

Abstract
The brain operates through the coordinated activation and the dynamic communication of neuronal assemblies. A major open question is how a vast repertoire of dynamical motifs, which underlie most diverse brain functions, can emerge out of a fixed topological and modular organization of brain circuits. Compared to in vivo studies of neuronal circuits which present intrinsic experimental difficulties, in vitro preparations offer a much larger possibility to manipulate and probe the structural, dynamical and chemical properties of experimental neuronal systems. This work describes an in vitro experimental methodology which allows growing of modular networks composed by spatially distinct, functionally interconnected neuronal assemblies. The protocol allows controlling the two-dimensional (2D) architecture of the neuronal network at different levels of topological complexity.
A desired network patterning can be achieved both on regular cover slips and substrate embedded micro electrode arrays. Micromachined structures are embossed on a silicon wafer and used to create biocompatible polymeric stencils, which incorporate the negative features of the desired network architecture. The stencils are placed on the culturing substrates during the surface coating procedure with a molecular layer for promoting cellular adhesion. After removal of the stencils, neurons are plated and they spontaneously redirected to the coated areas. By decreasing the inter-compartment distance, it is possible to obtain either isolated or interconnected neuronal circuits. To promote cell survival, cells are co-cultured with a supporting neuronal network which is located at the periphery of the culture dish. Electrophysiological and optical recordings of the activity of modular networks obtained respectively by using substrate embedded micro electrode arrays and calcium imaging are presented. While each module shows spontaneous global synchronizations, the occurrence of inter-module synchronization is regulated by the density of connection among the circuits.
Keywords: Neuroscience, Issue 98, In vitro, patterning, PDMS stencils, SU8-2075, silicon wafer, calcium imaging, Micro Electrode Array
Introduction
Experimental and theoretical evidences support the possibility that the brain operates through coordinated activation of cell assemblies1-5, which can be regarded as dynamic functional units that transiently interact with each other, shaping and underlying different brain states. Functional modularity is also dependent on and associated with the structural modular organization of the brain circuits6,7. How function and structure of brain circuits mutually shape each other is still one of the main open questions in neuroscience. To provide a deeper understanding of this question, it is important to identify optimal experimental frameworks where it is possible to address, at least partially, those issues. Since controlled manipulation of the spatio-temporal dynamics of neuronal networks in in vivo experiments is challenging, the development of in vitro neuronal networks models is of significant interest due to their easy accessibility, monitoring, manipulation and modeling8,9. In recent years, in vitro technologies supported by advanced substrate patterning methods have allowed to induce neuronal networks to develop a range of predefined modular structures3 and to study the functional properties of networks with imposed topologies10. In particular, methods were recently used to organize networks by imposing physical constraints4,11. Indeed, to study the link between structure and function in neuronal networks and to provide a simplified but plausible representation of interacting neuronal assemblies, in vitro systems should provide inter-connected neuronal sub-populations. Widely studied 2D homogenous neuronal cultures do not impose any spatial constraints on the self-organized emergent wiring of the circuits. Therefore a possible approach to shape artificially interconnected cell assemblies is to position different neuronal populations in spatially distinct areas. The distance among these areas does not prevent the inter assemblies connections. This approach, while ensuring a considerable control over network complexity, has been shown to provide a richer repertoire of synchronization models6,7,12.
In order to facilitate a reproducible culturing of modular neuronal assemblies, a protocol to assemble the self-organization of networks into neuronal clusters linked by axons and dendrites is presented and described. The polymeric structure for the physical confinement of neuronal cultures has been created from polydimtheylsiloxane (PDMS). PDMS is an elastomer widely used for biomedical applications owing to its biocompatibility, transparency and permeability to gases13. The PDMS is prepared and excluded from the micromachined SU8 207514,15 structures by spin-coating a liquid PDMS onto a "master" as described previously in Jackman et al.16 The achieved patterned neuronal networks are composed of inter-connected modules of different size and they have been successfully obtained on both coverslips and Micro Electrode Arrays (MEAs)17-20. The density of connections between the modules can change the features of the network synchronization, from a fully synchronized network, typical of uniform cultures, to transient states of synchronization among modules.
Protocol
The procedure was done in accordance with the NIH standards for care and use of laboratory animals and was approved by the Tel-Aviv University Animal Care and Use Committee (permit number – L-14-019).
1. Preparation of Instruments and PDMS
Prepare the wafer (Table of Materials, or order the wafer from a microfabrication lab), a scalpel, and a pair of tweezers – sterilization is not needed.
Make poly-D-lysine (PDL) solution according to the following conditions: 4 mg/ml in 0.1 M borate buffer, pH 8, and store at -20 °C.
- Prepare PDMS stencils according to the following procedure:
- Prepare polydimethylsiloxane (PDMS, silicone elastomer) by mixing together the base and the curing agent with a ratio of 10:1, then stir the 2 substances for approximately 5 min.
- Place in vacuum chamber twice for 15 min each time and verify bubbles are gone.
- When the PDMS is ready, prepare spin coater: open nitrogen knobs to allow gas usage, place the silicon wafer (Figure 1A) on spinner, and use the vacuum to prevent it from moving.
- Pour PDMS over wafer and activate the spinner for 1 min at 1,000 rpm (creates approximately 100 µm height).
- Place wafer on a hot plate at 100 °C for 30 min.
- When the PDMS has hardened, outline the stencil's borders with PDMS, using Pasteur pipette. Place wafer on a hot plate at 100 °C for 30 min.
- When the PDMS borders have hardened, use a scalpel to cut stencils according to the borders and peel off the silicon stencil from the wafer (a squared PDMS containing a single pattern).
2. Petri Dish and Cover Slip Preparation
- Prepare 23 mm square glass cover slips and clean them according to the following order:
- Wash with distilled water, 70% ethanol, and acetone.
- Wash with isopropanol and fast dry with nitrogen.
Place patterned PDMS stencils on coverslip, and gently press to verify that they are firmly attached to the glass surface. Place in vacuum chamber for 15 min.
Drop 1 ml of PDL on stencil. Insert the coverslip into the vacuum chamber twice for 20 min each time, and leave PDL O/N to dry.
- Prepare Petri dish to create a supporting cell network:
- Cover the surface of the dish (3.5 cm) with 1 ml of PDL for 2 hr in RT. Remove the PDL drop using a pipette.
- Wash with distilled water and leave to dry. Place a very small drop of silicone grease on each corner of the coverslip.
Place the coverslip on the center of the Petri dish (i.e., the PDMS should be facing up) and press gently to verify attachment.
Gently remove PDMS from coverslip using tweezers. For sterilization, expose to UV illumination for 7 min.
3. Multi-Electrode Array (MEA) Preparation
- Clean MEA in this order (Table of Materials):
- Wash with water under tap and sonicate in a concentrated, enzymatic detergent 3 times.
- Sonicate in distilled water 3 times.
- Wash with distilled water (in hood, 1,000 µl tip) for 3 times and place under UV for 30 min.
- Supporting network preparation:
- Prepare the designed support network mold.
- Pour PDMS into a 12-well plate (22 mm diameter).
- When the PDMS is hardened take out the mold and using a scalpel, cut a hole in the middle to create a ring shape.
- Place a designed support network mold on the center of the MEA (Figure 2A) and cover the rest of the surface with PDL for 2 hr at RT.
- Remove PDL using pipette and wash with distilled water.
- Remove the mold and leave to dry (Figure 2A).
- Align stencil to MEA in the following way:
- Place stencil on designated micro manipulator. Use an inverted microscope to accurately align patterned structure to electrodes, and lower the stencil until it is placed on the MEA surface (Figure 2B). Note: Contact with a well cleaned MEA provides competitive adhesion between the PDMS and the MEA itself.
- Lift the micromanipulator and if needed, use tweezers to apply a small amount of pressure above the PDMS to prevent it detaching from the MEA.
- Gently press stencil to MEA surface, and use microscope to verify it is firmly attached, and well aligned. (Figure 2C-D).
Place MEA in vacuum chamber for 15 min; put a 1 ml drop of PDL on stencil.
Insert into vacuum chamber 2 times for 20 min each. Leave PDL to dry O/N in the incubator.
Before plating, remove the PDMS stencils from MEAs and wash using sterile water. Place under UV illumination for 7 min.
4. Dissection and Culture
Prepare cultures as described in Herzog et al. 2011 21.
5. Plating
- Calculate number of cells needed for plating, using a hemocytometer (optimal density according to the size of the circuit as discussed in the result).
- Make sure the counting chamber (hemocytometer) is clean and place a cover slip on it (use alcohol to clean).
- Dilute 10 µl of the cell suspension in 190 µl of plating medium (dilution 1:20).
- Load 10 µl of the diluted cells onto the edge of the counting chamber and slowly pipette the cells out allowing the chamber to fill itself.
- Using an inverted microscope, visualise the haemocytometer grid. Determine the number of cells in the chamber by direct counting (healthy cells should be round). Count the cells within the large square without those crossing the edges.
- Calculate the concentration of cells: Total number of cells/1000 µl = Total cells counted × dilution factor × 104 (area of the hemocytometer). Note: For example, if the dilution factor was 20 and the total cells counted were 100: 100 × 20 × 104 = 0.1 x 107 cells/1,000 µl or 1 x 106/100 µl. In order to plate 0.75 x 106 cells, take 75 µl out of the cells.
Take the number of cells needed for plating per single MEA or Petri dish, resuspend the cells to prevent aggregation, and plate them at the center, on top of the patternate area (if needed, it is possible to dilute the cells in the medium). For the MEA, place a 100 µl drop in the middle. For cover slip, place a 1,000 µl drop in the middle.
Incubate the plated cells at 37 °C for 40 min. Add plating medium to the plated cells, up to 1 ml per MEA and 2 ml per cover slip.
Keep at 37 °C and, every 4 days, dilute with fresh growth medium enriched with 0.5 Pen-Strep, 2% B-27 and 0.75% glutamax.
From 6/7 DIV, after seeing connections between the islands, dilute medium with 10 µl/ml FUDR (25 mg deoxyuridine + 62.5 mg uridine in 12.5 ml MEM (Minimum Essential Medium Eagle)) or any other anti-mitotic agent to prevent glial overgrowth.
Representative Results
A SU8-2075 mold on a silicon wafer with a feature thickness of approximately 100 µm was used to shape the PDMS. The pattern was composed of squares of several dimensions, with a side length and distance varying between 200 and 700 µm (Figure 1B). The size of the square was chosen to fit the field of view of a 10X (for islands with a side length <800 µm) and of a 20X objective (for islands with a side length <400 µm). Three parameters, namely cell plating density, distance between circuits, circuits’ size are determinant for obtaining monolayers or clustered circuits as well as to impose and shape their connectivity. However, these parameters are not independent since, given a fixed distance between two circuits, the probability that they spontaneously establish a connection increase with the circuits’ size. In the example discussed in this work, for small neuronal modules of ~300 x 300 µm, connections were established when the distance was not larger than ~300 - 400 µm, while for large neuronal modules of ~700 x 700 µm, connections were established when the distance was smaller than ~700 µm. In general, by increasing the distance between the squares it was possible to change the probability to establish spontaneously generated inter-connections among the modules, passing from highly connected modules to isolated ones.
Neuronal modules of ~600 x 600 µm are shown at 4 days in vitro (DIV; Figure 3A). After few days in vitro neurons are mostly located within the coated areas while it is not possible to observe developed neuronal processes and connections. On the contrary, at 14 DIV (Figure 3B-C) neurons establish connections within and among modules. While larger neuronal circuits of ~600 x 600 µm could self-organize into monolayers (Figure 3A-C), circuits of smaller dimensions (e.g., ~300 x 300 µm in Figure 3D) tended to cluster. Therefore, in order to obtain the best calibration for two-dimensional circuits, cells were plated with different density varying from 0.25 x 106 to 1 x 106 cells/23 mm cover slip attached in a 35 mm Petri dish. For larger circuits of ~700 x 700 µm we found that 0.75 x 106 cells/23 mm cover slip attached in a 35 mm Petri dish is the optimal density which induces the formation of monolayer circuits not preventing their spontaneous inter-connection (Figure 3C). The same result of monolayer circuit was obtained in smaller circuits of ~300 x 300 µm by plating 0.5 x 106 cells/23 mm cover slip attached in a 35 mm Petri dish. In general, when considering cell plating density, it is important to highlight that, as previously discussed7, neuronal and glial cells have an innate tendency to cluster, so some degree of clustering is almost unavoidable after some time in culture. However, we observed that increasing the area destined to the supporting network would enhance the probability of circuits’ survival and their mono-layered organization, probably due a larger concentration of nutrients in the extracellular space. In any case, a trial and error approach is required to identify the optimal cell plating density to create mono-layered circuits for calcium imaging. Every 4 days the medium was diluted with fresh growth medium. From 6/7 DIV, after seeing connections between the islands, 10 µl/ml FUDR has been added to prevent glial overgrowth.
Figure 4 shows the dynamic of a modular network recorded using calcium imaging (performed as described in Bonifazi et al. 20138). Calcium-fluorescence images were acquired using a 4X magnification objective, which enabled monitoring the activity of the entire network (Figure 4A). Analysis of the calcium imaging was performed as described in Bonifazi et al. 20138. In Figure 4B raster plot of the calcium events onset displaying spontaneous activity is presented (colors correspond to modules marked in Figure 4A). Calcium imaging was performed at 30 Hz, and image size is 1,000 x 1,000 pixels.
The green and pink modules are highly synchronized as shown in the raster plot (Figure 4B) in agreement with their thick connecting bundles, possibly corresponding to a large number of connections (Figure 4A). The blue and red modules are weakly connected to the green and pink modules and therefore they occasionally synchronized with them (Figure 4B). A video of the recording is available online (movie M1).
The image of a modular network grown on a MEA at 21 DIV is shown in Figure 5A. In order to obtain the best calibration for two-dimensional circuits, we plated cortical cells with different density varying from 250 x 103 to 500 x 103 cells/per MEA. 250 x 103 cells/per MEA (30 mm ring) gave better results in terms of the formation of monolayer circuits, while not preventing their spontaneous inter-connection. Once patterned neuronal networks have been achieved on MEAs substrate (Figure 5A), electrophysiological activity has been monitored using a commercial system used for acquiring and recording electrophysiological signals. Spontaneous activity of cultures is detected using the Precise Timing Spike Detection (PTSD) algorithm22.
Figure 5B shows a raster plot corresponding to 5 min of spontaneous activity (only active electrodes are visible), colored coded according to the cluster number. By looking at these plots, it is possible to make qualitative assessments of the activity of single clusters and of the whole network at the same time. In particular, it is possible to observe a strong synchronization among the activities recorded within each cluster. A video of a representative recording from patterned cultures over MEA is available online (movie M2).
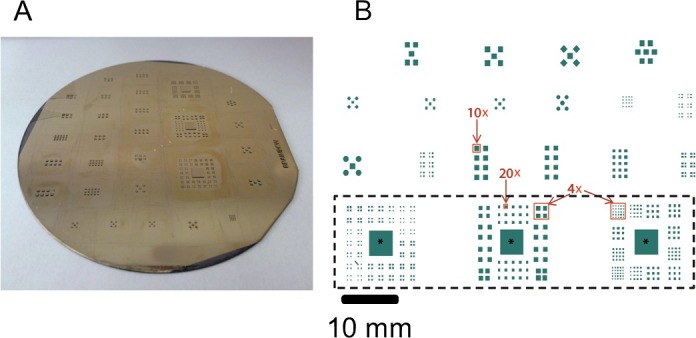 Figure 1. Silicon wafer. (A) The silicon wafer (~4 inch diameter) used to mold the PDMS stencils. Different SU8 structures represent different modular networks. (B) Representative feature design implemented on a silicon wafer for modular networks construction. Each green spot define an SU8 pillar structure and therefore the area for a single module. The designs within the dashed black rectangle correspond to three different stencils which can be used on standard square culture coverslips of 23 mm side length. In each stencil, an asterisk marks a macro-region for the supporting network. Depending on the distance between the spots, the finite size circuit or modules might be able to establish spontaneous neuronal connections among them. The design of the features was chosen to create different module size and inter-module distance to achieve isolated or interconnected neuronal circuits which could fit the field of view in different objective magnifications (4X, 10X and 20X; the field of view for each magnification is represented by the red squares). The features out of the dashed black rectangle are designed to fit the simultaneous use of MEA and calcium imaging. Please click here to view a larger version of this figure.
Figure 1. Silicon wafer. (A) The silicon wafer (~4 inch diameter) used to mold the PDMS stencils. Different SU8 structures represent different modular networks. (B) Representative feature design implemented on a silicon wafer for modular networks construction. Each green spot define an SU8 pillar structure and therefore the area for a single module. The designs within the dashed black rectangle correspond to three different stencils which can be used on standard square culture coverslips of 23 mm side length. In each stencil, an asterisk marks a macro-region for the supporting network. Depending on the distance between the spots, the finite size circuit or modules might be able to establish spontaneous neuronal connections among them. The design of the features was chosen to create different module size and inter-module distance to achieve isolated or interconnected neuronal circuits which could fit the field of view in different objective magnifications (4X, 10X and 20X; the field of view for each magnification is represented by the red squares). The features out of the dashed black rectangle are designed to fit the simultaneous use of MEA and calcium imaging. Please click here to view a larger version of this figure.
 Figure 2. PDMS structures deposition on a MEA. (A) PDMS mold with a ring shape (marked with the red arrow) mounted on a MEA. The mold is used to coat the area destined to the supporting neuronal network and located at the periphery of the culturing area (this is limited by the ring mounted on the MEA and marked by the green arrow). (B) Alignment of a PDMS stencil on the MEA using a micro manipulator. (C) PDMS stencil deposited on the on MEA after alignment. (D) Image of a PDMS stencil on MEA using a 10x magnification (inter-electrode distance is 500 µm). It is possible to note the holes on the stencils in correspondence of the electrodes. The adhesive layer favoring cellular adhesion will be deposited only on the opened areas. Please click here to view a larger version of this figure.
Figure 2. PDMS structures deposition on a MEA. (A) PDMS mold with a ring shape (marked with the red arrow) mounted on a MEA. The mold is used to coat the area destined to the supporting neuronal network and located at the periphery of the culturing area (this is limited by the ring mounted on the MEA and marked by the green arrow). (B) Alignment of a PDMS stencil on the MEA using a micro manipulator. (C) PDMS stencil deposited on the on MEA after alignment. (D) Image of a PDMS stencil on MEA using a 10x magnification (inter-electrode distance is 500 µm). It is possible to note the holes on the stencils in correspondence of the electrodes. The adhesive layer favoring cellular adhesion will be deposited only on the opened areas. Please click here to view a larger version of this figure.
 Figure 3. Modular neuronal networks at different DIV. Bright field image of cortical modules of ~600 x 600µm (A) after 4 DIV and (B) after 14 DIV. (C) Note the spontaneous interconnections among the circuits after 14 DIV. Cells were plated at the optimal cell density for large circuits to organize into monolayers, i.e., 750 x 103 cells/23 mm cover slip attached in a 35 mm Petri dish. (D) Using PDMS features of half size and same cell concentration, neuronal circuits organized into clustered structures. Please click here to view a larger version of this figure.
Figure 3. Modular neuronal networks at different DIV. Bright field image of cortical modules of ~600 x 600µm (A) after 4 DIV and (B) after 14 DIV. (C) Note the spontaneous interconnections among the circuits after 14 DIV. Cells were plated at the optimal cell density for large circuits to organize into monolayers, i.e., 750 x 103 cells/23 mm cover slip attached in a 35 mm Petri dish. (D) Using PDMS features of half size and same cell concentration, neuronal circuits organized into clustered structures. Please click here to view a larger version of this figure.
 Figure 4. Calcium imaging of modular networks. (A) Cortical cells (18 DIV) loaded with the calcium indicator OGB. Different colours mark different modules. The field of view is 2 x 2 mm. (B) Raster plot of spontaneous activity. The different colors correspond to the cells within different modules as shown in panel A. Please click here to view a larger version of this figure.
Figure 4. Calcium imaging of modular networks. (A) Cortical cells (18 DIV) loaded with the calcium indicator OGB. Different colours mark different modules. The field of view is 2 x 2 mm. (B) Raster plot of spontaneous activity. The different colors correspond to the cells within different modules as shown in panel A. Please click here to view a larger version of this figure.
 Figure 5. MEA recording of modular networks. (A) Modular network (cortical neurons, 21 DIV) composed of 3 distinct circuits grown on a 4Q MEA. Neuronal circuits are distant ~600 µm interconnected to each other. (B) Raster plot showing 5 min of spontaneous electrophysiological activity recording. C1, C2 and C3 correspond, respectively, to the upper left, the lower right and the lower left cluster’s activities, highlighted in different colors. Please click here to view a larger version of this figure.
Figure 5. MEA recording of modular networks. (A) Modular network (cortical neurons, 21 DIV) composed of 3 distinct circuits grown on a 4Q MEA. Neuronal circuits are distant ~600 µm interconnected to each other. (B) Raster plot showing 5 min of spontaneous electrophysiological activity recording. C1, C2 and C3 correspond, respectively, to the upper left, the lower right and the lower left cluster’s activities, highlighted in different colors. Please click here to view a larger version of this figure.
Discussion
A protocol to grow 2D modular neuronal networks in vitro composed of functionally inter-connected circuits is described. The procedure is based on patterning a cellular adhesive layer. Patterning is achieved with PDMS stencils reproducing the negative feature of the desired network architecture. PDMS stencils define the areas where the cellular adhesive layer is deposited. Once cells are plated, they spontaneously assemble to the coated islands and self-organize into active inter-connected circuits. Recordings of functional modular networks recorded using MEAs and calcium imaging was presented.
The presented protocol has been duly modified compared to the initially followed procedure. Firstly, problems with PDMS stencils’ alignment had to be addressed. Since the alignment has to be achieved in the first attempt (once the stencil has touched the surface, it is not recommended to relocate it), this issue has been overcome by abandoning the manual procedure and opting for a new method which consists of using a micro manipulator for precise stencil deposition on the MEA recording area. Another issue that had to be addressed consists in preventing the PDL drop from passing beneath the aligned stencil (otherwise the patterned design could not be obtained). To do so, once the stencil has been aligned and before putting the PDL drop on it, the considered device (MEA or coverslip) has to be left in the vacuum chamber for a proper period of time thus providing a good attachment of it on the surface. The same has been done once the PDL drop is placed on the stencil in order to eliminate the air bubbles that form between the device and the drop itself thus allowing the PDL to get in touch with the surface.
The critical steps of the described protocol are represented by: i) the spin coating phase which should guarantee to get “open replicas” or, at least, a sufficiently thin film over the SU8 structures that can be easily removed using tweezers; ii) the treatment of the culturing surface (cover slip or MEA); iii) the placement of the PDMS stencils on it; iv) the coating phase since there are cases in which, if the PDMS mask doesn’t seal properly against the substrate on which is positioned, it could happen that the adhesive protein passes underneath the mask thus compromising the patterning. So, the cleaning process of the cover slips or MEAs is of primary importance as well as the alignment of the PDMS stencil. Proper performances of these two steps ensure best patterning results.
The limitations of this technique have also to do with both the electrophysiological and the calcium imaging acquisition. In the first case, a very low spatial resolution (4 - 8 electrodes are included in each module) is present due to both the dimensions of the single circuit and the inter-electrode distance. In the second case, the risk consists in not finding the optimal cellular density which allows obtaining a two-dimensional cellular layer, thus preventing a single cell resolution acquisition.
This strategy, that required a careful optimization of the coating, assembly and sterilization procedures and the creation of a custom micro-alignment system, gave us some major advantages with respect to previously reported works (e.g., the long-term survival of our cells, i.e., up to 8 weeks in vitro). Specifically, it provided us with a more stable and reliable long-term confinement24 with respect to strategies based on chemical patterning23-28.
The described procedure relies on plating cortical cells on PDL-coated modules, which assures the self-organization of networks into neuronal assemblies linked by axons and dendrites bundles. To allow wide-field calcium imaging of the modular networks with a single cell resolution, it is essential that the neuronal system organizes into a 2D layer. Therefore it is critical to optimize the density of plated cells both to avoid high density, cellular clusters or at the other extreme low density modules which reduce inter-module connection probability.
Although neuronal-glial networks in culture lose their in-vivo structural organization, they spontaneously self-organize into active functional circuits offering the possibility to carry out studies under well controlled experimental conditions. The two dimensional modular neuronal networks composed of functionally inter-connected circuits described in this protocol provide a powerful tool for investigating the dynamics of communications among neuronal assemblies and the capacity of structurally defined neuronal networks to generate large repertoire of motifs.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the European Project BRIANBOW (FP7- Young Explorers, The authors would like to thank Dr. Jacopo Tessadori for useful comments on the manuscript, and Silvia Chiappalone for her help in producing the graphics used in the video.
References
- Buzsaki G. Neural syntax: cell assemblies, synapsembles, and readers. Neuron. 2010;68:362–385. doi: 10.1016/j.neuron.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier D, Lambiotte R, Bullmore ET. Modular and hierarchically modular organization of brain networks. Frontiers in Neuroscience. 2010;4:200. doi: 10.3389/fnins.2010.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O, Ziv NE, Marom S. Enhancement of neural representation capacity by modular architecture in networks of cortical neurons. European Journal of Neuroscience. 2012;35:1753–1760. doi: 10.1111/j.1460-9568.2012.08094.x. [DOI] [PubMed] [Google Scholar]
- Berdondini L, et al. A microelectrode array (MEA) integrated with clustering structures for investigating in vitro neurodynamics in confined interconnected sub-populations of neurons. Sensors and Actuators B-Chemical. 2006;114:530–541. [Google Scholar]
- Bisio M, Bosca A, Pasquale V, Berdondini L, Chiappalone M. Emergence of bursting activity in connected neuronal sub-populations. PloS One. 2014;9:e107400. doi: 10.1371/journal.pone.0107400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein Idelson M, Ben-Jacob E, Hanein Y. Innate synchronous oscillations in freely-organized small neuronal circuits. PloS One. 2010;5:e14443. doi: 10.1371/journal.pone.0014443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein-Idelson M, Ben-Jacob E, Hanein Y. Engineered neuronal circuits: a new platform for studying the role of modular topology. Frontiers in Neuroengineering. 2011;4:10. doi: 10.3389/fneng.2011.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifazi P, et al. In vitro large-scale experimental and theoretical studies for the realization of bi-directional brain-prostheses. Front Neural Circuits. 2013;7:40. doi: 10.3389/fncir.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungblut M, Knoll W, Thielemann C, Pottek M. Triangular neuronal networks on microelectrode arrays: an approach to improve the properties of low-density networks for extracellular recording. Biomedical Microdevices. 2009;11:1269–1278. doi: 10.1007/s10544-009-9346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi E, et al. Emergent functional properties of neuronal networks with controlled topology. PloS One. 2012;7:e34648. doi: 10.1371/journal.pone.0034648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Jeon NL. Microfluidic and compartmentalized platforms for neurobiological research. Critical Reviews in Biomedical Engineering. 2011;39:185–200. doi: 10.1615/critrevbiomedeng.v39.i3.20. [DOI] [PubMed] [Google Scholar]
- Sorkin R, et al. Compact self-wiring in cultured neural networks. J Neural Eng. 2006;3:95–101. doi: 10.1088/1741-2560/3/2/003. [DOI] [PubMed] [Google Scholar]
- Mata A, Fleischman AJ, Roy S. Characterization of polydimethylsiloxane (PDMS) properties for biomedical micro/nanosystems. Biomedical Microdevices. 2005;7:281–293. doi: 10.1007/s10544-005-6070-2. [DOI] [PubMed] [Google Scholar]
- Campo AGC. SU-8: a photoresist for high-aspect-ratio and 3D submicron lithography. Journal of Micromechanics and MicroengineeringEmail alert RSS feed. 2007;17:81–95. [Google Scholar]
- Liu GTY. Kan Y Fabrication of high-aspect-ratio microstructures using SU8 photoresist. Microsystem Technologies. 2005;11:343–346. [Google Scholar]
- Jackman RJ, Duffy DC, Cherniavskaya O, Whitesides GM. Using elastomeric membranes as dry resists and for dry lift-off. Langmuir. 1999;15:2973–2984. [Google Scholar]
- Pine J. Recording action potentials from cultured neurons with extracellular microcircuit electrodes. Journal of Neuroscience Methods. 1980;2:19–31. doi: 10.1016/0165-0270(80)90042-4. [DOI] [PubMed] [Google Scholar]
- Gross GW, Rieske E, Kreutzberg GW, Meyer A. New Fixed-Array Multi-Microelectrode System Designed for Long-Term Monitoring of Extracellular Single Unit Neuronal-Activity In vitro. Neuroscience Letters. 1977;6:101–105. doi: 10.1016/0304-3940(77)90003-9. [DOI] [PubMed] [Google Scholar]
- Gross GW, Williams AN, Lucas JH. Recording of spontaneous activity with photoetched microelectrode surfaces from mouse spinal neurons in culture. Journal of Neuroscience Methods. 1982;5:13–22. doi: 10.1016/0165-0270(82)90046-2. [DOI] [PubMed] [Google Scholar]
- Thomas CA, Springer PA, Loeb GE, Berwald-Netter Y, Okun LM. A miniature microelectrode array to monitor the bioelectric activity of cultured cells. Experimental Cell Research. 1972;74:61–66. doi: 10.1016/0014-4827(72)90481-8. [DOI] [PubMed] [Google Scholar]
- Herzog N, Shein-Idelson M, Hanein Y. Optical validation of in vitro extra-cellular neuronal recordings. J Neural Eng. 2011;8:056008. doi: 10.1088/1741-2560/8/5/056008. [DOI] [PubMed] [Google Scholar]
- Maccione A, et al. A novel algorithm for precise identification of spikes in extracellularly recorded neuronal signals. Journal of Neuroscience Methods. 2009;177:241–249. doi: 10.1016/j.jneumeth.2008.09.026. [DOI] [PubMed] [Google Scholar]
- Dworak BJ, Wheeler BC. Novel MEA platform with PDMS microtunnels enables the detection of action potential propagation from isolated axons in culture. Lab Chip. 2009;9:404–410. doi: 10.1039/b806689b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georger JH, et al. Coplanar Patterns of Self-Assembled Monolayers for Selective Cell-Adhesion and Outgrowth. Thin Solid Films. 1992;210:716–719. [Google Scholar]
- Torimitsu K, Kawana A. Selective Growth of Sensory Nerve-Fibers on Metal-Oxide Pattern in Culture. Developmental Brain Research. 1990;51:128–131. doi: 10.1016/0165-3806(90)90265-z. [DOI] [PubMed] [Google Scholar]
- Branch DW, Corey JM, Weyhenmeyer JA, Brewer GJ, Wheeler BC. Microstamp patterns of biomolecules for high-resolution neuronal networks. Medical & Biological Engineering & Computing. 1998;36:135–141. doi: 10.1007/BF02522871. [DOI] [PubMed] [Google Scholar]
- Petrelli A, et al. Nano-volume drop patterning for rapid on-chip neuronal connect-ability assays. Lab on a Chip. 2013;13:4419–4429. doi: 10.1039/c3lc50564b. [DOI] [PubMed] [Google Scholar]
- Boehler MD, Leondopulos SS, Wheeler BC, Brewer GJ. Hippocampal networks on reliable patterned substrates. Journal of Neuroscience Methods. 2012;203:344–353. doi: 10.1016/j.jneumeth.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]