontotemporal dementias; hence, technically speaking, these mice are not a model of sporadic pathology. These alterations by STZ suggest that it alters multiple metabolic and cell signaling pathways in the brain. One of the report by Zhang et al. (2015) suggested that STZ induced insulin deficiency in an AD transgenic mouse model produces β-amyloidogenic processing of APP in vitro and in vivo models. Thus from the above reports it inferred that STZ produces most prevalence type of AD.
Relevance of STZ induced AD model: The majority of AD cases are sporadic in origin and nature which are less clearly influenced by a single mutation of gene known as sAD. The etiology of sAD remains unclear but numerous risk factors that have been identified to increase the chance of AD progression. Among these risk factors, neuroinflammation of the brain has been implicated as leading risk factors. Subsequently neuroinflammation is one of the leading causes of sAD; however underlying molecular mechanism is not so clear. Since STZ produces similar characteristic pathology of sAD, so it can be used to explore the underlying pathophysiological mechanism.
STZ mediated neuroinflammation: Neuroinflammation is a pathological hallmark of AD and precedes plaque and tangle formation during AD progression (Kamat et al., 2012). There is a close association of neuroinflammation with the pathogenesis of AD and involves the activation of glial cells in neurodegenerative diseases such as AD (Rai et al., 2014). Rai et al. (2013) suggest that STZ treatment caused enhanced neuroinflammatory mediators and altered redox stress that contribute to the neurodegenerative processes. These free radicals also further trigger the neuronal damage via formation of pro-inflammatory mediators and associated cytotoxic products during neuroinflammation that can be detrimental to neuronal function. Reposts suggested that neuronal insulin protects from neuroinflammation and redox stress thus any impaired insulin function in brain hamper neuronal function (Figure 1).
Figure 1.
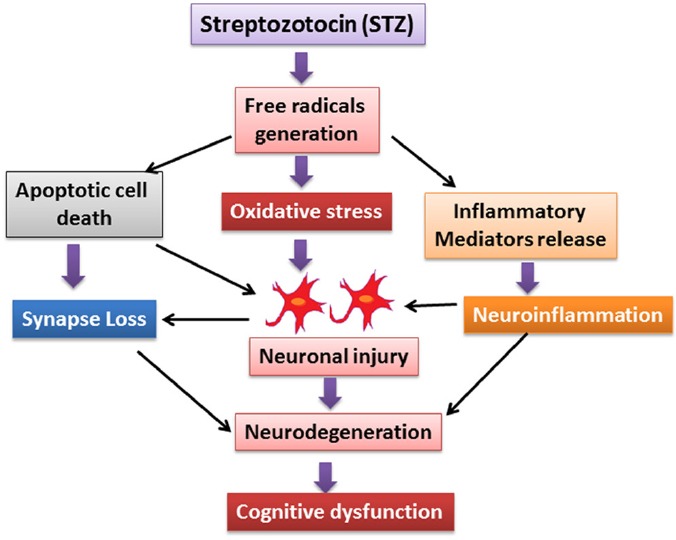
The flow diagram represents the streptozotocin (STZ) mediated free radical generation and their influence on oxidative stress, apoptotic cell death and neuroinflammation. These entire pathological factors collectively affect the neuronal function which may further implicate in Alzheimer's disease pathology.
Neuroprotective role of insulin in brain: Interestingly, insulin can directly modulate synaptic plasticity and learning and memory, and disturbances in insulin signaling pathways in the periphery and in the brain have recently been implicated in AD and aging brain. Insulin also negatively regulates the metabolism of Aβ and tau proteins which are key building blocks of amyloid plaques and neurofibrillary tangles, and are well documented neuropathological hallmarks of AD (Figure 2). These negative regulations by insulin lead to more devastating for neurological function. Conversely, insulin injected into the brain intracerebroventricularly can improve performance of memory tasks in animals (Grunblatt et al., 2006). Also, insulin delivered intranasally increased the performance of attention-related tasks in humans (Holscher, 2011). A recent study reported that insulin receptor levels are down-regulated in the brains of AD patients (Long-Smith et al., 2013). Insulin receptors were found to be internalized in neurons, and both insulin receptors (IRs)-1 and IRS-2 were reduced which taken together leads to reduced insulin signaling activity. It is becoming increasingly apparent that alteration of signaling molecules that are known to be involved in insulin signal transduction may play a role in the pathogenesis of AD (Stohr et al., 2013). Although the actions of brain insulin are not fully understood, binding of insulin to its receptor initiates autophosphorylation of the receptor's β-subunit, leading to binding and tyrosine phosphorylation of multiple insulin receptor substrates including IRS-1 and IRS-2, which play a role in synaptic plasticity and memory formation.
Figure 2.
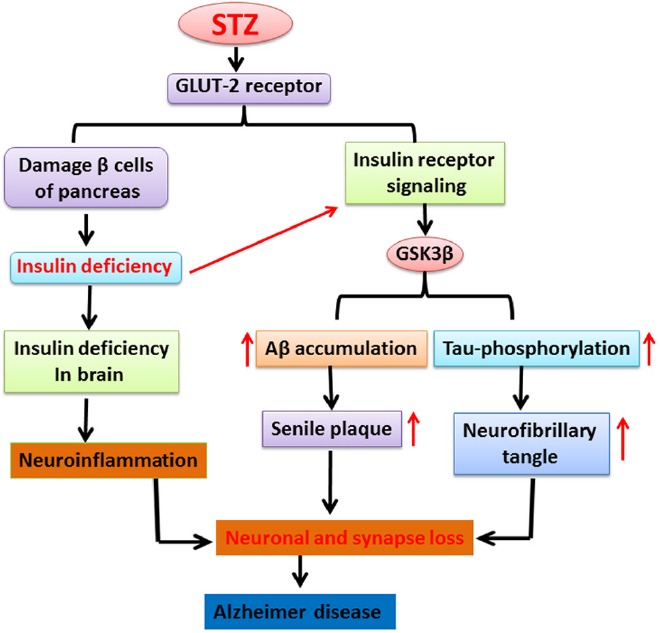
The cartoon illustrates the role of streptozotocin (STZ) in damage of pancreatic beta cells which lead to insulin deficiency. On the other hand, STZ also enhances low insulin level and impaired insulin receptor in brain that causes alteration of glycogen synthase kinase. Therefore, these alterations stimulate increased tau phosphorylation, formation of neurofibrillary tangle and finally implicated in neuronal and synapse dysfunction resulting into Alzheimer's disease like pathology.
GLUT-2: Glucose transporter 2; GSK3β: glycogen synthase kinase β; Aβ: amyloid beta.
STZ induced diabetic like condition and AD pathology: STZ is a naturally occurring chemical that is particularly toxic to the insulin-producing beta cells of the pancreas in mammals. It is used in medical research to produce an animal model for type 1 diabetes mellitus at large doses and for type 2 diabetes mellitus. There is accumulating evidence for a pathogenetic link between sAD and diabetes mellitus. Interestingly, a few studies also showed that type 2 diabetes mellitus exhibited Aβ accumulation in similar regions in AD and diabetes mellitus postmortem brains. STZ induced experimental models in rodents approximately mimic the age-related pathology of sAD in humans. STZ induced animal model is immensely important for challenging the novel therapeutic approaches for sAD treatment. Comprehensively used transgenic rodents models of AD have provided valuable perceptions into the underlying molecular mechanisms and pathophysiology such as Aβ-related gene manipulation. The transgenic models are thus less suitable to study the sAD pathology and therefore STZ is being used for AD model. Furthermore, several other reports have indicated that GSK-3 modulates splitting of APP into small fragments. On the other hand, at sub-diabetogenic doses i.e., intracerebral injection of the diabetogenic substance STZ induces an insulin-resistant brain state in rodent. Results showed that, in transgenic rodent's tg2576 APP-overproducing mice insulin resistant brains state, induced by STZ, may aggravate AD like pathological changes such as behavioral changes and increase the formation of pathological AD hallmarks via the GSK-3 alpha/beta pathway.
STZ and insulin resistance in brain: Insulin resistance occurs when the tissues fails responding against insulin. Clinical reports suggest that reduced expression of genes such as insulin like growth factor-1 (IGF-1), and IGF-1 receptors is present in the frontal cortex, hippocampus and hypothalamus of AD post mortem brains. Lee et al. (2014) demonstrated that expression of insulin/IGF signaling-related genes is mainly impaired in the cerebrum, frontal cortex and hippocampus in STZ injected monkey brain and that is similar to the early stage of sAD. The changes were also accompanied by the loss of oligodendrocytes and neurons. Concurrent with this observation, a recent study suggested that insulin deficiency and insulin resistance in brain are strongly correlated with late onset of sAD. It is a very well-known fact that STZ injection selectively destroys insulin reducing/secreting β cells in the pancreas, causing type I diabetes mellitus in adult animals. Intracerebroventricular injection of STZ causes central insulin resistance by damaging IR signaling and thus, increases desensitization of IRs. Protein kinase C, a down regulating molecule in insulin signaling pathway associated with IR signaling, may also contribute to insulin resistance by phosphorylating the IR substrates required for proper insulin function. On contrary, reports suggest that STZ injection in the brain at subdibetogenic dose creates the insulin resistant state which eventually leads to the development of Aβ and tau pathology (Shingo et al. 2012).
Conclusion and future direction: A large number of adaptable features have been reported to be associated with risk factor for AD such as diabetes and age associated free radical generation which promotes neuronal degeneration. Administration of STZ through different routes leads to brain insulin resistance, a central disorder appeared in diabetes brain and several AD-like pathologies including accumulation of Aβ, tau hyperphosphorylation, free radical generation and progressive impairment of cognitive function. Since STZ produces similar characteristic pathology of sporadic type AD which is highly prevalence type AD. Thus STZ induced AD pathology could bring new therapeutic paradigm and can be used to explore the underlying neural regeneration and degeneration mechanism.
This work was supported in part by the Council of Scientific and Industrial Research (CSIR), India and financial support to Pradip Kumar Kamat is greatly acknowledged.
References
- Chen S, An FM, Yin L, Liu AR, Yin DK, Yao WB, Gao XD. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience. 2014;256:137–146. doi: 10.1016/j.neuroscience.2013.10.038. [DOI] [PubMed] [Google Scholar]
- Gao C, Liu Y, Jiang Y, Ding J, Li L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3beta pathway in streptozotocin-induced Alzheimer rat model. Brain Pathol. 2014;24:261–269. doi: 10.1111/bpa.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C. Diabetes as a risk factor for Alzheimer's disease: insulin signalling impairment in the brain as an alternative model of Alzheimer's disease. Biochem Soc Trans. 2011;39:891–897. doi: 10.1042/BST0390891. [DOI] [PubMed] [Google Scholar]
- Kamat PK, Rai S, Swarnkar S, Shukla R, Nath C. Mechanism of synapse redox stress in okadaic acid (ICV) induced memory impairment: role of NMDA receptor. Neurochem Int. 2014;76:32–41. doi: 10.1016/j.neuint.2014.06.012. [DOI] [PubMed] [Google Scholar]
- Kamat PK, Tota S, Rai S, Swarnkar S, Shukla R, Nath C. A study on neuroinflammatory marker in brain areas of okadaic acid (ICV) induced memory impaired rats. Life Sci. 2012;90:713–720. doi: 10.1016/j.lfs.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Lee CH, Ahn JH, Park JH, Yan BC, Kim IH, Lee DH, Cho JH, Chen BH, Lee JC, Cho JH, Lee YL, Won MH, Kang IJ. Decreased insulin-like growth factor-I and its receptor expression in the hippocampus and somatosensory cortex of the aged mouse. Neurochem Res. 2014;39:770–776. doi: 10.1007/s11064-014-1269-3. [DOI] [PubMed] [Google Scholar]
- Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, O’Neill C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer's disease. Neuromolecular Med. 2013;15:102–114. doi: 10.1007/s12017-012-8199-5. [DOI] [PubMed] [Google Scholar]
- Rai S, Kamat PK, Nath C, Shukla R. A study on neuroinflammation and NMDA receptor function in STZ (ICV) induced memory impaired rats. J Neuroimmunol. 2013;254:1–9. doi: 10.1016/j.jneuroim.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Rai S, Kamat PK, Nath C, Shukla R. Glial activation and post-synaptic neurotoxicity: the key events in Streptozotocin (ICV) induced memory impairment in rats. Pharmacol Biochem Behav. 2014;117:104–117. doi: 10.1016/j.pbb.2013.11.035. [DOI] [PubMed] [Google Scholar]
- Shingo AS, Kanabayashi T, Kito S, Murase T. Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats. Behav Brain Res. 2013;241:105–111. doi: 10.1016/j.bbr.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Stohr O, Schilbach K, Moll L, Hettich MM, Freude S, Wunderlich FT, Ernst M, Zemva J, Bruning JC, Krone W, Udelhoven M, Schubert M. Insulin receptor signaling mediates APP processing and beta-amyloid accumulation without altering survival in a transgenic mouse model of Alzheimer's disease. Age. 2013;35:83–101. doi: 10.1007/s11357-011-9333-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yin F, Liu J, Liu Z, Guo L, Xia Z, Zidichouski J. Geniposide attenuates insulin-deficiency-induced acceleration of beta-amyloidosis in an APP/PS1 transgenic model of Alzheimer's disease. Neurochem Int. 2015 doi: 10.1016/j.neuint.2015.04.002. pii: S0197-0186(15)00063-7. [DOI] [PubMed] [Google Scholar]