

Abstract
Laser capture microdissection (LCM) allows the isolation of specific cells from thin tissue sections with high spatial resolution. Effective LCM requires precise identification of cells subpopulations from a heterogeneous tissue. Identification of cells of interest for LCM is usually based on morphological criteria or on fluorescent protein reporters. The combination of LCM and rapid immunolabeling offers an alternative and efficient means to visualize specific cell types and to isolate them from surrounding tissue. High-quality RNA can then be extracted from a pure cell population and further processed for downstream applications, including RNA-sequencing, microarray or qRT-PCR. This approach has been previously performed and briefly described in few publications. The goal of this article is to illustrate how to perform rapid immunolabeling of a cell population while keeping RNA integrity, and how to isolate these specific cells using LCM. Herein, we illustrated this multi-step procedure by immunolabeling and capturing dopaminergic cells in brain tissue from one-day-old mice. We highlight key critical steps that deserve special consideration. This protocol can be adapted to a variety of tissues and cells of interest. Researchers from different fields will likely benefit from the demonstration of this approach.
Keywords: Molecular Biology, Issue 98, Laser capture, microdissection, mRNA, immunolabeling, gene expression, dopamine neurons, RNA sequencing, qRT-PCR
Introduction
The brain is composed of a large variety of different neuron types forming complex networks. These neurons are organized in distinct groups and subgroups according to their morphology, connectivity and gene expression pattern1. The development of microarrays, next-generation sequencing, and qRT-PCR offered the possibility to compare gene expression profiles of neuronal populations in different biological contexts1,2. These sensitive analyses require the precise identification and isolation of cell types of interest while keeping RNA integrity2-4. Fluorescent activated cell sorting (FACS) technique has been widely used to isolate specific cell types based on cell-surface markers and/or morphology. FACS requires a cell dissociation step prior to sorting, which results in a complete loss of spatial resolution5. Many neuronal subpopulations are distinguished from one another according to their anatomical distribution in the brain. Laser capture microdissection applied on thin brain sections provides an appropriate option for specific isolation of cells with high spatial resolution6-8. A major limitation of LCM has been the need to identify cells of interest based on morphological criteria or on fluorescent protein reporters genetically engineered in animal models. The development of new techniques to perform quick immunolabeling methods that preserved RNA integrity, in combination with LCM, now allows the isolation of cell subpopulations to proceed with gene-profiling experiments.
This approach has been previously performed and briefly described in few publications1,9-13. Here, we demonstrate a detailed procedure to obtain high-quality RNA from a specific subset of cells in a complex tissue structure by combining quick immunolabeling with LCM. We show how to perform key critical steps to obtain maximal RNA recovery and avoid RNA degradation as it may significantly impact gene expression profiling.
For the demonstration of this protocol, dopaminergic neurons from one-day-old mouse midbrain were targeted. Dopaminergic neurons can be immunolabeled using an antibody directed against tyrosine hydroxylase (TH), the rate-limiting enzyme for dopamine synthesis. Following TH immunostaining, individual or groups of dopaminergic neurons can then be isolated using LCM. Microdissected cells are collected in the lysis buffer, and RNA is extracted using a RNA isolation kit. Quality and quantity of extracted RNA are measured using a bioanalyzer5. Further analysis of gene expression using: RNA sequencing, microarray, or qRT-PCR can subsequently be performed2,4,6. As an example, a two-step qRT-PCR is demonstrated on the laser captured isolated dopaminergic domains. Relative quantification of expression levels of two dopaminergic neuron marker genes illustrates the selectivity of this protocol.
Protocol
NOTE: The experiments were performed in accordance with the Canadian Guide for the Care and Use of Laboratory Animals and were approved by the Université Laval Animal Protection Committee.
1. Sample Preparation
NOTE: In this example, we used mouse brains at postnatal day 1.
Use hypothermia anesthesia in crushed ice for 3 to 4 min for pups, then dissect brains as quickly as possible in ice-cold L15 medium. Perform this step within 2 min.
Transfer dissected brains in embedding mold (cf. list of materiel) and fill with frozen tissue embedding media taking care to orient the specimen in the desired position. Freeze specimens immediately in liquid nitrogen and store at -80 °C. Perform this step within 30 sec. NOTE: Specimens can be stored few months without compromising the RNA quality.
2. Sectioning
Treat membrane coated glass slides (cf. list of material) with surface RNAse decontamination solution to eliminate any trace of RNAses. Wash the slides in DEPC water and let them dry. Alternatively, bake slides overnight at 200 °C.
Transfer frozen samples in a cryostat previously cleaned with a surface RNAse decontamination solution. Set cryostat chamber temperature at -20 °C and slice specimens at 10 µm thickness. Collect tissue sections on the membrane coated slides and let them dry 10 min before staining. Alternatively, store slides at -80 °C until staining.
3. Preparation of Staining Solutions
Prepare all solutions just before the start of the experiment, and use RNAse inhibitor in all staining solutions (except for washing solutions) to prevent RNA degradation.
Prepare the buffer solution. Use the same buffer in all solutions (composed of RNAse-free phosphate-buffered saline (PBS) supplemented of 1% BSA, 0.2% triton and 2% RNAse inhibitor). For each slide, prepare 400 µl of buffer solution (200 µl for the primary and 200 µl for the secondary antibodies).
Prepare the fixative solution: use 70% ethanol kept at -20 °C. Keep tissue fixation procedure minimal to avoid a drastic reduction in the RNA extraction yield. NOTE: Alternatively, use 95% ethanol solution supplemented of 5% acetic acid kept at -20 °C.
Prepare the primary antibody solution by diluting the primary antibody in the buffer solution. For dopaminergic neurons labeling, use an antibody targeting the tyrosine hydroxylase (TH, rate limiting enzyme for dopamine production). Use a high concentration of antibody to compensate for the quick incubation time. Use TH antibody at a dilution of 1:25. NOTE: In normal immunofluorescence protocol, this antibody gives a strong signal when incubated overnight at a dilution of 1:1,000. In this protocol, it is used 40X more concentrated due to the short incubation time. Due to the high concentration of antibody used for this method, perform a standard immunostaining in parallel in order to check any increase in non-specific labeling.
Prepare the secondary antibody solution by diluting the secondary antibody in the buffer solution. Use a biotinylated secondary antibody at a concentration of 1:100. NOTE: In this example, we used an anti-rabbit biotinylated antibody.
Prepare the avidin-biotin complex (ABC) solution for the detection of biotinylated secondary antibodies. Make ABC solution by adding the compound A and the compound B at a concentration of 1:100 diluted in DEPC PBS supplemented with 2% RNAse inhibitor. Prepare the Avidin-Biotin complex 30 min before adding it to the slides.
4. Staining
- Thaw one or two slides at the time from -80 °C by flicking them in the air quickly (or use an air blower). Surround sections with a hydrophobic barrier pen and let dry for a few sec.
- Put the slides in the cold fixative solution for no more than 5 min at -20 °C. Shake once in the air to remove excess of fixative solution then, quickly dip slides in DEPC PBS 6-8 times for washing.
- Flick the slides to remove excess of DEPC PBS. Ensure that the slides defrosting step does not exceed 5 min.
Place slides on a clean tray (pretreated with a surface RNAse decontamination solution to eliminate any trace of RNAses) and put 200 µl of the primary antibody solution on each slide (rabbit anti-TH) for 10 min at room temperature. Dip slides quickly in DEPC PBS 3 times for washing and flick the slides once to remove the excess of DEPC PBS.
Put 200 µl of the secondary antibody solution on each slide for 6 min at room temperature. Dip slides quickly 3 times in DEPC PBS for washing and flick the slides to remove the excess of DEPC PBS.
Put 200 µl of the ABC solution on each slide for 4 min at room temperature. Dip slides quickly in DEPC PBS 3 times for washing and flick the slides to remove the excess DEPC PBS.
Prepare the DAB (3,3’ diaminobenzidine) solution during the 4 min of incubation in the ABC solution. Use the reagents provided in the kit (DAB Peroxidase Substrate Kit). Mix 1 drop of provided buffer, 1 drop of DAB solution and 1 drop of 30% H2O2 in 2.5ml of DEPC water. Mix the solution by inverting.
Take 196 µl of the DAB solution and add 4 µl (2%) of RNAse inhibitor solution just before spreading on slides. Place slides on a clean tray (pretreated with a surface RNAse decontamination solution) and put the 200 µl of DAB solution on slides. NOTE: DAB solution should react within1 or 2 min.
Dip slides quickly in DEPC PBS 3 times for washing and put slides for few seconds in absolute ethanol. Dry the slides by flicking them in the air (or use an air blower). NOTE: Slides can be stored at -80 °C for future use or processed for LCM.
5. Laser Capture Microdissection
Defrost slides quickly by flicking them in the air (or use an air blower). Ensure that the slides defrosting step does not exceed 5 min.
- Proceed to LCM. Place the slide under the microscope with the sample side facing the collecting tube cap. Choose the best magnification for the sample. Adjust the focus to see the cells of interest.
- Define the cutting area and start cutting with the laser. Collect dissected cells or piece of tissue in the collecting tube cap filled with 50 µl of lysis buffer (from RNA extraction kit). Ensure that the step of LCM does not exceed 30 min.
- Extract total RNA from isolated cells using an RNA extraction kit (cf. list of materials). Follow the manufacturer’s protocol.
- Incubate collected cells for 30 min at 42 °C to lyse the cells. Pre-condition the RNA purification column by adding 250 μl of conditioning buffer. Load 50 μl of ethanol to lysed cells. Load the 100 μl of lysed cells onto a preconditioned purification column.
- Centrifuge the column at 16,000 x g for 2 min at room temperature. Wash the column twice with washing buffers. Elute in the minimal recommended volume (11 µl) of RNAse-free water (not DEPC treated as it may interfere with the bioanalyser). Keep 2 µl to test quality. NOTE: All solutions and reagents are provided in the kit.
6. cDNA Synthesis and Quantitative RT-PCR
Reverse-transcribe RNA samples from laser captured dopaminergic cells into complementary DNA using reverse transcriptase and oligo(dT). Follow the manufacturer’s protocol (cf. list of material).
- Use 1 µl of cDNA for qRT-PCR amplification with gene-specific primers. Set up PCR reactions in 20 µl volume with a one-component hot start reaction mix for quantitative PCR as recommended by the manufacturer. Carry out each reaction in triplicate.
- For the experiments described here, use the following primer pairs: Gapdh-F(5’-CCA CCC AGA AGA CTG TGG AT-3’)/Gapdh-R (5’-GGA TGC AGG GAT GAT GTT CT-3’); Rpl13a-F (5’-ACA GCC ACT CTG GAG GAG AA-3’) / Rpl13a-R (5’-CTG CCT GTT TCC GTA ACC TC-3’) ; Th-F (5’-CAG TGG AGG ATG TGT CTC -3’) / Th-R (5’-GAA AAT CAC GGG CAG ACA G-3’); Aadc-F (5’- CAT GAG AGC TTC TGC CCT TC -3’)/Aadc-R (5’- GGA TGT GGT CCC CAG TGT AG -3’).
Perform quantitative RT-PCR amplification using the following rapid cycling parameters: 95 °C for 5 min followed by 45 cycles of 10 sec 95 °C, 10 sec at 60 °C and 10 sec at 72 °C. Perform melting curve analysis using the default setting of the instrument to determine homogeneous product formation.
- Analyze the data using the built-in software.
- Normalize the expression values of DA markers genes Th and Aadc with the expression of the two reference genes Gapdh and Rpl13a to obtain relative expression levels as described previously14.
Representative Results
This rapid immunolabeling procedure allows visualization of cells of interest while keeping RNA integrity. Figure 1 illustrates the steps of tissue preparation before RNA extraction. After cryosectioning, dopaminergic neurons are labeled using an antibody directed against the tyrosine hydroxylase (labeled neurons appear in brown). Depending of the experimental question, single cells or larger region of interest can be isolated using LCM (Figure 1D-E). It is important to assess the quality of RNA extracted, as degraded RNA will have a considerable impact on the quality of the following analysis. Each sample is thus processed on a microfluidics-based platform bioanalyser for quantification and to check RNA integrity. Figure 2 shows typical bioanalyser results of degraded and good-quality samples. Both digital gel and electropherograms demonstrate the importance of using RNAse inhibitor in each solution to protect RNA. The expected amount of RNA extracted from 500 neurons is around 1-2 ng. Microdissected cells from several tissue sections can be pooled in the same tube to increase RNA quantity.
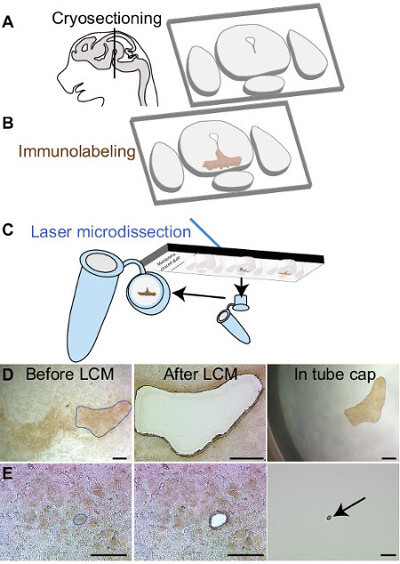 Figure 1:Rapid immunolabeling and laser capture microdissection procedure. (A) Schematic showing sagittal view of a mouse brain at embryonic day 15.5. Black line indicates the location of the anteroposterior regions where midbrain dopamine neurons are located. Thin sections are made using a cryostat and are collected on membrane coated glass slides. Following immunolabeling, midbrain dopaminergic neurons are visible in brown (B). Laser microdissected samples are collected in a tube cap filled with lysis buffer (C). Region of interest (blue line in D) or individual cell (E) can be dissected and retrieved in the tube cap. Scale bars: (D) 250μm, (E) 50μm. Please click here to view a larger version of this figure.
Figure 1:Rapid immunolabeling and laser capture microdissection procedure. (A) Schematic showing sagittal view of a mouse brain at embryonic day 15.5. Black line indicates the location of the anteroposterior regions where midbrain dopamine neurons are located. Thin sections are made using a cryostat and are collected on membrane coated glass slides. Following immunolabeling, midbrain dopaminergic neurons are visible in brown (B). Laser microdissected samples are collected in a tube cap filled with lysis buffer (C). Region of interest (blue line in D) or individual cell (E) can be dissected and retrieved in the tube cap. Scale bars: (D) 250μm, (E) 50μm. Please click here to view a larger version of this figure.
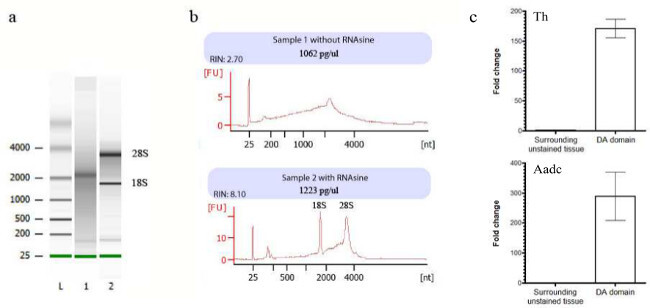 Figure 2:Quality control of total RNA sample isolated after laser capture microdissection using a bioanalyzer chip kit. (A) The digital gel obtained with the bioanalyzer (L, ladder; 1, sample 1 without RNAse inhibitor (RNAsine), sample 2 with RNAsine). (B) Electropherograms of sample 1 (upper panel) showing partially degraded RNA and sample 2 (lower panel) typical of good-quality RNA (RIN >8 is acceptably good) in which the 18S/28S rRNA peaks are clearly visible. (C) Normalized relative expression levels represented in fold change for two DA marker genes (TH and AADC) between surrounding unstained tissue and DA domain. Please click here to view a larger version of this figure.
Figure 2:Quality control of total RNA sample isolated after laser capture microdissection using a bioanalyzer chip kit. (A) The digital gel obtained with the bioanalyzer (L, ladder; 1, sample 1 without RNAse inhibitor (RNAsine), sample 2 with RNAsine). (B) Electropherograms of sample 1 (upper panel) showing partially degraded RNA and sample 2 (lower panel) typical of good-quality RNA (RIN >8 is acceptably good) in which the 18S/28S rRNA peaks are clearly visible. (C) Normalized relative expression levels represented in fold change for two DA marker genes (TH and AADC) between surrounding unstained tissue and DA domain. Please click here to view a larger version of this figure.
Discussion
The most critical point of this method consists in labeling cells of interest while preventing RNA degradation. As RNAses are active in aqueous solutions, decreasing incubation times improves RNA conservation11,15. All solutions used must be treated with DEPC to inactivate RNAses. All materials and surfaces need to be treated with a surface RNAse decontamination solution to prevent RNAses contamination. Finally, in these conditions, the addition of RNAse inhibitor in all solutions is effective in preserving RNA from being degraded. The use of high salt conditions applied during antibody incubation has been previously shown to be effective in preserving RNA9,10. However, this alternative method remains limited to quick staining using robust antibodies. The thickness of the sections represents another critical point and depends on the size of the cell of interest. Since the average size of the cell body of dopaminergic neurons is around 10µm, we select this section thickness to obtain one layer of cells.
The major limitation of this technique compared to FACS is the low number of cells that can be microdissected. The use of RNA amplification kit is required for gene profiling experiments using RNA-sequencing or microarray16. However, qRT-PCR can be performed directly without the amplification step (Figure 2c). Qualitative assessment of the staining obtained with the quick immunolabeling protocol should always be performed and should not differ from the staining obtained using the normal immunolabeling protocol.
Until now, laser capture experiments were mostly used to isolate populations of cells from transgenic animals expressing markers like GFP or using quick histological colorations such as Nissl staining. The method presented here allows visualizing cells of interest using immunohistochemistry. Isolating and acquiring the gene expression profile of chemically identified populations of cells is now possible17. The protocol described here can be applied to any tissues and used with any suitable antibodies.
In the brain, neurons can be divided by their geographic localization but most brain regions contain a variety of different subpopulations based on their chemical identity. The gene expression pattern of certain types of neurons can be largely different from another cell type in the same cerebral area. According to the biological or pathological context, gene expression profile of a specific cell population can also change considerably. Using the procedure described here, it is possible to monitor these changes, paving the way to new discoveries8,18,19.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work is supported by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC: 418391-2012). AC receives a scholarship from the Centre thématique de recherche en Neurosciences (CTRN). HDB is funded by a scholarship from the Fonds de Recherche en Santé du Québec (FRSQ) and ML is a FRSQ Chercheur-Boursier.
References
- Chung CY, et al. The transcription factor orthodenticle homeobox 2 influences axonal projections and vulnerability of midbrain dopaminergic neurons. Brain : a journal of neurology. 2010;133(Pt 7):2022–2031. doi: 10.1093/brain/awq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, et al. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications. Applied immunohistochemistry & molecular morphology : AIMM / official publication of the Society for Applied Immunohistochemistry. 2013;21(1):31–47. doi: 10.1097/PAI.0b013e31824d0519. [DOI] [PubMed] [Google Scholar]
- Decarlo K, Emley A, Dadzie OE, Mahalingam M. Laser capture microdissection: methods and applications. Methods in molecular biology. 2011;755:1–15. doi: 10.1007/978-1-61779-163-5_1. [DOI] [PubMed] [Google Scholar]
- Espina V, Heiby M, Pierobon M, Liotta LA. Laser capture microdissection technology. Expert review of molecular diagnostics. 2007;7(5):647–657. doi: 10.1586/14737159.7.5.647. [DOI] [PubMed] [Google Scholar]
- Fend F, Raffeld M. Laser capture microdissection in pathology. Journal of clinical pathology. 2000;53(9):666–672. doi: 10.1136/jcp.53.9.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadi G, Maximino JR, de Oliveira GP. The importance of molecular histology to study glial influence on neurodegenerative disorders. Focus on recent developed single cell laser microdissection. Journal of molecular histology. 2009;40(4):241–250. doi: 10.1007/s10735-009-9235-0. [DOI] [PubMed] [Google Scholar]
- Liu A. Laser capture microdissection in the tissue biorepository. Journal of biomolecular techniques : JBT. 2010;21(3):120–125. [PMC free article] [PubMed] [Google Scholar]
- Baskin DG, Bastian LS. Immuno-laser capture microdissection of rat brain neurons for real time quantitative PCR. Methods in molecular biology. 2010;588:219–230. doi: 10.1007/978-1-59745-324-0_23. [DOI] [PubMed] [Google Scholar]
- Brown AL, Smith DW. Improved RNA preservation for immunolabeling and laser microdissection. RNA. 2009;15(12):2364–2374. doi: 10.1261/rna.1733509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Brown AL, Day TA, Dayas CV, Smith DW. Purity and enrichment of laser-microdissected midbrain dopamine neurons. BioMed research international. 2013;2013:747938. doi: 10.1155/2013/747938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Liotta L, Star RA. IF-LCM: laser capture microdissection of immunofluorescently defined cells for mRNA analysis rapid communication. Kidney international. 2000;58(3):1346–1353. doi: 10.1046/j.1523-1755.2000.00295.x. [DOI] [PubMed] [Google Scholar]
- Greene JG, Dingledine R, Greenamyre JT. Gene expression profiling of rat midbrain dopamine neurons: implications for selective vulnerability in parkinsonism. Neurobiology of disease. 2005;18(1):19–31. doi: 10.1016/j.nbd.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Endo S, Isacson O. An Endogenous Serine/Threonine Protein Phosphatase Inhibitor, G-Substrate, Reduces Vulnerability in Models of Parkinson's Disease. Journal of Neuroscience. 2007;27(31):8314–8323. doi: 10.1523/JNEUROSCI.1972-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. AZ of quantitative PCR. La Jolla, CA: International University Line (IUL); 2004. Chapter 3, Quantification strategies in real-time. [Google Scholar]
- Smolinski D, Blessenohl M, Neubauer C, Kalies K, Gebert A. Validation of a novel ultra-short immunolabeling method for high-quality mRNA preservation in laser microdissection and real-time reverse transcriptase-polymerase chain reaction) The Journal of MolecularDiagnostics. 2006;8(2):246–253. doi: 10.2353/jmoldx.2006.050096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podgornyĭ OV, Lazarev VN, Govorun VM. Laser microdissection for biology and medicine. Tsitologiia. 2012;54(5):381–389. [PubMed] [Google Scholar]
- Vandewoestyne M, ewoestyne & Deforce D. Laser capture microdissection in forensic research: a review. International journal of legal medicine. 2010;124(6):513–521. doi: 10.1007/s00414-010-0499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JP, et al. A quantitative proteomic workflow for characterization of frozen clinical biopsies: laser capture microdissection coupled with label-free mass spectrometry. Journal of. 2012;77:433–4340. doi: 10.1016/j.jprot.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet B, Maclennan GT, Lopez-Beltran A, Montironi R, Cheng L. Laser capture microdissection in the genomic and proteomic era: targeting the genetic basis of cancer. International journal of clinical and experimental pathology. 2008;1(6):475–488. [PMC free article] [PubMed] [Google Scholar]