

Abstract
Hereditary non-polyposis colorectal cancer (HNPCC) was previously synonymous with Lynch syndrome; however, identification of the role of germline mutations in the DNA mismatch repair (MMR) genes has made it possible to differentiate Lynch syndrome from other conditions associated with familial colorectal cancer (CRC). Broadly, HNPCC may be dichotomized into conditions that demonstrate defective DNA MMR and microsatellite instability (MSI) vs those conditions that demonstrate intact DNA MMR. Conditions characterized by MMR deficient CRCs include Lynch syndrome (germline MMR mutation), Lynch-like syndrome (biallelic somatic MMR mutations), constitutional MMR deficiency syndrome (biallelic germline MMR mutations), and sporadic MSI CRC (somatic biallelic methylation of MLH1). HNPCC conditions with intact DNA MMR associated with familial CRC include polymerase proofreading associated polyposis and familial colorectal cancer type X. Although next generation sequencing technologies have elucidated the genetic cause for some HNPCC conditions, others remain genetically undefined. Differentiating between Lynch syndrome and the other HNPCC disorders has profound implications for cancer risk assessment and surveillance of affected patients and their at-risk relatives. Clinical suspicion coupled with molecular tumor analysis and testing for germline mutations can help differentiate the clinical mimicry within HNPCC and facilitate diagnosis and management.
Keywords: Hereditary non-polyposis colorectal cancer, Lynch syndrome, Lynch-like syndrome, Familial colorectal cancer, DNA mismatch repair, Microsatellite instability, Familial colorectal cancer type X, Constitutional mismatch repair deficiency syndrome, Hereditary colorectal cancer
Core tip: Clinical criteria and phenotypic presentation of patients and families with hereditary non-polyposis colorectal cancer (HNPCC) do not adequately differentiate the several genetic diseases now classified under HNPCC. Tumor analysis for microsatellite instability (MSI) can dichotomize for the clinician conditions with MSI or without MSI, allowing a focused differential diagnosis. Individual or panel germline genetic testing can further differentiate HNPCC into its genetically defined syndromes or its phenocopies.
INTRODUCTION
About one-third of patients diagnosed with colorectal cancer (CRC) have a family history of cancer, placing them and other family members at elevated risk for this disease. Only 5% of all patients with CRC have identifiable causes for their cancer predisposition; most of which are inherited mutations in genes that regulate growth processes in colonic stem cells, and/or are caretakers of the genome to ensure the fidelity of DNA passed onto progeny cells. The most common of these inherited CRC syndromes is Lynch syndrome, identified and defined by heritable germline mutations of DNA mismatch repair (MMR) genes[1-5]. The term hereditary non-polyposis colorectal cancer (HNPCC), previously used interchangeably with Lynch syndrome, now refers to a broader spectrum of familial CRC encompassing disorders that can mimic some clinical features of Lynch syndrome, but without germline mutations in MMR genes characteristic of Lynch syndrome (Figure 1).
Figure 1.
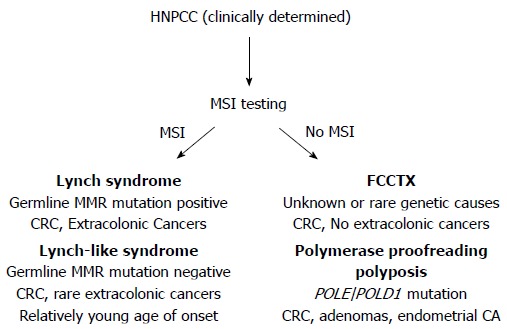
Hereditary non-polyposis colorectal cancer conditions can be dichotomized via microsatellite instability testing and/or DNA mismatch repair protein immunohistochemistry. When MSI [or loss of mismatch repair (MMR) protein expression] is present in the colorectal cancer indicating loss of functional DNA MMR, Lynch syndrome and Lynch-like syndrome remain in the differential. Germline testing for DNA MMR gene mutation can differentiate these two syndromes. When MSI is absent meaning DNA MMR remains intact and functional, consideration for polymerase proofreading associated polyposis and familial colorectal cancer type X should be undertaken. Performing germline testing for POLE and POLD1 mutations might help differentiate these two syndromes. HNPCC: Hereditary non-polyposis colorectal cancer; MSI: Microsatellite instability; CRC: Colorectal cancer.
Distinguishing among the HNPCC disorders is important clinically, as the approach to surveillance for patients and their at-risk family members differs according to risks for colonic and extracolonic cancers associated with each syndrome[5]. Health care providers should be observant for “red flags” suggestive of genetic predisposition to CRC, such as strong personal or family history of cancer, diagnoses of colorectal neoplasia at young ages, and histopathologic and molecular tumor features that are characteristic of specific syndromes. Screening of CRC tumors for microsatellite instability (MSI) and expression of DNA MMR genes (Figure 1) is an effective strategy to facilitate identification of patients at risk for Lynch syndrome[6]. Individuals whose personal and/or family history raises suspicion for a hereditary cancer syndrome should undergo clinical genetic evaluation, which includes genetic counseling and evaluation of patient health records[5,7]. Even if a genetic mutation is not identified, the outcome of the genetic evaluation may help guide decision making regarding surveillance and other interventions to reduce future cancer risk.
Here, we review several HNPCC conditions that may mimic Lynch syndrome and present distinguishing features and tests that can help differentiate among them. Next generation sequencing approaches will facilitate discovery of novel genetic events that will define the clinical and molecular phenotypes of familial CRC without germline MMR gene mutations.
FAMILIAL CRC WITH DEFECTIVE DNA MISMATCH REPAIR
The DNA MMR system provides recognition of post-DNA synthetic polymerase mistakes in the DNA strand at single base mispairs, chemotherapy-induced nucleotide alterations, and slippage mistakes at repetitive sequences termed microsatellites[1,3,8-12]. The DNA MMR recognition complexes, MutSα and MutSβ, consists of heterodimers of the MMR proteins MSH2-MSH6 and MSH2-MSH3, respectively, with MutSα recognizing single base mispairs and short insertion/deletion loops (I/D loops) of ≤ 2 nucleotides, and MutSβ recognizing ≥ 2 I/D loops[1,12,13]. Once a mispair or I/D loop is recognized, the execution complex MutLα (heterodimer of the MMR proteins MLH1 and PMS2) binds to MutSα or MutSβ to signal other proteins for excision and re-synthesis of the affected DNA, or commits the cell to programmed cell death if repair is futile[1,13,14].
There are several mechanisms that can inactivate DNA MMR function[1,4,13,15-29]. Abrogation of DNA MMR function generates a hypermutated tumor that accumulates hundreds of random point mutations and frameshifts in the cell’s genome[30], and transition from an adenoma to CRC occurs at a rapid pace compared to MMR-intact tumors (1-3 years vs 1-2 decades, respectively)[1,3,31]. The biomarker assay that is used to determine loss of MMR function clinically is microsatellite instability (MSI)[1,3,32], which detects acquired new frameshift length changes of microsatellites in neoplastic tissue compared to non-neoplastic tissue when MMR function is defective (Figure 2). Additionally, the use of immunohistochemistry (IHC) to detect expression of DNA MMR proteins in neoplastic tissue is highly correlative to MMR function, with absence of MMR proteins predictive of MSI within the tumor[1,3,32]. The finding of MSI and/or absence of DNA MMR protein expression identifies that a tumor has lost DNA MMR function, and is the basis for differentiating familial CRC cases associated with Lynch syndrome from other HNPCC conditions (Figure 1).
Figure 2.
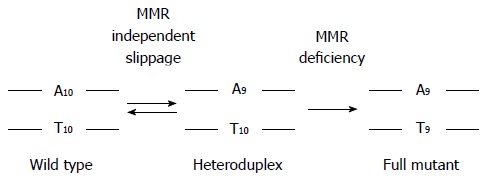
Loss of DNA mismatch repair forces polymerase slippage mistakes to become permanent frameshift mutations at microsatellite sequences. Depicted is a mononucleotide microsatellite of 10 adenines. During DNA replication, occasional polymerase mistakes allow slippage at microsatellite sequences, creating a heteroduplex structure often with one nucleotide as a loop. With intact DNA mismatch repair (MMR), the deletion loop is recognized, excised, and resynthesized correctly such that daughter cells will maintain fidelity of the proper microsatellite length. In the absence of DNA MMR, the deletion loop becomes a permanent frameshift in daughter cells. Frameshift mutations can occur in non-coding as well as in coding microsatellites; coding frameshifts cause the transcription and ultimate translation of truncated proteins that can act as neoantigens to the immune system.
CRCs with defective DNA MMR comprise only 15% of all CRCs and are associated with specific clinicopathologic features. MSI tumors are more likely to be located in the colon proximal to the splenic flexure, often exhibit poor differentiation and mucinous features, possess sub-epithelial lymphoid aggregates as response to neoantigens induced by truncated proteins produced from frameshifted coding microsatellite mutations (Figure 2), and demonstrate better survival compared to same-staged patients without MSI CRCs[1-3,33-37]. Because intact DNA MMR can recognize chemotherapy-induced nucleotide alterations to trigger cell demise, and in particular incorporated 5-fluorodeoxyuracil as a result of 5-fluorouracil (5-FU) therapy, loss of DNA MMR function renders the CRC resistant to 5-FU, and 5-FU treatment does not improve survival of patients with MSI CRCs[8-12,38-42]. However, the MSI CRC somatic mutational load is high, making it more susceptible to immune killing when the immune checkpoint inhibitor to PD-1 is administered to patients[43]. While the majority of MSI CRCs represent sporadic tumors which develop as a consequence of somatic events (BRAF mutation, MLH1 promoter hypermethylation), a minority develop as a consequence of germline mutations in MMR genes associated with Lynch syndrome.
Lynch syndrome
Lynch syndrome can be identified in 2%-3% of all CRC patients, and approximately 2% of all endometrial cancer patients, the two most common cancers observed with this syndrome[2,44,45]. Lifetime risk for CRC approaches 80% and lifetime risk for endometrial cancer in women approaches 50%[2,5]. Patients can develop synchronous and metachronous cancers at relatively young ages, and Lynch-associated CRCs demonstrate accelerated neoplastic progression, with reports of cancers developing within 3 years after colonoscopy[46]. While CRCs are the most common tumors, risks for malignancies of the endometrium and ovaries, gastrointestinal tract (stomach and small intestine, pancreas, biliary tract), urinary tract, brain (glioblastomas), and skin (keratoacanthomas and sebaceous adenomas) are also increased[2,5].
Lynch syndrome is associated with germline mutations in one of the DNA MMR genes (MSH2, MLH1, MSH6, PMS2, EPCAM) (Table 1), and is transmitted in an autosomal dominant fashion[2,4,5]. Germline testing for mutations in the MMR genes is the gold standard for characterizing Lynch syndrome, and can be identified in > 80% of Lynch kindreds. The two most commonly mutated genes, MSH2 and MLH1, account for approximately 90% of mutations found in Lynch kindreds, and can be point mutations, deletions, or rearrangements. MSH2 and MLH1 are critical components of the MMR recognition complexes and MMR execution complexes, respectively. Germline mutation of MSH6 and PMS2 are identified in < 10% of Lynch kindreds, and deletion of EPCAM, upstream of MSH2 on chromosome 2 that causes allele specific methylation of the promoter of MSH2, is a relatively rare cause for loss of MSH2 expression and Lynch syndrome[2,4,5]. Germline MSH3 mutations have only rarely been identified in any families with Lynch syndrome[13,47]. Specific mutations of DNA MMR genes are associated with differences in phenotype of Lynch patients. For instance, MLH1 and MSH2 mutation carriers present with cancers at younger ages (40-50 years) whereas MSH6 mutation carriers tend to be older at CRC diagnosis (age 50-65 years) with higher prevalence of endometrial cancer[2,3,5].
Table 1.
Germline and cancer-specific genetic and epigenetic features for hereditary non-polyposis colorectal cancer conditions
| Lynch syndrome | CMMRD | Lynch-like syndrome | Sporadic MSI CRC and sessile serrated polyps | FCCTX | PPAP | HBOC | |
| Germline mutation | One allele of a MMR gene: MSH2, MLH1, MSH6, PMS2, EPCAM | Both alleles of a MMR gene: MSH2, MLH1, MSH6, PMS2, EPCAM | None | None | RPS20, SEMA4A, HNRNPA0, WIF1, likely others | POLE (L424V) POLD1 (S478N) (other exonuclease domain mutations) | BRCA1 or BRCA2 |
| Somatic mutation | 2nd allele of MMR gene | None | Both alleles of a MMR gene | BRAF | Various | 2nd allele of POLE, POLD1 and > 100 × somatic mutations (hypermutated) compared with other MSS tumors | 2nd allele of BRCA1 or BRCA2 |
| Tumor MMR phenotype | MMR deficient (MSI) | MMR deficient (MSI) | MMR deficient (MSI) | MMR deficient, (MSI) | MMR proficient (MSS) | MMR proficient (MSS) | MMR proficient (MSS) |
| Epigenetic | Germline deletion in 3’ end of EPCAM leads to Somatic Allele-specific MSH2 methylation in tissues | None reported | None reported | Somatic biallelic promoter methylation for MLH1 | None reported | None reported | None reported |
MSS: Microsatellite stable; CMMRD: Constitutional mismatch repair-deficiency; FCCTX: Familial colorectal cancer type X; PPAP: Polymerase proofreading associated polyposis syndrome; HBOC: Hereditary breast and ovarian cancer syndrome; MSI: Microsatellite instability; CRC: Colorectal cancer.
Family history-based clinical criteria, such as the Amsterdam criteria (3 relatives with CRC, across 2 generations, with one case diagnosed at age < 50 years) and/or Bethesda guidelines, have limited sensitivity and identify only a portion of MMR mutation carriers[2,5,44]. Given that intensive surveillance with colonoscopy every 1-2 years has been shown to decrease morbidity and mortality in families with Lynch syndrome, universal testing of all CRC tumors for MMR deficiency has been proposed as a cost-effective strategy to screen for Lynch syndrome[44,48]. Tumors associated with germline MMR gene mutations can be differentiated from sporadic MSI CRCs on the basis of the absence of somatic BRAF mutations and absence of methylation of MLH1)[1,3,5,33]. Loss of expression of MSH2 and/or MSH6 was previously thought to be diagnostic of germline mutations in the MMR gene corresponding to the absent protein, but recent data demonstrate that absent expression of these MMR proteins can also be observed in Lynch-like CRCs (Table 1).
Lynch-like syndrome
Lynch-like syndrome may account for as many as 60%-70% of cases in which Lynch syndrome is clinically suspected, but genetic testing fails to identify a germline MMR gene mutation[4]. Patients with Lynch-like syndrome resemble those with Lynch syndrome in that their tumors manifest MSI and immunohistochemical absence of a DNA MMR protein. Patients with Lynch-like syndrome present with cancer at younger ages, similar to Lynch syndrome (53.7 years vs 48.5 years of age), fueling the speculation that undiagnosed germline mutations may be implicated in at least some of these cases[22,23,49]. However, analysis of cancers among probands and families demonstrate heterogeneity in risks, with standardized incidence ratio (SIR) for CRC lower in Lynch-like (2.12) compared to Lynch syndrome (6.04), and with SIR for extracolonic cancers also lower in Lynch-like (1.69) vs Lynch syndrome (2.81) families[23].
From a clinical perspective, the key feature differentiating Lynch syndrome from Lynch-like syndrome is that the former is associated with presence of germline DNA MMR gene mutations, while the latter lacks identifiable germline mutations. There are several potential explanations for Lynch-like syndrome. First, it is possible that some Lynch-like patients could actually have Lynch syndrome, as there may be some germline mutations in DNA MMR genes that are not detectable by current testing, such as those occurring in areas of intronic sequences and promoters[4]. However, an alternative explanation is that there are other mechanisms that inactivate DNA MMR (aside from germline mutation in MMR genes) which could also result in tumor phenotype that closely resembles Lynch syndrome. Unlike sporadic MSI-H CRCs, Lynch-like CRCs do not show epigenetic inactivation of the DNA MMR gene MLH1 or mutation in BRAF. However, 50%-60% of Lynch-like CRCs do exhibit the biallelic somatic inactivation of DNA MMR genes within the tumor[20,22,23,49-51] (Table 1). Somatic mutation in any one allele of the DNA MMR genes coupled with loss of heterozygosity (LOH) of the other allele is the most common pattern (with second most common mechanism being two somatic sequence mutations). Finally, it is also possible that patients with Lynch-like syndrome may harbor germline mutations in genes other than the DNA MMR genes known to be associated with Lynch syndrome. Consequently, Lynch-like syndrome cases might represent a spectrum of HNPCC conditions with heterogeneous etiologies; however until we have more information regarding cancer risks and rates of neoplastic progression, intensive surveillance for cancer similar to Lynch syndrome guidelines is recommended.
Constitutional mismatch repair deficiency syndrome
Constitutional mismatch repair-deficiency (CMMRD) is a rare condition in which biallelic germline mutations in DNA MMR genes predispose to development of multiple cancers, often at very early ages[26,27] (Table 1). Affected individuals inherit a germline MMR mutation from each parent, with PMS2 and MSH6 mutations most commonly implicated. Although initial descriptions of CMMRD cases involved consanguineous families, a significant proportion of cases involve offspring of unrelated parents not previously diagnosed with Lynch syndrome. CMMRD patients often present in childhood, with brain tumors, colorectal and/or other gastrointestinal cancers (including in some cases multiple colonic adenomas), with hematological malignancies such as leukemias and lymphomas also commonly reported[27]. Other tumors such as rhabdomyosarcoma, Wilms tumor, and neuroblastomas have also been reported[27,52]. The presentation can be variable, but the scope of tumors and the extremely young age at presentation can provide clues to the diagnosis. Since nearly all CMMRD patients exhibit cutaneous café-au-lait spots[27], there may be some phenotypic overlap with other syndromes such as neurofibromatosis type 1, Li-Fraumeni syndrome, and familial adenomatous polyposis[27,53].
The diagnosis of CMMRD is confirmed with detection of biallelic germline mutations in MMR genes[27] (Table 1); however the diagnosis is not always straightforward. PMS2 in particular has approximately 20 pseudogenes that make identifying one allele mutation, let alone both alleles, challenging. Additionally, variants of uncertain significances (VUSs) in one or both alleles of MSH6 or PMS2 or any other MMR gene are found in over 30% of suspected CMMRD patients, making the genetic confirmation of this syndrome difficult[3,5,27].
Like tumors which develop in Lynch syndrome and Lynch-like syndrome, CMMRD CRCs display MSI, and immunohistochemistry will demonstrate absence of staining of the mutated MMR protein. A differentiating feature of CMMRD patients is that the surrounding normal colon tissue may also demonstrate absence of the MMR protein corresponding with the germline mutation[27], given that both alleles are inactivated in every cell of the CMMRD patient’s body even before cancer formation.
Sporadic microsatellite unstable CRC
Microsatellite unstable (MSI) is found in approximately 15% of all sporadic CRCs with MSI tumors more likely to develop at older ages (≥ 70 years of age) and more often in females[1,3,34]. Most of these sporadic MSI CRCs are associated with biallelic hypermethylation of the promoter of the MLH1 gene (Table 1), which prevents its transcription[15-19]. An additional common finding among sporadic MSI CRCs is the presence of BRAFV600E, an activating mutation that causes incessant mitogenic pathway signaling[1,3,5,33] (Table 1). The older presentation for CRC, lack of family history of cancer, as well as the presence of BRAFV600E and/or methylation of MLH1 help distinguish patients with sporadic MSI CRC from those with Lynch or Lynch-like syndrome. Likewise, patients with sessile serrated polyps and adenomas (SSAs) exhibit multiple methylated DNA loci including that of the MLH1 promoter, and manifest MSI and BRAFV600E[33,54]. Although patients with SSAs often present with proximal colon location of the lesion similar to Lynch syndrome tumors, the MLH1 hypermethylation and presentation of BRAFV600E show that the SSA is not part of Lynch syndrome.
FAMILIAL CRC WITH INTACT DNA MISMATCH REPAIR
Polymerase proofreading associated polyposis syndrome
Polymerase proofreading associated polyposis syndrome (PPAP) is a rare autosomal dominantly inherited syndrome in which the exonuclease domain of POLE (encoding DNA polymerase ε) or POLD1 (encoding DNA polymerase δ1) is mutated in the germline[55,56]. Two highly penetrant mutations are described (POLE p.Leu424Val and POLD1 p.Ser478Asn)[55], but other mutations in the exonucleoase domain could also be causal[57] (Table 1). Individuals with germline POLE mutations exhibit colonic oligopolyposis (generally between 5-70 adenomas) as early as 20 years of age, CRCs, and duodenal adenomas and carcinomas[55,56]. POLD1 mutation carriers exhibit colonic oligopolyposis (generally 3-50 adenomas) and CRC as young as 20 years of age as well, but in addition exhibit increased risk for endometrial cancers and brain tumors[55] which overlaps with the spectrum of tumors seen in Lynch syndrome. Indeed, the clinical phenotype of PPAP patients can be highly variable, ranging from an attenuated polyposis picture resembling that seen in association with germline mutations in APC or MYH to oligopolyposis or non-polyposis resembling HNPCC and Lynch syndrome[55,56].
Interestingly, although CRCs from PPAP patients are hypermuted or ultramutated (with 100-fold more mutations than sporadic microsatellite stable tumors) due to loss in polymerase function that then generates multiple random mutations in the cancer cell genome, these tumors are microsatellite stable and do not exhibit loss of expression of DNA MMR proteins[30,55,56,58] (Table 1). The absence of MSI in these CRCs is a distinguishing feature, and should raise the clinical suspicion of PPAP in families with cancer histories suggestive of Lynch syndrome.
Familial colorectal cancer type X syndrome
Familial colorectal cancer type X (FCCTX) is the designation for patients with family history of CRC meeting Amsterdam Criteria for Lynch syndrome, but whose tumors lack MSI and whose germline lacks DNA MMR gene mutations[59]. Nearly half of Amsterdam Criteria-positive CRC families fit the description of FCCTX. Clinically, however, CRC risk among FCCTX patients is increased approximately 2-fold over the general population (compared to > 6-fold for Lynch syndrome patients), and FCCTX families lack extracolonic cancers[59].
As the “type X” label implies, the genetic etiology for FCCTX is largely unknown. Recent investigations suggest that FCCTX may be a heterogeneous condition, since gene finding studies have uncovered mutations in several genes, each affecting only one or a few families. Germline mutations in RPS20, encoding an rRNA maturation protein[60], as well as in SEMA4A, HNRNPA0 and WIF1, whose encoded proteins are involved in the regulation of PI3 Kinases and MAPK/ERK signaling and NAD biosynthesis, have been identified[61,62] (Table 1). There is potential for phenotypic overlap between FCCTX and other known genetic conditions (such as PPAP), and germline mutations in BMPR1A (associated with juvenile polyposis) have been identified in some individuals with the clinical diagnosis of FCCTX[63]. Even so, genetic testing is clinically uninformative in the vast majority of patients with MMR proficient tumors without polyposis phenotypes.
Potential overlap with other hereditary cancer syndromes: Hereditary breast and ovarian cancer syndrome
CRCs are common, and thus may be seen in association with other hereditary cancer syndromes not typically associated with increased risk for colorectal neoplasia. Patients with hereditary breast and ovarian cancer syndrome (HBOC) possess a germline mutation in BRCA1 or BRCA2, two genes involved in DNA double strand break repair (Table 1). The spectrum of cancers in kindreds with HBOC can include not only breast and ovarian cancer, but also pancreatic cancer and prostate cancer[64]. Additionally, some reports suggest that breast and prostate cancers may also be overrepresented in Lynch syndrome kindreds[64,65]. Thus, there is potential for phenotypic overlap between HBOC and Lynch syndrome, especially with regard to ovarian and pancreatic cancers[66,67], with Lynch syndrome potentially accounting for 13%-15% of hereditary ovarian cancers[68]. The lifetime risk for ovarian cancer in Lynch syndrome patients is approximately 8%[68-70]. Consequently, Lynch syndrome and HBOC should each be considered in the differential diagnosis for kindreds with ovarian and/or pancreatic cancers.
In evaluating suspected Lynch syndrome families, gene panel testing covering 25 cancer-causing genes yielded 9% of suspected families with germline DNA MMR gene mutations[71]. Surprisingly, another 1% demonstrated germline mutations in BRCA1 or BRCA2, with 93% of these patients meeting NCCN guidelines for Lynch syndrome testing while only 33% meeting NCCN guidelines for BRCA1 or BRCA2 analysis[71]. This study demonstrates the phenotypic overlap between HBOC and HNPCC. This study also demonstrates that the use of broader panel testing can be highly informative in differentiating these syndrome genetically.
CONCLUSION
HNPCC encompasses a spectrum of conditions that have significant phenotypic overlap, and making a genetic diagnosis in familial CRC cases can be clinically challenging. Since risks for CRC and extracolonic cancers differ among the various conditions, genetic confirmation of the diagnosis can help direct surveillance recommendations for the patient and their at-risk family members. As clinical criteria have limited sensitivity and specificity, analysis of tumor tissue for the presence or absence of MSI can be effective for identifying individuals at risk for Lynch syndrome. Genetic testing for germline mutations in individual genes (or panels of genes) can further differentiate HNPCC into specific, defined syndromes. Even in the absence of an informative genetic test result, clinical suspicion should remain high, and specialized surveillance is justified for at-risk individuals from families affected with HNPCC. Next generation sequencing will likely uncover additional genetic defects in HNPCC kindreds. Documentation of phenotypes and cancer incidence through longitudinal studies will provide valuable clinical information regarding the natural history of disease which will help differentiate the Lynch syndrome mimics and guide diagnosis and management for the heterogeneous conditions currently grouped under the category of familial CRC.
Footnotes
Supported by The United States Public Health Service (DK067287 and CA162147); and the A. Alfred Taubman Medical Research Institute of the University of Michigan.
Conflict-of-interest statement: No potential conflicts of interest are disclosed. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 12, 2015
First decision: June 2, 2015
Article in press: July 8, 2015
P- Reviewer: Lakatos PL, Roncucci L S- Editor: Ji FF L- Editor: A E- Editor: Wang CH
References
- 1.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boland CR, Koi M, Chang DK, Carethers JM. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Fam Cancer. 2008;7:41–52. doi: 10.1007/s10689-007-9145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–2087.e3. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carethers JM. Differentiating Lynch-like from Lynch syndrome. Gastroenterology. 2014;146:602–604. doi: 10.1053/j.gastro.2014.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoffel EM, Kastrinos F. Familial colorectal cancer, beyond Lynch syndrome. Clin Gastroenterol Hepatol. 2014;12:1059–1068. doi: 10.1016/j.cgh.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11:42–65. doi: 10.1097/GIM.0b013e31818fa2db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carethers JM. DNA testing and molecular screening for colon cancer. Clin Gastroenterol Hepatol. 2014;12:377–381. doi: 10.1016/j.cgh.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tajima A, Hess MT, Cabrera BL, Kolodner RD, Carethers JM. The mismatch repair complex hMutS alpha recognizes 5-fluorouracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology. 2004;127:1678–1684. doi: 10.1053/j.gastro.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Tajima A, Iwaizumi M, Tseng-Rogenski S, Cabrera BL, Carethers JM. Both hMutSα and hMutSß DNA mismatch repair complexes participate in 5-fluorouracil cytotoxicity. PLoS One. 2011;6:e28117. doi: 10.1371/journal.pone.0028117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwaizumi M, Tseng-Rogenski S, Carethers JM. DNA mismatch repair proficiency executing 5-fluorouracil cytotoxicity in colorectal cancer cells. Cancer Biol Ther. 2011;12:756–764. doi: 10.4161/cbt.12.8.17169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwaizumi M, Tseng-Rogenski S, Carethers JM. Acidic tumor microenvironment downregulates hMLH1 but does not diminish 5-fluorouracil chemosensitivity. Mutat Res. 2013;747-748:19–27. doi: 10.1016/j.mrfmmm.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamaya Y, Guarinos C, Tseng-Rogenski SS, Iwaizumi M, Das R, Jover R, Castells A, Llor X, Andreu M, Carethers JM. Efficacy of Adjuvant 5-Fluorouracil Therapy for Patients with EMAST-Positive Stage II/III Colorectal Cancer. PLoS One. 2015;10:e0127591. doi: 10.1371/journal.pone.0127591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carethers JM, Koi M, Tseng-Rogenski SS. EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression. Genes (Basel) 2015;6:185–205. doi: 10.3390/genes6020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carethers JM, Hawn MT, Chauhan DP, Luce MC, Marra G, Koi M, Boland CR. Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N’-nitro-N-nitrosoguanidine. J Clin Invest. 1996;98:199–206. doi: 10.1172/JCI118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, Igarashi S, Kotake K, Koyama Y, Nagai H. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology. 2001;121:1300–1309. doi: 10.1053/gast.2001.29616. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa H, Nuovo GJ, Zervos EE, Martin EW, Salovaara R, Aaltonen LA, de la Chapelle A. Age-related hypermethylation of the 5’ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001;61:6991–6995. [PubMed] [Google Scholar]
- 17.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 20.Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, Goossens M, Ouchene H, Hendriks-Cornelissen SJ, Kwint MP, Hoogerbrugge N, Nagtegaal ID, Ligtenberg MJ. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146:643–646.e8. doi: 10.1053/j.gastro.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Diouf B, Cheng Q, Krynetskaia NF, Yang W, Cheok M, Pei D, Fan Y, Cheng C, Krynetskiy EY, Geng H, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298–1303. doi: 10.1038/nm.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, Pritchard CC. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147:1308–1316.e1. doi: 10.1053/j.gastro.2014.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, Zapater P, Castillejo A, Barberá VM, Juárez M, Bessa X, Xicola RM, Clofent J, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013;144:926–932.e1; quiz e13-e14. doi: 10.1053/j.gastro.2013.01.044. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crépin M, Dieu MC, Lejeune S, Escande F, Boidin D, Porchet N, Morin G, Manouvrier S, Mathieu M, Buisine MP. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat. 2012;33:180–188. doi: 10.1002/humu.21617. [DOI] [PubMed] [Google Scholar]
- 26.Scott RH, Mansour S, Pritchard-Jones K, Kumar D, MacSweeney F, Rahman N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol. 2007;4:130–134. doi: 10.1038/ncponc0719. [DOI] [PubMed] [Google Scholar]
- 27.Bakry D, Aronson M, Durno C, Rimawi H, Farah R, Alharbi QK, Alharbi M, Shamvil A, Ben-Shachar S, Mistry M, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014;50:987–996. doi: 10.1016/j.ejca.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Tseng-Rogenski SS, Chung H, Wilk MB, Zhang S, Iwaizumi M, Carethers JM. Oxidative stress induces nuclear-to-cytosol shift of hMSH3, a potential mechanism for EMAST in colorectal cancer cells. PLoS One. 2012;7:e50616. doi: 10.1371/journal.pone.0050616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng-Rogenski SS, Hamaya Y, Choi DY, Carethers JM. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology. 2015;148:579–589. doi: 10.1053/j.gastro.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, Tavaré S, Shibata D. Genetic reconstruction of individual colorectal tumor histories. Proc Natl Acad Sci USA. 2000;97:1236–1241. doi: 10.1073/pnas.97.3.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 33.Carethers JM. One colon lumen but two organs. Gastroenterology. 2011;141:411–412. doi: 10.1053/j.gastro.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carethers JM, Murali B, Yang B, Doctolero RT, Tajima A, Basa R, Smith EJ, Lee M, Janke R, Ngo T, et al. Influence of race on microsatellite instability and CD8+ T cell infiltration in colon cancer. PLoS One. 2014;9:e100461. doi: 10.1371/journal.pone.0100461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carethers JM. Screening for colorectal cancer in African Americans: determinants and rationale for an earlier age to commence screening. Dig Dis Sci. 2015;60:711–721. doi: 10.1007/s10620-014-3443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 37.Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, von Knebel Doeberitz M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134:988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jo WS, Carethers JM. Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomark. 2006;2:51–60. doi: 10.3233/cbm-2006-21-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126:394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 41.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung H, Chaudhry J, Lopez CG, Carethers JM. Cyclin E and histone H3 levels are regulated by 5-fluorouracil in a DNA mismatch repair-dependent manner. Cancer Biol Ther. 2010;10:1147–1156. doi: 10.4161/cbt.10.11.13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 45.Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 46.Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 47.Yang X, Wu J, Lu J, Liu G, Di G, Chen C, Hou Y, Sun M, Yang W, Xu X, et al. Identification of a comprehensive spectrum of genetic factors for hereditary breast cancer in a Chinese population by next-generation sequencing. PLoS One. 2015;10:e0125571. doi: 10.1371/journal.pone.0125571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology. 2014;147:502–526. doi: 10.1053/j.gastro.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 49.Geurts-Giele WR, Leenen CH, Dubbink HJ, Meijssen IC, Post E, Sleddens HF, Kuipers EJ, Goverde A, van den Ouweland AM, van Lier MG, et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol. 2014;234:548–559. doi: 10.1002/path.4419. [DOI] [PubMed] [Google Scholar]
- 50.Sourrouille I, Coulet F, Lefevre JH, Colas C, Eyries M, Svrcek M, Bardier-Dupas A, Parc Y, Soubrier F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer. 2013;12:27–33. doi: 10.1007/s10689-012-9568-9. [DOI] [PubMed] [Google Scholar]
- 51.Kang SY, Park CK, Chang DK, Kim JW, Son HJ, Cho YB, Yun SH, Kim HC, Kwon M, Kim KM. Lynch-like syndrome: characterization and comparison with EPCAM deletion carriers. Int J Cancer. 2015;136:1568–1578. doi: 10.1002/ijc.29133. [DOI] [PubMed] [Google Scholar]
- 52.Kratz CP, Holter S, Etzler J, Lauten M, Pollett A, Niemeyer CM, Gallinger S, Wimmer K. Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome. J Med Genet. 2009;46:418–420. doi: 10.1136/jmg.2008.064212. [DOI] [PubMed] [Google Scholar]
- 53.Jasperson KW, Samowitz WS, Burt RW. Constitutional mismatch repair-deficiency syndrome presenting as colonic adenomatous polyposis: clues from the skin. Clin Genet. 2011;80:394–397. doi: 10.1111/j.1399-0004.2010.01543.x. [DOI] [PubMed] [Google Scholar]
- 54.Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 55.Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spier I, Holzapfel S, Altmüller J, Zhao B, Horpaopan S, Vogt S, Chen S, Morak M, Raeder S, Kayser K, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015;137:320–331. doi: 10.1002/ijc.29396. [DOI] [PubMed] [Google Scholar]
- 57.Valle L, Hernández-Illán E, Bellido F, Aiza G, Castillejo A, Castillejo MI, Navarro M, Seguí N, Vargas G, Guarinos C, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. 2014;23:3506–3512. doi: 10.1093/hmg/ddu058. [DOI] [PubMed] [Google Scholar]
- 58.Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, Gorman M, Martin L, Howarth KM, Hodgson SV, et al. DNA polymerase Ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22:2820–2828. doi: 10.1093/hmg/ddt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, Gallinger S, Bapat B, Aronson M, Hopper J, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293:1979–1985. doi: 10.1001/jama.293.16.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nieminen TT, O’Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, Ellonen P, Abdel-Rahman WM, Valo S, Mecklin JP, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology. 2014;147:595–598.e5. doi: 10.1053/j.gastro.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schulz E, Klampfl P, Holzapfel S, Janecke AR, Ulz P, Renner W, Kashofer K, Nojima S, Leitner A, Zebisch A, et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat Commun. 2014;5:5191. doi: 10.1038/ncomms6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei C, Peng B, Han Y, Chen WV, Rother J, Tomlinson GE, Boland CR, Chaussabel D, Frazier ML, Amos CI. Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Fam Cancer. 2015;14:297–306. doi: 10.1007/s10689-014-9758-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nieminen TT, Abdel-Rahman WM, Ristimäki A, Lappalainen M, Lahermo P, Mecklin JP, Järvinen HJ, Peltomäki P. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2011;141:e23–e26. doi: 10.1053/j.gastro.2011.03.063. [DOI] [PubMed] [Google Scholar]
- 64.Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269–275. doi: 10.1002/cncr.29041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buerki N, Gautier L, Kovac M, Marra G, Buser M, Mueller H, Heinimann K. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer. 2012;51:83–91. doi: 10.1002/gcc.20935. [DOI] [PubMed] [Google Scholar]
- 66.Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, Syngal S, Cooney KA, Gruber SB. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013;31:1713–1718. doi: 10.1200/JCO.2012.44.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, Bandipalliam P, Stoffel EM, Gruber SB, Syngal S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–1795. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakamura K, Banno K, Yanokura M, Iida M, Adachi M, Masuda K, Ueki A, Kobayashi Y, Nomura H, Hirasawa A, et al. Features of ovarian cancer in Lynch syndrome (Review) Mol Clin Oncol. 2014;2:909–916. doi: 10.3892/mco.2014.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.South SA, Vance H, Farrell C, DiCioccio RA, Fahey C, Piver MS, Rodabaugh KJ. Consideration of hereditary nonpolyposis colorectal cancer in BRCA mutation-negative familial ovarian cancers. Cancer. 2009;115:324–333. doi: 10.1002/cncr.24012. [DOI] [PubMed] [Google Scholar]
- 70.Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Järvinen HJ, Myrhøj T, Sunde L, Wijnen JT, Lynch HT. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–449. doi: 10.1002/ijc.23508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, Roa BB, Wenstrup RJ, Hartman AR, Syngal S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients with Suspected Lynch Syndrome. Gastroenterology. 2015:Epub ahead of print. doi: 10.1053/j.gastro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]