

Abstract
Chemokine (C-C motif) ligand 2 (CCL2) has recently been found to be a key player in the pathology of many human glomerular and tubulointerstitial diseases. CCL2 has also been found to be expressed in various cancers, including human hepatoma cells, human cancer progression, and human multiple myeloma cells. Thus, the inhibition of elevated CCL2 production may provide a new avenue for therapeutic intervention in CCL2-mediated cancer diseases. A previous study has indicated that knockdown of human p53 has a strong negative impact on CCL2 induction. We therefore are interested in how p53 regulates CCL2 gene expression. In the following study, our findings indicate that p53 binds to CCL2, consequently significantly downregulating CCL2 promoter activity. Furthermore, injection of CCL2-promoting cancer cells (CCL2/A549) in p53-deficient mice for 3 weeks strongly induced subcutaneous xenograft tumor growth compared with the control. Overall, the research results support the novel role of p53 in suppression of chemokine (such as CCL2)-mediated cancer diseases.
Keywords: p53, Suppression, Downregulates, CCL2 promoter activity, Xenograft
Introduction
It is well known that p53 regulates apoptotic signaling pathways. The isoforms of human p53 with the alternative translation sites have been studied [1, 2]. Aberrant expression of these isoforms occurs in a variety of tumors [3, 4]. In addition, deficiency or mutation of p53 has been linked to autoimmune disorders [5] and lung inflammation. As certain cancers are caused by chronic inflammation, regulation of this inflammation may ultimately lead to tumor suppression [6, 7]. However, the potential role of p53 in regulating the inflammatory genes that cause chronic lung disease [8] needs to be investigated further.
It is well known that chemokines and chemokine receptors are involved in a variety of inflammatory disorders [9–11]. Chemokine (C-C motif) ligand 2 (CCL2; also named MCP-1) has been suggested as a potential target in inflammatory diseases due to the gene’s several-fold production increase in patients’ peripheral blood, synovial fluid, and synovial tissue [12]. CCL2 has also been suggested to be a potent mediator of monocytes/macrophages, as these cells have been shown to be directly involved in the induction and perpetuation of synovitis and the subsequent joint destruction in rheumatoid arthritis [13]. Macrophages/monocytes modulated by the CCL2/CCR2 axis have recently been identified as key players in the pathology of many human glomerular and tubulointerstitial diseases [14]. CCL2 has been found to be regulated at the protein level and elevated in cancer cells [15]. Inhibition of CCL2 during tumor development resulted in decreased tumor volume in tumor-bearing mice [16]. Thus, the inhibition of elevated CCL2 production may provide a new therapeutic intervention in CCL2-induced inflammatory and cancer diseases [17].
We have previously shown that p53 is involved in inflammatory gene expression [18, 19]. Additionally, a recent study has indicated that the knockdown of p53 leads to a strong negative regulation of CCL2 induction [20]. Therefore, we are interested in a model to test how p53 suppresses CCL2-mediated cancer disease.
In the present study, we found that UV-induced p53 accumulation in cells had significantly decreased CCL2 promoter activity. In order to further investigate the mediation of CCL2 transcriptional activity by p53, we analyzed CCL2 5′UTR&promoter along with its derived DNA constructs. We found that p53 interacts with CCL2. Furthermore, the growth of cancer cell-induced subcutaneous tumor xenograft in p53-deficient mice was fast after subcutaneous injection of CCL2/A549 cells (CCL2-promoting cancer cells) compared with the control. These results may pave the way for future studies linking p53 and intervention of cancer diseases mediated by CCL2 or other chemokines.
Material and methods
Cell culture
All bacterial cloning constructs used Escherichia coli strain Top10 (Invitrogen). Cells of A549 (human lung cancer cells) or CRL-2280 (Mus musculus) were obtained from ATCC and grown in RPMI medium 1640 supplemented with 10 % fetal bovine serum (FBS) and maintained in a humidified atmosphere of 5 % CO2 at 37 °C.
Mice
Mice with p53 heterozygous type (129S2-Trp53tm1TYj/J) or with wild type as control (obtained from The Jackson Laboratory) were maintained following BU IACUC instructions (protocol number, AN-15,138).
DNA constructs
Primer pairs used for PCR of DNA constructs were shown in Table 1 and all cloned DNA were confirmed by DNA sequencing. 1. The clone pcp53WT, which contains a full-length mouse p53 gene (aa 1~391), was provided by Open Biosystems. 2. The clone pcCCL2, which contains a full-length mouse CCL2 gene (BC145867), was generated by PCR with 1 ng of mouse complementary DNA (cDNA) (provided by Open Biosystems) as template. Both DNA fragments above were inserted into protamine complementary DNA 3HA (pcDNA3HA) [21]. 3. A series of truncated mouse CCL2 5′UTR&promoter DNAs (GQ917241) were generated by PCR with mouse genomic DNA (Clontech) as template and appropriate primer pairs (Table 1) for mcl2pwt (a full length), mcl2p315, mcl2p115, or mcl2p53m (a mutation of a putative p53 binding site: Δ16–35 bp). All PCR products of reporter DNAs were purified and inserted into pGL3-basic vectors. 4. Recombinant lentivirus, a PCR-generated mouse CCL2 5′UTR&promoter DNA from mcL2pwt, a mouse CCL2 in-frame DNA from pcCCL2, or a luciferase in-frame DNA (pGL3-basic plasmid, Promega) was digested with by enzymes (BamHI, XhoI, HindIII, or ApaI) and ligated together with T4 ligase, then treated with pLenti6.3/V5-TOPO vectors (Invitrogen). The cloned DNA of the right size and orientation was screened and confirmed by DNA sequencing. The cloned DNA (mcl2p115/pcCCL2/luciferase/green fluorescent protein (GFP)) was co-transfected with ViraPower Packaging Mix (Invitrogen) into 293FT producer cells by using Lipofectamine 2000 (Invitrogen) and cultured at 37 °C, 5 % CO2 for 2–5 days. The viral pellet from its supernatant was harvested and suspended in an appropriate volume of PBS. The titer (1 × 108 pfu) of viral particles (named LentiCLG) was measured following manufacturer’s instruction.
Table 1.
Primer pairs used for PCR of DNA constructs
| DNA clone | 1st PCR amplification | 2nd PCR amplification | |
|---|---|---|---|
| No. | Name | Primer pair | Primer pair |
| 1 | Pcp 53wt | 5′-ATGACTGCCATGGAGGAGTC-3′+5′-TCAGTCT GAGTCAGGCCCCA-3′ |
|
| 2 | pcCCL2 | 5′-CTCGAGCCATGCAGGTCCCTGTCATG-3′+5′- AAGCTTCTAGTTCACTGTCACACTG-3′ |
|
| 3 | mcL2pwt | 5′-ACTCAGACAGTTCATATCAA-3′+5′-GGTGGT GGAGGAAGAGAGAGAG-3′ |
|
| 4 | mcl2p115 | 5′-CAACTTCCACTTTCCATCAC-3′+5′-GGTGGTG GAGGAAGAGAGAGAG-3′ |
|
| 5 | mcl2p315 | 5′-GCAGAGCCACTCCATTCACA-3′+5′-GGTGGTG GAGGAAGAGAGAG-3′ |
|
| 6 | mcl2p53m | 5′-CAACTTCCACTTTCCATCACTTATCCAGGGTG ATGCTACTCCTTGGCACCAAGCACCCTGCCT GACTCCACCCCCCTGGCTTACAATAAAAGGC TGCCTC-3′+5′-GGTGGTGGAGGAAGAGAGAG CTGGCTTCAGTGAGAGTTGGCTGGTGCTGGC GTCTGGCTCTCTGCACTTCTGGCTGCTCTGAG GCAGCCTTTTATTGTAA-3′ |
5′-CAACTTCCACTTTCCATCAC-3′+5′- GGTGGTGGAGGAAGAGAGAG-3′ |
Establishment of a stable cell line
A total of 1 × 106 A549 (human lung adenocarcinoma epithelial cell line) cells were infected with MOI: two of LentiCLG (as described in Table 1, for 3 days. A single cell containing a fusion DNA of CCL2 5′UTR&promoter/CCL2 cDNA/luciferase cDNA/GFP integrated into the chromosome was screened and transferred into a new plate until enough cells (about 10 ~ 100) were grown. The stable lung cancer cell line with biological markers (luciferase and GFP, driven by CCL2 5′UTR&promoter enhance element) was confirmed by luciferase assay, microscopy-based observation, and PCR. Cells were collected and named CCL2/A549. Cells were grown in DMEM with 10 % FBS and maintained in a 37 °C humidified atmosphere containing 5 % CO2.
Luciferase assay
A commercial kit was used (Cat# E1500, luciferase reporter assay system, Promega), and assay was performed according to the protocol provided by the manufacturer.
Western blot analysis
Cell lysate from each experimental group was detected by Western blot with antibodies against to actin (C-11, Santa Cruz), luciferase (sc28525, Santa Cruz), or p53 (FL-393-G, Santa Cruz). Protein band intensity was analyzed using VersaDoc Imaging System model 4000MP with Quantity One quantitation software version 4.6.3 (Bio-Rad).
IP-p53 constructions
A549 cells (1 × 106) were respectively transfected with individual p53 construction DNA using Lipofectamine 2000 (Invitrogen) for 3 h, washed with PBS, and cultured overnight. The protein from treated cells was extracted with lysis buffer (Promega) and immunoprecipitated (IP) with hemagglutinin (HA) plus IgG and a Protein A/G Plus-Agarose (sc-2003, Santa Cruz Biotechnology) following the manufacturer’s instructions. The IP protein from each concentration was used for electrophoresis mobility shift assay (EMSA).
EMSA
A commercial kit, Gel Shift Assay System (Promega), was used. IP proteins used for EMSA were prepared as described above. The protein level of each IP protein was confirmed by Western blot. A reaction mixture for EMSA contained 1 × 105 cpm/μL of radiolabeled double-stranded DNA probe (ccl2/p53oligo), 1 mM DTT, 1 μg each of IP protein (omitted from control), 2 μL of 5× binding buffer (Promega), and nuclease-free water to achieve a final volume of 10 μL. Mixtures were added with cold competitor or T7 primer as control and incubated at room temperature for 20 min, followed by electrophoresis on nondenaturing 6 % polyacrylamide gels in Tris-borate-EDTA buffer [90 mmol/L Tris-borate/2 mmol/L EDTA HEPES (pH 8)]. The gel was dried and exposed to a SynGene bio imaging system (Frederick, MD). The signal was further improved by Photoshop (Adobe).
Isolation of chromosomal DNA
DNA was isolated from A5CLG cells, or A549 cells as control, using a commercial kit (QuickGene DNA tissue kit, Fujifilm) based on the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP)
Assay was performed with some modifications using a commercial kit (Cat# 53,009, ChIP-IT Express Enzymatic, Active Motif). A total of 1 × 106 A5CLG cells or A549 cells as control were transfected with 1 μg of p53 DNA overnight. The 10 μg nuclear extracts (NE) as input from the cross-linked cells was IP with 1 μg p53 antibody (FL-393-G, Santa Cruz) or 1 μg normal IgG as control at 4 °C for 4 h. DNA from each experimental group (input or IP) was isolated by elution, reverse cross-linking, and Proteinase K treatment according to the manufacturer’s instructions. The DNA was then used as a template to perform PCR with primer pairs, primerA + primerB for amplification of a 210-bp DNA fragment of CCL2 5′UTR&promoter, or primerA + primerD for amplification of a 410-bp DNA fragment of CCL2 5′UTR&promoter + cDNA or GAPDH primer pairs (Invitrogen) as control.
Statistical analysis
All experiments were repeated at least three times. All the data were normally distributed. In case of multiple mean comparisons, data were analyzed by analysis of variance (ANOVA). In case of single mean comparison, data were analyzed by Student’s t test. In case of time course study, data were analyzed by two-way repeated measure ANOVA and P values less than 0.05 were regarded as significant.
Results
Involvement of p53 in CCL2 production
In order to investigate whether endogenous p53 affects CCL2 gene expression, A549 cells were transfected with mcl2pwt and exposed to ultraviolet (UV) radiation according to the conventional method [22]. As shown in Fig. 1a, UV-induced p53 accumulation significantly decreased CCL2 promoter activity compared to the control at time point 2 to 4 h after UV exposure. We therefore hypothesized that CCL2 is regulated via p53 binding activity. To address our hypothesis, A549 cells were co-transfected with mcl2pwt plus pcp53WT and their proteins were analyzed by luciferase assay. As shown in Fig. 1b, the transient transfection of increasing concentrations of pcp53WT from 0.1 to 1 μg caused a concomitant decrease in mcl2pwt-promoting luciferase activity, suggesting that overexpression of p53 downregulates CCL2 promoter activity. Furthermore, the sequence of CCL2 5′UTR&promoter was analyzed and a putative region of CCL2 5′UTR&promoter for p53 binding was proposed (Fig. 2).
Fig. 1.

Regulation of CCL2 production by the accumulation of endogenous p53. a A549 cells were transfected with 0.5 μg reporter DNA (mcl2pwt) for 2 h and exposed to UV radiation. Cells were then cultured. The treated cells were harvested at different times (0, 1, 2, 4, 6, 8, 16, or 24 h), and the lysate from cells at each time point was purified and analyzed by Western blot with antibodies against p53, luciferase, or actin as control. b Regulation of CCL2 promoter activity by overexpression of p53. A549 cells were transfected with mcl2pwt DNA alone or co-transfected with 0.5 μg mcl2pwt DNA and different concentrations (0, 0.1, 0.25, 0.5, or 1 μg) of pcp53WT DNA. Cells were cultured overnight. The lysate from cells in each group was assessed by luciferase assay (n = 3). Data are presented as mean ± SEM
Fig. 2.

Analysis of CCL2 sequence. Mouse CCL2 5′UTR&promoter sequence from −555 to +85. BLAST search of the sequence showed that it was identical to mouse genomic DNA (AL713839) and ended before the start codon of CCL2. The putative binding sites for p53 in CCL2 5′UTR&promoter were analyzed by PROMO version 3.0.2 (UPC) and displayed in bold characters. The binding sites for other potential transcription factors have not been analyzed. The oligonucleotide, including p53 binding site (underlined sequence, named CCl2/p53oligo) and its complementary oligo, was synthesized. Both synthetic oligonucleotides were annealed and labeled with [32P]ATP as a double-stranded DNA probe for EMSA
Analysis of p53-CCL2 binding activity
To determine which region of CCL2 5′UTR&promoter, deletions (mcl2pwt, mcl2p315, mcl2p115, or mcl2p53m) were subcloned into pGL3-basic vector (Fig. 3a). These cloned DNAs were transiently transfected into A549 cells to confirm their enhancement of luciferase production. The luciferase activity for mcl2pwt was assigned a value of 100 % as the baseline, and the relative luciferase activities for the others were 76 % for mcl2p315, 77.5 % for mcl2p115, and 63.4 % for mcl2p53m. Next, A549 cells were co-transfected with these clones plus pcp53WT or pcDNA3 (Control) overnight, and their proteins were assessed by luciferase assay. As shown in Fig. 3b, the site of CCL2 5′UTR&promoter for p53 binding activity was located in the region from −115 to 85 because luciferase activity induced by mcl2p115 was significantly downregulated by p53 overexpression. However, no suppression of mcl2p53m-induced luciferase activity was observed in cells when treated with pcp53WT compared to the control, suggesting that the region of +16~+35 of CCL2 5′UTR is specific to p53 binding (Fig. 3b).
Fig. 3.

Analysis of p53 binding site in CCL2 5′UTR&promoter. a Diagram of mouse CCL2 5′UTR&promoter DNA constructs and its promoter activity. Gray box: region representing full length of CCL2 5′UTR&promoter (mcl2pwt). White boxes: region representing CCL2 5′UTR&promoter deletions from 85 to −315 (mcl2p315), 85 to −115 (mcl2p115), or 85 to −115 with deletion of 16–35 (mcl2p53m). DNA deletions were confirmed by sequencing. The relative promoter activity from each construct was shown. b Determination of p53 binding site in CCL2 5′UTR&promoter. A549 cells were transfected with pcDNA3 alone or pcp53WT alone without reporter genes as controls, with various reporter genes alone (white bars), mcl2pwt, mcl2p315, mcl2p115, and mcl2p53m, or co-transfected with reporter genes plus 0.5 μg pcDNA3 (gray bars) or plus 0.5 μg pcp53WT (black bars) overnight. The lysate from each experimental group was assessed by luciferase assay (n = 3). Values were normalized with respect to protein concentrations. Data are presented as mean ± SEM
Determination of p53-CCL2 binding site
To further determine the binding domain on p53, pcp53WT DNA was transfected into A549 cells. The p53 fusion protein from treated cells was immunoprecipitated and used for EMSA. The [32P]ATP-labeled double-stranded nucleotide (ccl2/p53oligo) was treated with IP-p53 fusion protein to test its specific binding activities. The shifted DNA bands are indicated by arrows (Fig. 4). To further analyze DNA-protein interaction of CCL2 5′UTR&promoter and p53 in cells, we established a stable cell line (named CCL2/A549) containing the CCL2 promoter-enhanced cDNAs of a full-length CCL2, a full-length luciferase, and GFP in its chromosome (Fig. 5a). These integrated DNAs were confirmed by PCR: a CCL2 5′UTR&promoter DNA fragment (210 bp), a CCL2 DNA fragment (200 bp), CCL2 5′UTR&promoter plus its cDNA (410 bp), or a partial length of luciferase cDNA (513 bp). CCL2/A549 cells were used to analyze DNA-protein interaction of CCL2 promoter and p53 using chromatin immunoprecipitation (ChIP). As shown in Fig. 5b, the PCR-amplified DNA fragment of CCL2 5′UTR&promoter (210 bp) was detected in both CCL2/A549 and control cells, suggesting that p53 occurs in a cell through interaction with the CCL2 promoter.
Fig. 4.
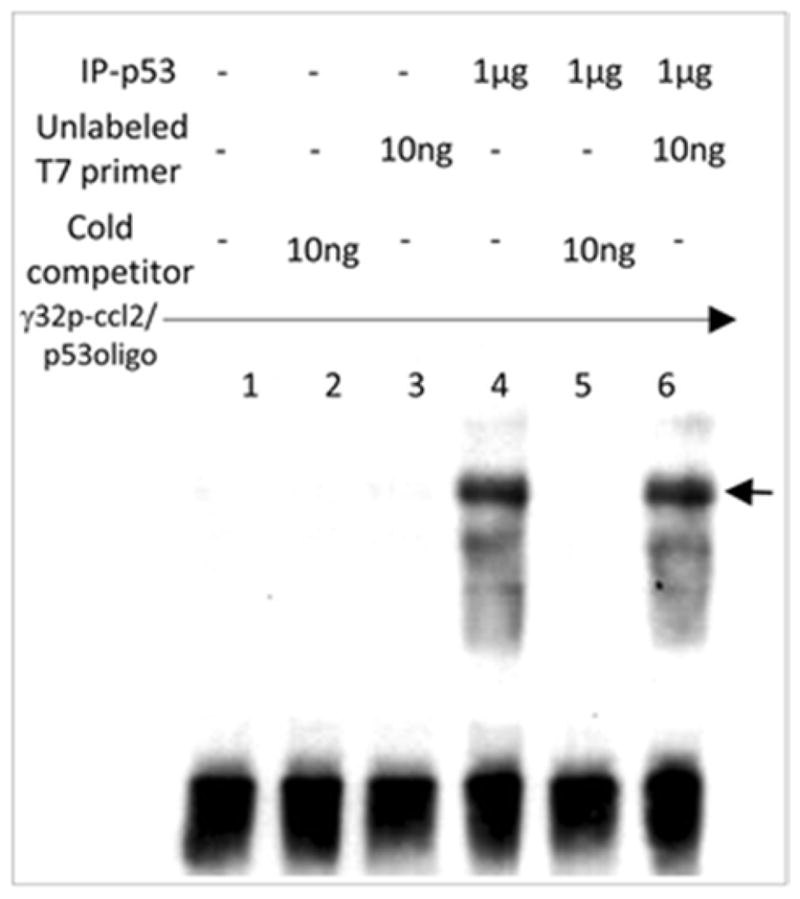
EMSA. A probe consisting of [32P]ATP-labeled ccl2/p53oligo was added to each reaction buffer. Probe was alone (lane 1) or mixed with 10 ng (about 100-fold of probe) cold competitor (lane 2), 10 ng unlabeled T7 primer (Invitrogen) as control (lane 3), or 1 μg anti-HA immunoprecipitation (IP-p53 fusion protein, lanes 4–6) plus 10 ng cold competitor (lane 5) or 10 ng unlabeled T7 primer (lane 6). The band shifts are indicated by arrows
Fig. 5.
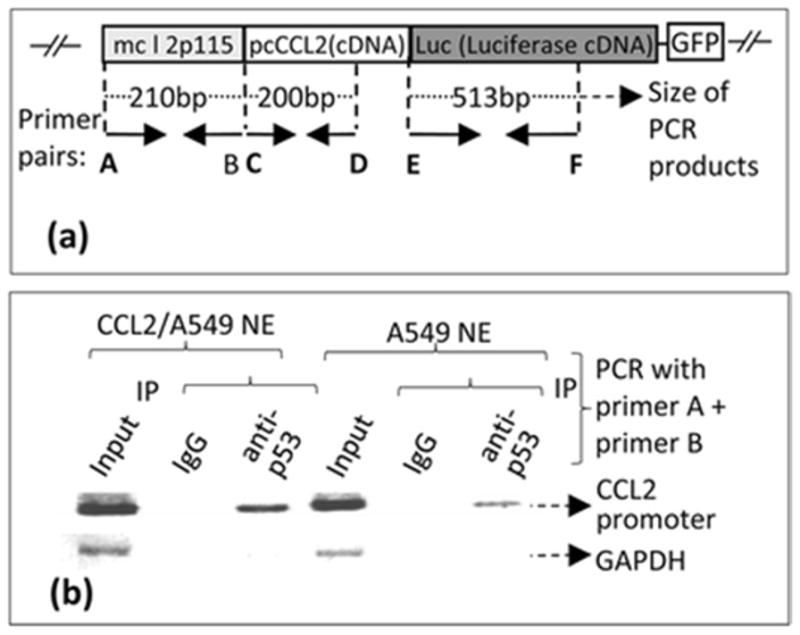
Confirmation of a stable cell line integrated with a cloned DNA (mcl2p115/pcCCL2/Luc/GFP). a. A. Schematic diagram of the cloned DNA (mcl2p115/pcCCL2/Luc/GFP) integrated into the chromosome of A549 cells (Named CCL2/A549). The arrows indicate the location and size of primers (Named primers A–F) used in PCR amplification of coding regions. These primer pairs were used for ChIP as described (Panels B). b. ChIP-detection of protein-DNA interaction between p53 and CCL2 5′UTR&promoter. The PCR-amplified specific DNA fragments are indicated with arrows.
Xenograft mouse model
In order to investigate whether p53 is involved in suppression of CCL2-mediated tumor growth, a xenograft model for therapeutic testing was performed. Three groups (three mice/group) were subcutaneously (in the mouse forehead) injected with 1 × 103 cells/100 μL CCL2/A549 alone for groups 1 and 2 or 1 × 104 cells/100 μL CRL-2280 alone as control for group 3. Mice were sacrificed by cervical dislocation 3 weeks post injection, and their subcutaneous tissues were immediately harvested and cross-sectioned. As shown in Fig. 6, a fast-growing subcutaneous tumor with massive tumor vessels began to form in tissue or section (No. 1–3, 4–6, or 7–9). In contrast, the preneoplastic lesion was seen with tumor vessel growth (tumor development is considered to be suppressed, No. 10–12, 13–15, or 16–18), and the control tissue remained normal (No. 19–21, 22–24, or 25–27). These results suggest that CCL2/A549 injection induces subcutaneous tumor in all three mice of group 1 (No. 1–9), but the xenograft tumor is almost suppressed in group 2 (No. 10–18) compared to the control group (No. 19–27).
Fig. 6.

Image analysis of xenograft mouse model. p53-deficient mice (No. 1–18, groups 1 and 2) were treated with CCL2/A549 cells or wild-type mice (No. 10–27, group 3) were treated with CRL-2280 cells as control. Treated mice were monitored daily for health care and maintained for 3 weeks. Tissues and sections from treated mice (No. 1–27, groups 1–3) were observed and photographed in visible light for structure identification of tissues (No. 1, 4, 7, 10, 13, 16, 19, 22, or 25) or sections (No. 2, 5, 8, 11, 14, 17, 20, 23, or 26). Fluorescent light was used for signal location of sections (No. 3, 6, 9, 12, 15, 18, 21, 24, or 27) using an Olympus BX40 microscope at ×200 magnification. Image analysis was performed with Image-Pro plus 5.0. The paired panels of tissues and sections (No. 1–3, 4–6, 7–9, 10–12, 13–15, 16–18, 19–21, 22–24, or 25–27) were obtained from each mouse. The phase contrast panels (No. 2 and 3, 5 and 6, 8 and 9, 11 and 12, 14 and 15, 17 and 18, 20 and 21, 23 and 24, 26 and 27) are pairs of sections from each mouse
Discussion
CCL2 is implicated in a wide range of diseases, playing an important role in inflammatory responses as well as cancer- and HIV-related disorders. Patients with autoimmune conditions [23] and rheumatoid arthritis [24] are found to have elevated levels of CCL2, and studies have demonstrated that CCL2 is involved in atherosclerotic plaque formation [25], pulmonary hypertension [26], insulin resistance [9, 11, 27], and allergic asthma [28]. Furthermore, CCL2 is linked to the development of carcinomas such as those in prostate, colorectal, and breast cancer as well as metastasis and tumor recurrence [29–32]. In addition, CCL2 has been found to be involved in HIV-mediated diseases [33, 34]. Recently, p53 was linked to the regulation of CCL2 production in HPV-positive cells [35]. However, it remains unclear how p53 regulates CCL2. It is known that tetramerization of p53 protein contributes to its activation and high binding affinity to DNA and proteins [36]. Studies have shown that the tetramerization domain on p53 C-terminal is important in protein-protein interaction [37]. However, it was found that p53 mutants lacking the C-terminal still bound to DNA and maintained significant transactivation and growth suppressor activity [38, 39]. Consistent with these studies, we reported a similar result: without its C-terminal, a short peptide of p53 alone is able to suppress the CCL2 gene expression via its binding activity [20].
We have investigated the effect of UV-induced p53 accumulation on CCL2 promoter activity by a time course study (0–24 h). The time course variation was regular only in the first two time points due to the fact that the accumulation of p53 in cells induced by UV treatment usually occurs in the first 2 h [22], and p53 is degraded thereafter to reach baseline levels, therefore not requiring a regular time course variation once degradation has started. We show that under these conditions, p53 significantly downregulates CCL2 promoter-enhanced luciferase production. Furthermore, we established a stable CCL2-promoting cancer cell line (CCL2/A549). CCL2/A549 cells were found to induce a significant CCL2 promoter activation and an upregulation of CCL2 expression as confirmed independently by protein array [20]. Using CCL2/A549 cells, we have confirmed in vitro the role of p53 in the regulation of CCL2 and found in vivo that injection of these cancer cells into p53-deficient mice strongly induced subcutaneous xenograft tumor growth compared with the control. Additionally, CCL2 overexpression in CCL2/A549 cells did not have any major impact of the morphology of cells, but it remains unknown whether this CCL2 protein increase is endogenous or a result of CCL2 transfection.
Overall, these results pave the way for future studies on the role of p53 in suppression of chemokine (such as CCL2)-mediated cancer development.
Acknowledgments
This work was supported by NIH grants R01 HL76081 and R01DE014079 to SA.
Footnotes
Conflicts of interest
None
References
- 1.Marcel V, et al. Delta160p53 is a novel N-terminal p53 isoform encoded by Delta133p53 transcript. FEBS Lett. 2010;584(21):4463–8. doi: 10.1016/j.febslet.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Rohaly G, et al. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell. 2005;122(1):21–32. doi: 10.1016/j.cell.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 3.Avery-Kiejda KA, et al. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin Cancer Res. 2008;14(6):1659–68. doi: 10.1158/1078-0432.CCR-07-1422. [DOI] [PubMed] [Google Scholar]
- 4.Hofstetter G, et al. Alternative splicing of p53 and p73: the novel p53 splice variant p53delta is an independent prognostic marker in ovarian cancer. Oncogene. 2010;29(13):1997–2004. doi: 10.1038/onc.2009.482. [DOI] [PubMed] [Google Scholar]
- 5.Okuda Y, Okuda M, Bernard CC. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003;135(1–2):29–37. doi: 10.1016/s0165-5728(02)00428-9. [DOI] [PubMed] [Google Scholar]
- 6.Komarova EA, et al. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005;19(8):1030–2. doi: 10.1096/fj.04-3213fje. [DOI] [PubMed] [Google Scholar]
- 7.Staib F, et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005;65(22):10255–64. doi: 10.1158/0008-5472.CAN-05-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baum N, et al. The prolyl cis/trans isomerase cyclophilin 18 interacts with the tumor suppressor p53 and modifies its functions in cell cycle regulation and apoptosis. Oncogene. 2009;28(44):3915–25. doi: 10.1038/onc.2009.248. [DOI] [PubMed] [Google Scholar]
- 9.Kondo M, et al. Transcription factor activating protein-2beta: a positive regulator of monocyte chemoattractant protein-1 gene expression. Endocrinology. 2009;150(4):1654–61. doi: 10.1210/en.2008-1361. [DOI] [PubMed] [Google Scholar]
- 10.Schilling M, et al. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009;161(3):806–12. doi: 10.1016/j.neuroscience.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 11.Yang SJ, et al. Inhibition of the chemokine (C-C motif) ligand 2/chemokine (C-C motif) receptor 2 pathway attenuates hyperglycaemia and inflammation in a mouse model of hepatic steatosis and lipoatrophy. Diabetologia. 2009;52(5):972–81. doi: 10.1007/s00125-009-1309-8. [DOI] [PubMed] [Google Scholar]
- 12.Haringman JJ, et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(8):2387–92. doi: 10.1002/art.21975. [DOI] [PubMed] [Google Scholar]
- 13.Tak PP, Bresnihan B. The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum. 2000;43(12):2619–33. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 14.McIntosh LM, et al. Selective CCR2-targeted macrophage depletion ameliorates experimental mesangioproliferative glomerulonephritis. Clin Exp Immunol. 2009;155(2):295–303. doi: 10.1111/j.1365-2249.2008.03819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010;102(8):522–8. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abangan RS, Jr, et al. MCP1 directs trafficking of hematopoietic stem cell-derived fibroblast precursors in solid tumor. Am J Pathol. 2010;176(4):1914–26. doi: 10.2353/ajpath.2010.080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ajuebor MN, Swain MG, Perretti M. Chemokines as novel therapeutic targets in inflammatory diseases. Biochem Pharmacol. 2002;63(7):1191–6. doi: 10.1016/s0006-2952(02)00854-7. [DOI] [PubMed] [Google Scholar]
- 18.Tang X, Molina M, Amar S. p53 short peptide (p53pep164) regulates lipopolysaccharide-induced tumor necrosis factor-alpha factor/cytokine expression. Cancer Res. 2007;67(3):1308–16. doi: 10.1158/0008-5472.CAN-06-1600. [DOI] [PubMed] [Google Scholar]
- 19.Tang X, et al. p53 peptide prevents LITAF-induced TNF-alpha-mediated mouse lung lesions and endotoxic shock. Curr Mol Med. 2011;11(6):439–52. doi: 10.2174/156652411796268731. [DOI] [PubMed] [Google Scholar]
- 20.Tang X, et al. p53 is an important regulator of CCL2 gene expression. Curr Mol Med. 2012;12(8):929–43. doi: 10.2174/156652412802480844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang X, et al. LPS induces the interaction of a transcription factor, LPS-induced TNF-alpha factor, and STAT6(B) with effects on multiple cytokines. Proc Natl Acad Sci U S A. 2005;102(14):5132–7. doi: 10.1073/pnas.0501159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng H, et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419(6909):849–53. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 23.Goser S, et al. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation. 2005;112(22):3400–7. doi: 10.1161/CIRCULATIONAHA.105.572396. [DOI] [PubMed] [Google Scholar]
- 24.Scanu A, et al. High-density lipoproteins downregulate CCL2 production in human fibroblast-like synoviocytes stimulated by urate crystals. Arthritis Res Ther. 2010;12(1):R23. doi: 10.1186/ar2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Combadiere C, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117(13):1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda Y, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 2002;283(5):H2021–8. doi: 10.1152/ajpheart.00919.2001. [DOI] [PubMed] [Google Scholar]
- 27.Bernatoniene J, et al. Induction of CC and CXC chemokines in human antigen-presenting dendritic cells by the pneumococcal proteins pneumolysin and CbpA, and the role played by toll-like receptor 4, NF-kappaB, and mitogen-activated protein kinases. J Infect Dis. 2008;198(12):1823–33. doi: 10.1086/593177. [DOI] [PubMed] [Google Scholar]
- 28.Ip WK, Wong CK, Lam CW. Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2 but not c-Jun NH2-terminal kinase 1/2 signalling pathways. Clin Exp Immunol. 2006;145(1):162–72. doi: 10.1111/j.1365-2249.2006.03085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hembruff SL, et al. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia. 2010;12(5):425–33. doi: 10.1593/neo.10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu H, et al. Tumor cell-microenvironment interaction models coupled with clinical validation reveal CCL2 and SNCG as two predictors of colorectal cancer hepatic metastasis. Clin Cancer Res. 2009;15(17):5485–93. doi: 10.1158/1078-0432.CCR-08-2491. [DOI] [PubMed] [Google Scholar]
- 31.Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem. 2009;284(42):29087–96. doi: 10.1074/jbc.M109.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka K, et al. The expression of monocyte chemotactic protein-1 in papillary thyroid carcinoma is correlated with lymph node metastasis and tumor recurrence. Thyroid. 2009;19(1):21–5. doi: 10.1089/thy.2008.0237. [DOI] [PubMed] [Google Scholar]
- 33.Eugenin EA, et al. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and neuroAIDS. J Neurosci. 2006;26(4):1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez E, et al. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci U S A. 2002;99(21):13795–800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hacke K, et al. Regulation of MCP-1 chemokine transcription by p53. Mol Cancer. 2010;9:82. doi: 10.1186/1476-4598-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joerger AC, et al. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci U S A. 2009;106(42):17705–10. doi: 10.1073/pnas.0905867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaustov L, et al. The conserved CPH domains of Cul7 and PARC are protein-protein interaction modules that bind the tetramerization domain of p53. J Biol Chem. 2007;282(15):11300–7. doi: 10.1074/jbc.M611297200. [DOI] [PubMed] [Google Scholar]
- 38.Shaulian E, et al. Tight DNA binding and oligomerization are dispensable for the ability of p53 to transactivate target genes and suppress transformation. EMBO J. 1993;12(7):2789–97. doi: 10.1002/j.1460-2075.1993.tb05940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slingerland JM, Jenkins JR, Benchimol S. The transforming and suppressor functions of p53 alleles: effects of mutations that disrupt phosphorylation, oligomerization and nuclear translocation. EMBO J. 1993;12(3):1029–37. doi: 10.1002/j.1460-2075.1993.tb05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]