

Abstract
Necroptosis is characterized by programmed necrotic cell death and autophagic activation and might be involved in the death process of dopaminergic neurons in Parkinson's disease. We hypothesized that necrostatin-1 could block necroptosis and give protection to dopaminergic neurons. There is likely to be crosstalk between necroptosis and other cell death pathways, such as apoptosis and autophagy. PC12 cells were pretreated with necroststin-1 1 hour before exposure to 6-hydroxydopamine. We examined cell viability, mitochondrial membrane potential and expression patterns of apoptotic and necroptotic death signaling proteins. The results showed that the autophagy/lysosomal pathway is involved in the 6-hydroxydopamine-induced death process of PC12 cells. Mitochondrial disability induced overactive autophagy, increased cathepsin B expression, and diminished Bcl-2 expression. Necrostatin-1 within a certain concentration range (5–30 μM) elevated the viability of PC12 cells, stabilized mitochondrial membrane potential, inhibited excessive autophagy, reduced the expression of LC3-II and cathepsin B, and increased Bcl-2 expression. These findings suggest that necrostatin-1 exerted a protective effect against injury on dopaminergic neurons. Necrostatin-1 interacts with the apoptosis signaling pathway during this process. This pathway could be a new neuroprotective and therapeutic target in Parkinson's disease.
Keywords: nerve regeneration, neurodegeneration, necrostatin-1, necroptosis, apoptosis, cytotoxicity, 6-hydroxydopamine, Parkinson's disease, neuroprotection, autophagy, necrosis, programmed cell death, neurodegenerative disease, PC12 cells, neural regeneration
Introduction
Necroptosis is a unique type of cell death observed under different experimental conditions and pathological processes (Fayaz et al., 2014; Linkermann and Green, 2014; Re et al., 2014). It is characterized by programmed necrotic cell death accompanied by autophagic pathway activation (Chavez-Valdez et al., 2012; Jain et al., 2013). Necrostatin-1 (Nec-1) was discovered to be a specific inhibitor of this pathway (Degterev et al., 2005). Several studies have disrupted necroptosis to investigate the neuroprotective effect of Nec-1 and have identified a protective potential in different models (Smith et al., 2007; Degterev et al., 2008; Linkermann et al., 2012; Oerlemans et al., 2012). Recently, emerging evidence has indicated the involvement of this pathway in neurodegenerative diseases (Le, 2014). Does necroptosis play a role in the pathophysiology of Parkinson's disease? If yes, then, could Nec-1 be a preventive or therapeutic agent for this intractable disease? Oxidative stress has been demonstrated as the crucial mechanism of dopaminergic cell death in Parkinson's disease (Dias et al., 2013; Streck et al., 2013). Autophagy, which is downstream of necroptosis, is involved in dopaminergic cell death in Parkinson's disease (Lynch-Day et al., 2012; Arduino et al., 2013; Pan and Yue, 2014). Nec-1 can attenuate oxidative stress and mitochondrial dysfunction in neurons following neonatal hypoxia-ischemia (Chavez-Valdez et al., 2012), and can also play an important protective role in myocardial ischemia/reperfusion injury (Oerlemans et al., 2012). Hence, we hypothesize that Nec-1 can protect PC12 cells from oxidative cytotoxicity induced by 6-hydroxydopamine (6-OHDA) by interfering with necroptosis.
Materials and Methods
Cell culture and treatment
PC12 cells (Chinese Academy of Sciences Cell Bank, Shanghai, China) were cultivated in Dulbecco's modified Eagle's medium (DMEM) (Gibco, New York, NY, USA), supplemented with 100 U/mL penicillin, 100 mg/mL streptomycin and 10% heat-inactivated fetal bovine serum (Beijing Beyotime Institute of Biotechnology, Beijing, China). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 before collection at passages 10–20 and seeded on poly-L-lysine-coated plates (Sigma, St. Louis, MO, USA) at a density of 2 × 105 cells/mL. Cells in the Nec-1 treated group were pretreated with different concentrations of Nec-1 for 1 hour before being challenged by 6-OHDA (final concentration 100 μM; Sigma). Cells in the control groups were treated with medium followed by 6-OHDA or saline. All cells were harvested at 24 hours after drug administration for further investigation.
Growth-arrested PC12 cells were divided into four groups: normal DMEM group (NS group), 100 μM 6-OHDA DMEM group (6-OHDA group), 10 μM Nec-1 + 100 μM 6-OHDA DMEM group (6-OHDA + Nec-1 group), and 0.2% dimethyl sulfoxide + normal DMEM group (vehicle group).
Colorimetric MTT assay
Cell viability was measured by the MTT assay according to previous methods (Wang et al., 2011). Cell viability was expressed as a percentage of cells in the normal control. To determine the protective effect of Nec-1 against neurotoxicity, PC12 cells were exposed to Nec-1 at the concentrations of 5, 10, 20, 30, 60 and 90 μM before 6-OHDA treatment for 1 hour. The viability of the cells was assessed as described above.
Detection of autophagy by fluorescence staining
PC12 cells were seeded into 24-well plates containing cover slips pre-coated with 0.1% poly-lysine and then treated with 100 μM of 6-OHDA after cell synchronization (Wang et al., 2011). The cultivation medium was then replaced with fresh medium containing 50 μM monodansylcadaverine (Sigma) at 2, 4, 6, 12 and 24 hours after cell synchronization, and these cells were further cultured at 37°C in a humidified atmosphere containing 5% CO2 for 10 minutes. After two washes with D-Hank's solution, the cover slips were removed and placed onto gelatin pre-treated slides and observed under an inverted fluorescence microscope (Olympus, Tokyo, Japan).
Mitochondrial membrane potential assay
The disruption of active mitochondria, including changes in mitochondrial membrane potential, is a distinctive feature of the early stage of apoptosis. This change can be determined using the membrane permeant dye, JC-1 (Beijing Beyotime Institute of Biotechnology). After exposure to this dye, a potential-sensitive color shift from red to green suggests mitochondrial depolarization. At 24 hours after 6-OHDA treatment, PC12 cells were incubated with 5 μg/mL JC-1 dye at 37°C for 20 minutes. Cells were then rinsed twice with PBS. Mitochondrial membrane potentials were monitored by determining the relative amount of dual emissions from mitochondrial JC-1 monomers or aggregates using a fluorescence microscope (Olympus, Tokyo, Japan) equipped with an argon ion laser emitting at 488 nm.
Western blot assay
PC12 cells were seeded in 60 mm dishes at a density of 2 × 105 cells/mL. After removing medium, cells were washed twice with ice-cold PBS, and then lysed on ice using lysis buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM ethylenediamine tetraacetic acid, 1% Na3VO4, 0.5 μg/mL leupeptin, and 1 mM phenylmethanesulfonyl fluoride for 20 minutes. The collected lysates were centrifuged at 12,000 × g at 4°C for 5 minutes to give supernatants for western blot assay. Protein concentrations were determined by the Bradford protein assay. Equal amounts of protein (20 μg) from each sample were loaded onto 12.5% sodium dodecyl sulfate-polyacrylamide gels for electrophoresis, followed by electro-transfer onto nitrocellulose membranes (Amersham Biosciences Buckinghamshire, UK) at 0.8 mA/cm2 for 30–60 minutes. Membranes were blocked at room temperature for at least 1 hour with 5% skimmed milk in Tris-buffered saline and Tween 20 (50 mM Tris/HCl, 150 mM NaCl, pH 7.5, 0.2% v/v Tween-20) and then incubated overnight at 4°C with rabbit anti-light chain 3 (LC3) antibody (1:500; Sigma), mouse anti-casthepsin B antibody (1:500; Sigma), mouse anti-Bcl-2 antibody (1:500; Zhongshan Golden Bridge Biotechnology Company, Beijing, China) or rabbit or mouse β-actin (1:1,000; Sigma-Aldrich, Madrid, Spain) antibodies. After washing with Tris-buffered saline and Tween 20, membranes were reacted with alkaline phosphatase-conjugated anti-mouse IgG or anti-rabbit secondary antibodies (1:1,000; Zhongshan Golden Bridge Biotechnology Company) at room temperature for 2 hours. NBT/BCIP (Promega, Madison, WI, USA) was used to develop membranes. Membranes were scanned and analyzed on an image analyzer (Quantity One, Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analysis
One-way analysis of variance followed by the least significant difference test (two-tailed) was performed to compare the effects of Nec-1 on PC12 cells using SPSS 16.0 software (SPSS, Chicago, IL, USA). Data are expressed as the mean ± SD. A P value < 0.05 was considered statistically significant.
Results
Effects of Nec-1 on PC12 cell viability
The viability of PC12 cells in the 6-OHDA group was significantly lower than that in the NS group (P < 0.01), indicating that 6-OHDA caused injury to cells. Treatment with 5, 10, 20 and 30 μM Nec-1 could substantially reverse the decreased viability, although further cell deterioration occurred with Nec-1 treatment of 60 and 90 μM (P < 0.01). This finding suggests a potential neuroprotective effect of Nec-1 at concentrations of 5–30 μM (Figure 1).
Figure 1.
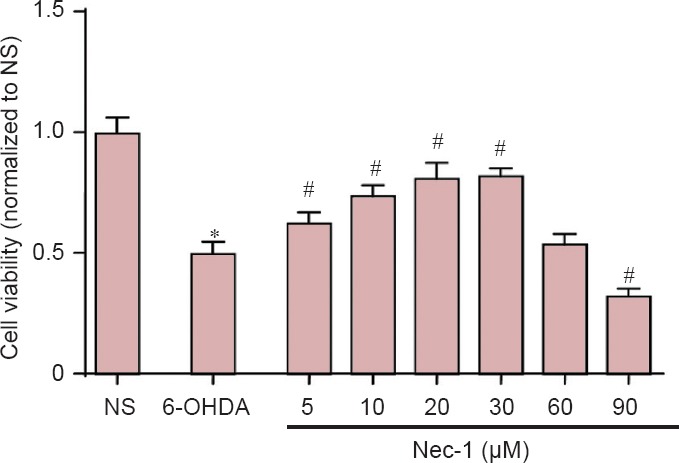
Effects of Nec-1 on PC12 cell viability assessed by MTT assay.
Cells were treated with 6-OHDA for 24 hours. *P < 0.01, vs. NS group; #P < 0.05, vs. 6-OHDA group. Data are expressed as the mean ± SD (The number of samples in each group was six). NS group: Normal DMEM media; 6-OHDA group: 100 μM 6-OHDA. Six different Nec-1 groups: 5, 10, 20, 30, 60 and 90 μM Nec-1 + 100 μM 6-OHDA. The viability of PC12 cells was represented as the ratio of the average absorbance value of three parallel experiments from treated groups to the control. Nec-1: Necrostatin-1; 6-OHDA: 6-hydroxydopamine; DMEM: Dulbecco's modified Eagle's medium; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide.
Changes to autophagic vacuoles in PC12 cells
After 2 hours of treatment with 6-OHDA (100 μM), autophagic vacuoles started to appear as fluorescent particles clearly scattered within the cytoplasm. This staining was not observed in the NS group. Large quantities of fluorescent particles were visible at 6 and 12 hours, which indicated their continuous production. However, at 24 hours after treatment, the number of fluorescent particles had declined, indicating fewer autophagic vacuoles in the cytoplasm (Figure 2).
Figure 2.
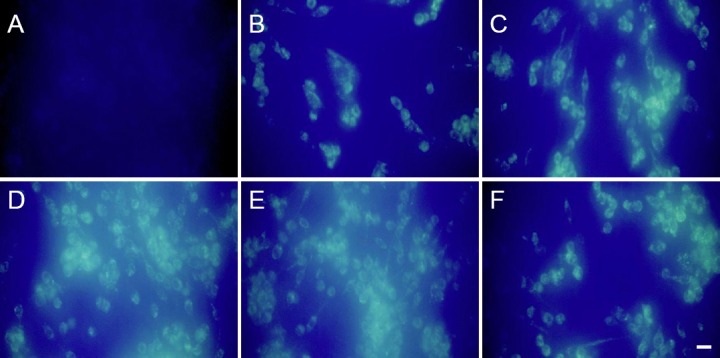
Changes in autophagic vacuoles in PC12 cells (monodansyl cadaverine fluorescence staining).
PC12 cells were cultured on cover slips. Fluorescent particles indicate autophagy vacuoles. After 2 hours of 6-OHDA treatment (100 μM), autophagic vacuoles started to appear, which were not seen in the NS group. Large quantities of autophagy vacuoles were observed at 6 and 12 hours after treatment. Scale bars: 20 μm. (A) NS group; (B–F) 2, 4, 6, 12 and 24 hours after treatment, respectively. 6-OHDA: 6-Hydroxydopamine; NS group: normal Dulbecco's modified Eagle's medium.
Nec-1 effects on mitochondrial membrane potential in PC12 cells treated with 6-OHDA
PC12 cells in the NS group stained with JC-1 dye displayed orange-red fluorescence in mitochondria. 6-OHDA treatment produced obvious green fluorescence, indicating remarkably reduced mitochondrial potentials. Fluorescence was shifted towards orange-red when cells were exposed to a combination of 6-OHDA and Nec-1 (P < 0.01; Figure 3).
Figure 3.
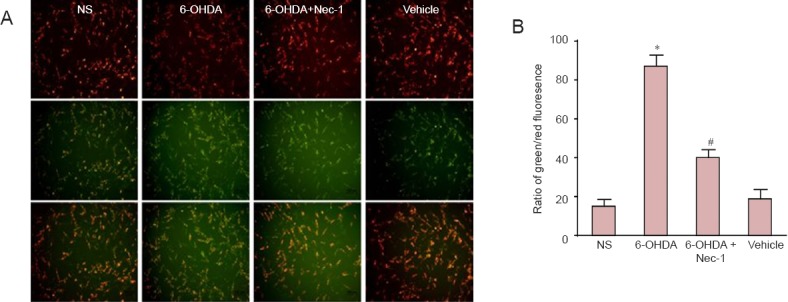
Nec-1 attenuated mitochondrial membrane potential reduction in PC12 cells induced by 6-OHDA.
Growth-arrested PC12 cells were exposed to NS, 6-OHDA, 6-OHDA + Nec-1, or vehicle for 24 hours. (A) Representative photographs of JC-1 staining in different groups. The cells were stained with JC-1 and imaged under a fluorescence microscope to analyze the ratio of green to red fluorescence. An increase in green/red fluorescence indicates mitochondrial depolarization. (B) Quantitative analysis of a shift from bright orange-red fluorescence to green. All the values are expressed as the mean ± SD (The number of samples in each group was six). There were ten parallel images in each group. *P < 0.01, vs. NS group; #P < 0.01, vs. 6-OHDA group. NS group: Normal DMEM media; 6-OHDA: 100 μM 6-OHDA; 6-OHDA + Nec-1: 10 μM Nec-1 + 100 μM 6-OHDA; vehicle: 0.2% dimethyl sulfoxide in normal DMEM medium. Nec-1: Necrostatin-1; 6-OHDA: 6-hydroxydopamine; DMEM: Dulbecco's modified Eagle's medium.
Effects of Nec-1 on LC3, cathepsin B and Bcl-2 expression in PC12 cells treated with 6-OHDA
Normally, cytosolic LC3-I is detected in PC12 cells, while LC3-II is not because it is bound to phosphatidylethanolamine in the autophagosome membrane. After 6-OHDA treatment, the activation of LC3-II gradually increased over 24 hours (Figure 4A). This activation was down-regulated if the cells were pre-treated with Nec-1 (Figure 4B). Pretreatment with Nec-1 suppressed the expression of cathepsin B and increased the level of Bcl-2 (Figure 4C and D).
Figure 4.
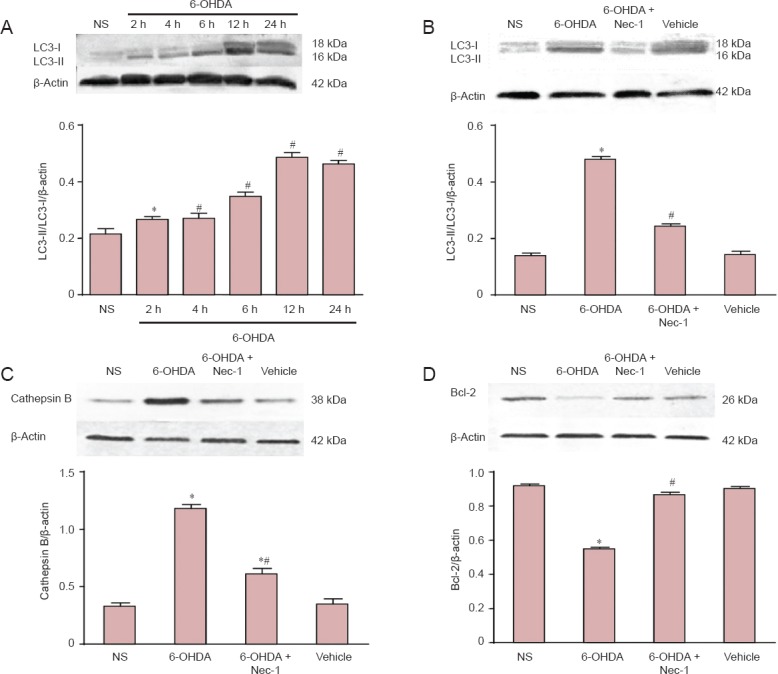
Effects of Nec-1 on the protein expression of LC3-II, Cathepsin B and Bcl-2 in PC12 cells (western blot assay).
Growth-arrested PC12 cells received the following treatments: (A) NS, 100 μM 6-OHDA for 2, 4, 6, 12 and 24 hours; (B–D) NS, 100 μM 6-OHDA, 10 μM Nec-1 + 6-OHDA, or vehicle. After the indicated exposure times (A), the cells were harvested and lysed. The relative absorbance is represented as a fold value compared with the NS group. Data are expressed as the mean ± SD (The number of samples in each group was six). *P < 0.05, vs. NS group; #P < 0.05, vs. 6-OHDA group. NS group: Normal DMEM medium; 6-OHDA: 100 μM 6-OHDA; 6-OHDA + Nec-1: 10 μM Nec-1 + 100 μM 6-OHDA; vehicle: 0.2% dimethyl sulfoxide in normal DMEM medium. Nec-1: Necrostatin-1; 6-OHDA: 6-hydroxydopamine; LC3: microtubule-associated protein 1A/1B-light chain 3.
Discussion
Apoptosis in the pathogenesis of Parkinson's disease has been widely studied. However, novel non-apoptotic pathways have been discovered in recent years (Cabon et al., 2013; Cho, 2014; Feoktistova and Leverkus, 2015). A general consensus of neurodegenerative disease is that complex signal transduction involving different pathways occurs in neurons during degeneration (Perier et al., 2012; Venderova and Park, 2012) and cross-talk among different cell death pathways has been intensively studied in Parkinson's disease. In the present study, 6-OHDA-induced PC12 cell death manifested both apoptotic and autophagic features. LC3 and cathepsin B levels increased in PC12 cells after 6-OHDA treatment, and a prosurvival effect of Nec-1 was found in this cytotoxic model. This indicated not only apoptosis but also non-apoptotic pathways were evoked by these experimental conditions. Nec-1 can attenuate oxidative stress and mitochondrial dysfunction in neurons following neonatal hypoxia-ischemia (Northington et al., 2011; Chavez-Valdez et al., 2012) and can produce protective effects against neuronal injury induced by ischemia/reperfusion (Xu et al., 2010). Here, supporting our hypothesis, blocking necroptosis signaling attenuated 6-OHDA cytotoxicity of PC12 cells. In this study, 5, 10, 20, and 30 μM of Nec-1 protected PC12 cells from 6-OHDA-induced cytotoxicity, and also reversed decreased mitochondrial membrane potentials. However, the results showed that 60 and 90 μM Nec-1 caused a substantial decrease in the viability of PC12 cells, demonstrating dual effects of Nec-1; protection at lower concentrations in comparison with toxicity at higher concentrations. This result was consistent with a point of discussion made by Smith and Yellon (2011).
Apoptosis plays an important role in the degeneration of substantia nigra dopaminergic neurons in Parkinson's disease (Duyckaerts et al., 2010). Animal models and post-mortem material have enabled morphological observations of changes in apoptosis, while immunohistochemical evidence indicates activation of caspases in substantia nigra dopaminergic neurons (Vila and Przedborski, 2003). Recent studies have focused on the crosstalk among apoptosis, neroptosis and other death pathways (Venderova and Park, 2012; Arduino et al., 2013; Nikoletopoulou et al., 2013). In the present study, Bcl-2 protein in PC12 cells was down-regulated by 6-OHDA, but was protected by pretreatment of Nec-1. This implies that Nec-1 pretreatment also interfered with apoptosis in this process. The changes in mitochondrial membrane potential and mitochondrial function in 6-OHDA-treated PC12 cells were inhibited by Nec-1, which indicates a potential anti-apoptotic mechanism of Nec-1 by stabilizing the mitochondrial membrane. Autophagy can be a survival strategy and also a cell death pathway (Tung et al., 2012). The results of the present study also suggest the activation of autophagy in PC12 cells after 6-OHDA exposure. The increased expression of LC3-II and cathepsin B induced by 6-OHDA treatment were reversed by Nec-1 pretreatment. This indicates that blocking necroptosis by Nec-1 could prevent PC12 cells from undergoing autophagic cell death and downstream necroptotic signaling. Inhibition of necroptosis could be a new target to protect cells from oxidative stress. Based on these limited data, we cannot answer the question of whether Nec-1 has a protective effect in animal models of dopaminergic neurotoxicity. In vivo testing of Nec-1 may give new clues for neuroprotection in Parkinson's disease.
Acknowledgments
We thank Dong HY from the Laboratory of Neurobiology, Wang RZ and Zhao XM from the Laboratory of Pathology, Xuzhou Medical College in China for their technical support.
Footnotes
Funding: This study was supported by grants from the Science and Technology Project of Xuzhou City in China, No. XM12B017; and the Priority Academic Program Development of Jiangsu Higher Education Institutions in China.
Conflicts of interest: None declared.
Copyedited by Allen J, Stow A, Wang J, Qiu Y, Li CH, Song LP, Zhao M
References
- Arduino DM, Esteves AR, Cardoso SM. Mitochondria drive autophagy pathology via microtubule disassembly: a new hypothesis for Parkinson disease. Autophagy. 2013;9:112–114. doi: 10.4161/auto.22443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabon L, Martinez-Torres AC, Susin SA. Programmed cell death comes in many flavors. Med Sci (Paris) 2013;29:1117–1124. doi: 10.1051/medsci/20132912015. [DOI] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ. Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience. 2012;219:192–203. doi: 10.1016/j.neuroscience.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS. Perspectives on the therapeutic modulation of an alternative cell death, programmed necrosis (review) Int J Mol Med. 2014;33:1401–1406. doi: 10.3892/ijmm.2014.1716. [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Chen IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Sazdovitch V, Seilhean D. Update on the pathophysiology of Parkinson’ disease. Bull Acad Natl Med. 2010;194:1287–1304. [PubMed] [Google Scholar]
- Fayaz SM, Suvanish KVS, Rajanikant GK. Necroptosis: who knew there were so many interesting ways to die. CNS Neurol Disord Drug Targets. 2014;13:42–51. doi: 10.2174/18715273113126660189. [DOI] [PubMed] [Google Scholar]
- Feoktistova M, Leverkus M. Programmed necrosis and necroptosis signalling. FEBS J. 2015;282:19–31. doi: 10.1111/febs.13120. [DOI] [PubMed] [Google Scholar]
- Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K, Schweizer F, Lengerke C, Davoodpour P, Palicharla VR, Maddika S, Los M. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J Cell Mol Med. 2013;17:12–29. doi: 10.1111/jcmm.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le VV. Familial and sporadic ALS astrocytes activate necroptosis in motor neurons. Med Sci (Paris) 2014;30:748–750. doi: 10.1051/medsci/20143008011. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–761. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009357. doi: 10.1101/cshperspect.a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, Sluijter JP. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107:270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- Pan PY, Yue Z. Genetic causes of Parkinson's disease and their links to autophagy regulation. Parkinsonism Relat Disord. 2014;20 Suppl 1:S154–157. doi: 10.1016/S1353-8020(13)70037-3. [DOI] [PubMed] [Google Scholar]
- Perier C, Bove J, Vila M. Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid Redox Signal. 2012;16:883–895. doi: 10.1089/ars.2011.4074. [DOI] [PubMed] [Google Scholar]
- Re DB, Le VV, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent. Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- Smith CC, Yellon DM. Necroptosis, necrostatins and tissue injury. J Cell Mol Med. 2011;15:1797–1806. doi: 10.1111/j.1582-4934.2011.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streck EL, Czapski GA, Goncalves dSC. Neurodegeneration, mitochondrial dysfunction, and oxidative stress. Oxid Med Cell Longev. 2013 doi: 10.1155/2013/826046. 826046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung YT, Wang BJ, Hu MK, Hsu WM, Lee H, Huang WP, Liao YF. Autophagy: a double-edged sword in Alzheimer's disease. J Biosci. 2012;37:157–165. doi: 10.1007/s12038-011-9176-0. [DOI] [PubMed] [Google Scholar]
- Venderova K, Park DS. Programmed cell death in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009365. doi: 10.1101/cshperspect.a009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:365–375. doi: 10.1038/nrn1100. [DOI] [PubMed] [Google Scholar]
- Wang J, Cheng YB, Yin JL, Lu Q, Xu XS, Yin XX. Protective effects of Ginkgo biloba extract on 6-hydroxydopamine-induced apoptosis in PC12 cells. Neural Regen Res. 2011;6:2565–2572. [Google Scholar]
- Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]