

Abstract
The correct topology and orientation of integral membrane proteins are essential for their proper function, yet such information has not been established for many membrane proteins. A simple technique called fluorescence protease protection (FPP) is presented, which permits the determination of membrane protein topology in living cells. This technique has numerous advantages over other methods for determining protein topology, in that it does not require the availability of multiple antibodies against various domains of the membrane protein, does not require large amounts of protein, and can be performed on living cells. The FPP method employs the spatially confined actions of proteases on the degradation of green fluorescent protein (GFP) tagged membrane proteins to determine their membrane topology and orientation. This simple approach is applicable to a wide variety of cell types, and can be used to determine membrane protein orientation in various subcellular organelles such as the mitochondria, Golgi, endoplasmic reticulum and components of the endosomal/recycling system. Membrane proteins, tagged on either the N-termini or C-termini with a GFP fusion, are expressed in a cell of interest, which is subject to selective permeabilization using the detergent digitonin. Digitonin has the ability to permeabilize the plasma membrane, while leaving intracellular organelles intact. GFP moieties exposed to the cytosol can be selectively degraded through the application of protease, whereas GFP moieties present in the lumen of organelles are protected from the protease and remain intact. The FPP assay is straightforward, and results can be obtained rapidly.
Keywords: Cellular Biology, Issue 98, Membrane protein, topology, GFP, fluorescence assay, protease, proteolysis, Digitonin
Introduction
The plasma membranes, as well as the numerous intracellular membranes, serve as barriers separating two aqueous compartments. In the case of the plasma membrane, the separation is between the outside and inside of the cell; for intracellular organelles it is between the cytoplasm and the organelle lumen. For example, the endoplasmic reticulum (ER) membrane separates an oxidizing environment within the lumen of the ER from a cytosolic reducing environment1. Membrane proteins are synthesized on ER-associated ribosomes, and achieve their final topology within the ER membrane2. The acquisition of appropriate membrane orientation and topology for proteins is critical for their normal function. Correct topology allows relevant domains of membrane proteins to interact with their binding partners, it allows critical post-translational modifications to occur, and in the case of plasma membrane proteins, allows the cell to interact with and respond to its environment. To fully appreciate the function of a membrane protein, it is clearly imperative to know how that protein is oriented with respect to the membrane within which it resides, i.e., its membrane topology. In addition to acquisition of basic scientific knowledge, understanding the topology of a membrane protein and which aspects of a protein surface are exposed to different environments has marked clinical implications since membrane proteins comprise the majority of pharmacological targets3. Until recently, approaches to determining membrane protein topology have required considerable investment in time and money or have required reagents that are difficult to come by.
Both experimental and in silico approaches have been employed to determine the membrane topology of proteins residing within the plasma membrane. Since the first predictions of membrane spanning domains based on the evaluated hydrophobicity of individual amino acids3, numerous predictive algorithms are now available on the internet, and simply require knowledge of the protein’s amino acid sequence. However, assumptions are often central to such modeling programs, assumptions that can lead to incorrect assignments of topology4,5. Moreover, while these computer-based predictions can tentatively assign membrane spanning regions, they do not always determine whether the amino or carboxy termini of proteins are in the cytoplasm, organelle lumen or cell exterior. Even with increased computational power, and the use of machine-learning algorithms6, such data is still a model, and must be validated using experimentally acquired data. Direct experimental determination of membrane topology has been undertaken using panels of monoclonal antibodies with known epitopes distributed throughout the protein, where assessment of their immunoreactivity has been made before and after cell permeabilization. This approach requires a set of antibodies, which may not be available for the protein of interest.
An alternative strategy is to engineer epitope tags such as myc or hemagglutinin (HA) into various locations throughout the protein, again followed by determination of immunoreactivity before and after membrane permeabilization. In addition to immunogenic tags, enzymatic tags (including alkaline phosphatase, β-galactosidase, or β-lactamase) and chemical modifications such as cysteine scanning have all been employed to determine membrane protein topology7,8. An additional method employed for topological mapping of plasma membrane proteins relies on a slightly different approach to epitope tags. In this method the tag sequence is the N-linked glycosylation consensus sequence NXS/T. Since glycosylation only occurs when such a sequence is present in the lumen of the biosynthetic pathway, the presence of the tag in a luminal versus a cytosolic compartment is easily observed as a mass shift on SDS-PAGE gels. Such an approach has been applied to the multispanning ion channel CFTR9. While all these approaches have been utilized, it is clear that they all require considerable investments in molecular biology to generate and sequence the manifold constructs.
To determine topological information regarding transmembrane proteins located within intracellular organelles has proven to be more challenging. The application of fluorescence based technologies however, has made the determination of membrane topology a lot simpler. The technique of bimolecular fluorescence complementation (BiFC) relies on the interaction between two non-fluorescent fragments of a fluorescent protein, restoring the fluorescent properties of that protein10. Although initially described for determining protein-protein interactions in vivo10, this approach has also been utilized to determine membrane protein topology in plant cells11. However this approach is also time intensive as it requires the generation not only of fusion proteins with the protein of interest but also a variety of fusion proteins targeted to the cytosol or organelle lumen, and requires knowledge of which intracellular membranes the protein of interest resides in.
An alternative simpler approach to determining membrane protein topology has been described12. The assay, Fluorescence Protease Protection (FPP) requires the generation of a fusion protein between GFP and the gene of interest. The approach is based on the relative accessibility of non-specific proteases to the GFP moiety; depending on whether GFP is protected from proteolysis by residing in the lumen of intracellular organelles or is exposed to proteolysis by being present in the cytosol. Thus, if the GFP moiety on the protein of interest faces the cytoplasm, it will be exposed to protease activity and the fluorescence signal lost. Conversely, if the GFP moiety on the protein of interest faces an environment 'protected' from the protease (such as the Golgi lumen) then the fluorescence signal will persist.
To allow proteases to enter the cell, but not enter intracellular membrane compartments the cholesterol binding drug digitonin is used. Cholesterol is the dominant sterol in vertebrates, and is specifically enriched in the plasma membrane relative to intracellular compartments. The glycoside toxin, digitonin, is extracted from the plant Digitalis purpurea. Digitonin has an affinity for cholesterol rich membranes, where it leads to selective membrane permeabiliztion14,16 (Figure 1). In addition to allowing exit of small cytosolic components, digitonin permeabilization also permits the entry of exogenous molecules, such as proteinase K or trypsin. The FPP protocol takes advantage of the fact that the plasma membrane contains up to 80% of cellular cholesterol17, whereas other organelles, such as ER, Golgi, endosomes, mitochondria, which have very low cholesterol content, remain intact14. The selective incorporation of cholesterol into the plasma membrane has been observed in many eukaryotic cells, permitting the use of digitonin-dependent plasma membrane permeabilization in such diverse eukaryotic species as S. cerevisiae to human14,18. The FPP assay provides a simple, rapid and fairly robust means of determining (a) whether a protein is membrane bound/associated or freely diffusible in the cytosol and (b) which domain of a membrane protein faces the cytosol or organelle lumen. Should a membrane protein have multiple orientations, the signal will arise from the dominant form and minor forms will not be detected. While there can be some concern that the addition of a GFP moiety to the protein on interest may affect its function and/or subcellular localization, this is actually more theoretical than actual. Indeed many studies have clearly shown that the GFP tag does not alter the properties of the protein19,20.
Protocol
1. Generation and Validation of Fluorescent Protein Chimeras
- Append green fluorescent protein (GFP) to the amino or carboxyl termini of the protein of interest using standard recombinant DNA strategies13. NOTE: Although we have used GFP extensively, other variants such as cyan fluorescent protein (CFP), yellow fluorescent protein (YFP), DsRed and mCherry can be employed. Improvements in fluorophore folding efficiency, brightness and photostability are constantly being made, and investigators should avail themselves of the latest generation constructs.
- Confirm correct DNA sequence and ensure that the GFP moiety is in frame with the protein of interest. Perform sequencing using standard recombinant DNA strategies21. NOTE: Many institutions have core DNA facilities, which the investigator is encouraged to contact for additional information.
- For single or double pass membrane proteins, generate both amino- and carboxyl terminal versions of the fusion proteins19.
- For multi-spanning proteins insert GFP within putative extramembrane loops, as well as the termini. Take special care when generating transmembrane chimeras with the fluorescent fusion protein attached to the amino terminus of the target protein as signal sequences are often cleaved by proteolysis once the protein has passed through the ER translocon.
- In the case of extant signal sequences, insert the fluorescent fusion protein distal to the cleavage site so as to generate a mature protein containing an intact amino-terminal tagged fluorescent protein. Employ standard recombinant DNA strategies for the generation of chimeras21.
- Determine the presence of sufficient appropriately localized fluorescent signal in the cell line of interest.
- Observe the cells under epifluorescence microscopy using the intrinsic fluorescence of GFP under appropriate filter set conditions. For GFP filter sets that encompass 510-560 nm, confirm that the correct filters are used for the fluorophore employed. NOTE: A sufficient signal is achieved when assessment by eye provides a strong enough signal that the cellular structure within which the signal resides is easily resolved above background. In addition the staining pattern of the GFP-protein should be consistent with that of markers for the appropriate organelle, and preferably identical to that of the native untagged protein.
Determine whether a stable or transient transfection approach will be utilized based upon the protein being evaluated. NOTE: Stable transfectants generally afford lower levels of expression than transient systems. Although transient expression yields high level expression of proteins and increase signal intensity (~5-24 hr post transfection depending on the protein), it can also lead to overexpression artifacts such as protein aggregation or saturation of protein targeting pathways, leading to erroneous subcellular localization.
2. Transfection of Cells and Cell Culture
NOTE: A variety of cells are amenable to FPP analysis, including Hek293, HeLa, COS-7 and CHO cells14. The following description is based on expressing membrane proteins in Hek293 cells.
Grow human embryonic kidney cells (Hek293) as adherent monolayers in 6 ml of Dulbecco’s Modified Eagles Medium (DMEM), supplemented with 5% fetal bovine serum (FBS), 1% sodium pyruvate, and 250 µg/ml penicillin/streptomycin at 37 °C with 5% CO2. Perform all work with cells in a laminar flow tissue culture hood. Grow cells in culture medium until ~80% confluent in a T-75 flask.
Transfect cells using any standard high-efficiency, low toxicity transfection method, including lipid-, viral- or electroporation-based technologies to generate transient or stable expression. Typically 2 µg of plasmid DNA per 1 x 106 cells gives good reproducible protein expression. Maintain cells for 5-48 hr in appropriate tissue culture media at 37 °C in a CO2 capable tissue culture incubator.
Monitor cells for expression of the GFP-fusion protein using epifluorescence microscopy, using the intrinsic fluorescence signal of GFP, using appropriate filter sets according to manufacturer’s protocol. Fluorescence signal usually reaches a maximum within 24-72 hr following transfection. Use cells when the fluorescence signal reaches a maximum level. Monitor protein expression levels by immunoblot analysis using antibodies directed against either the protein of interest or the GFP moiety.
- For experimental analysis plate cells on coated coverslips. View either under a live-cell configuration or following fixation and mount on microscopy slides. Plate cells to achieve 60%-80% confluence within 24-48 hr.
- Coat coverslips with poly-lysine solution (0.1 mg/ml), so that the entire surface is covered. Incubate for 30 min under an ultra violet light at room temperature, followed by 60 min in a cell culture incubator at 37 °C. Remove excess poly-lysine and rinse the coverslips in PBS 3 times.
- Alternatively, plate the cells on a chambered cover-glass consisting of cell culture plates with an optical glass bottom to allow direct imaging with oil-immersion objectives. The actual microscopy setup depends on the equipment available to the investigator, but should be capable of rapid solution changes. Depending on the cell type and cell shape, cells should be around 60-90% confluent at the time of the FPP assay.
3. Fluorescence Microscopy Setup
Use a high numerical aperture (NA) oil-immersion objective for maximal signal collection and spatial resolution, such as a 60X, 1.42 NA oil immersion objective. Use the following lasers to stimulate the fluorophores: Argon gas 488 nm (GFP and YFP) and 561 diode (RFP, mCherry).
Determine the optimal exposure, gain and capture rate of images for each construct. Adjust laser intensity and exposure time to minimize photobleaching (see step 7.1.2).
For studies using multiple fluorophores (e.g., a soluble marker for membrane permeabilization in concert with a membrane protein marker), choose appropriate filter sets to minimize signal cross-talk and maximize signal-to-noise ratios.
4. Establishing Optimal Conditions for Plasma Membrane Permeabilization
Remove the cell culture medium from cells expressing only soluble fluorescent proteins (e.g. EGFP, DsRed). Wash the cells 3 times (1 min each) in KHM buffer (110 mM potassium acetate, 3 mM MgCl2, 20 mM HEPES-NaOH, pH 7.2). Perform experiments with cells and all reagents at room temperature.
Place cells on coverslips (see step 2.4) in KHM buffer on a fluorescence microscope stage and collect the first image, which represents the control 'non-permeabilized' stage.
To selectively permeabilize the plasma membrane, add digitonin (30 µM) in KHM buffer to the cells. Perfuse cells with buffer (buffer + digitonin) at a rate of 5 ml/min for 10-60 sec.
- Determine the optimal digitonin concentration by gradually increasing the concentration of digitonin. The optimal digitonin concentration is achieved when there is complete loss of fluorescence signal from soluble EGFP within 10-60 sec of digitonin application. For many cells application of 10-50 μM digitonin is sufficient to permeabilize the cell membrane.
- For all applications use the lowest possible concentration of digitonin. Collect images after digitonin permeabilization for permeabilized background signal.
- Confirm that the GFP-protein chimera of interest is membrane integrated by confirming cellular retention of the fluorescence signal following digitonin permeabilization of the plasma membrane16.
- Express an organelle specific fluorescent protein23. Mitochondrial proteins work well in this regard16, e.g., pDsRed2-Mito vector. Determine optimal expression conditions as described in step 2.1.
- Confirm that the digitonin concentration is appropriate for the cell type employed by ensuring that the morphology of intracellular organelles remains constant over prolonged (up to 60 min) exposure to digitonin. NOTE: Use standard immunofluorescence microscopy protocols using antibodies against various cellular compartments, such as lysosomes (e.g., LAMP1), Golgi (e.g., TGN38), endoplasmic reticulum (e.g., calnexin), endosomes (e.g., Rab5), mitochondria (e.g., cytochrome c). If intracellular organelle morphology/integrity changes dramatically over 60 min, it is likely that the digitonin concentration was too high, and requires a reduction in digitonin concentration and/or exposure time. NOTE: Digitonin is toxic by inhalation and skin contact. Wear appropriate personal protection equipment (PPE).
5. Protease Treatment of Fluorescent Proteins
- Wash the cells in KHM buffer and then add protease (proteinase K; 50 μg/ml in KHM buffer) directly onto the cells. Perfuse cells at a flow rate of 5 ml/min using a gravity fed perfusion system to continuously bath the preparation.
- Store solutions in 50 ml syringes and polyethylene tubing connected independent reservoirs to a manifold with a single outlet at the bath (5 ml/min flow rate). Position a suction pipette to maintain a constant (0.5 ml) bath volume and achieve solution exchange by manually switching between perfusion reservoirs.
Collect images to determine whether the fluorescent signal persists or degrades. A loss of >90% of the initial signal intensity over 30-60 sec is consistent with proteolytic degradation of the GFP-protein chimera (see Representative Results).
If no signal degradation is observed with proteinase K, when such loss is anticipated, replace the proteinase K solution with trypsin (4-8 mM) in KHM buffer. If necessary, utilize a mixture of proteinase K and trypsin. There is no need to quench the protease activity.
6. Alternative Approach for Plasma Membrane Proteins with External Domains
For plasma membrane proteins, perform the assay for external fluorescent proteins chimeras prior to digitonin permeabilization. Wash the cells on coverslips in KHM buffer (5 ml/min x 3 min), and take an image for pre-protease treatment and quantify pre-protease fluorescence signal as in Step 7.
Expose cells to protease (proteinase K; 50 μg/ml or trypsin 4-8 mM in KHM buffer) in the absence of digitonin by perfusing the cells with KHM media containing protease (flow rate 5 ml/min). Collect images of the fluorescent protein, to determine how quickly the signal disappears or how long the signal persists.
Before digitonin permeabilization, quench all protease activity, by washing the cells three times (1 min each) in cell culture media containing 10% serum (FBS).
Wash the cells by perfusing with KHM buffer (flow rate of 5 ml/min) for 2 min. Acquire a pre-permeabilization image. Permeabilize the cell membrane as described in Step 4.
Collect images following protease addition as described in Step 5.
7. Image Analysis
- Use an inverted microscope capable of “live cell” recording, in which loss of fluorescence signal can be monitored in real-time. For continuous monitoring, employ identical parameters (exposure time, gain, laser intensity, etc.).
- Determine experimental parameters by observing the fluorescence signal from the GFP-protein of interest under control conditions (i.e., KBH buffer alone). Adjust the gain, laser intensity and exposure time settings on the computer-driven microscope to ensure that there is no appreciable loss of signal intensity over a 10-20 min period. NOTE: Signal intensity is not an absolute value, but is based on the microscope system and settings available to each PI. Monitor changes in relative fluorescence intensity.
- Alternatively, treat cells at set time-points in digitonin followed by protease, and fix cells prior to mounting on coverslips for imaging. Determine time points empirically following procedures in steps 4 and 5. For the initial investigation, start at 0, 30, 60, 120 and 300 sec post addition of digitonin or protease.
- Fix cells (20 min at room temperature) using a sufficient quantity of fixative (3.7% paraformaldehyde in PBS) to fully submerge the cells.
- Quench residual paraformaldehyde by rinsing cells with PBS containing 20 mM glycine (3 x 5 min).
- Mount coverslips by removing excess liquid from the coverslip and inverting the coverslip onto 50 µl mounting media on a glass microscope slides for imaging. Allow slides to dry overnight before imaging.
Collect fluorescence microscopy images (under the appropriate filters sets compatible with the fluorophore being used) by using the video image capture function on a computer controlled fluorescence microscope.
Quantify fluorescent signal intensities as a function of incubation condition. Obtain the mean pixel intensity within a region of interest (ROI) that encloses an individual cell.
Representative Results
Plasma Membrane Permeabilization
Efficient plasma membrane permeabilization is determined by the use of soluble fluorescent proteins (e.g., GFP, DsRed) (Step 4). These proteins, when expressed in cells, are free to diffuse in the cytosol, and are lost when the plasma membrane is permeabilized using digitonin (Figure 2). A complete disappearance of fluorescent signal should occur within 10-60 sec of digitonin application. Confirmation that the protein of interest is associated with intracellular organelles is achieved by noting the retention of fluorescent signal following digitonin permeabilization.
Protease Treatment
Following successful generation of fluorescent chimeras between the protein of interest and the fluorescent protein (GFP, YFP, etc.) (Step 1) and expression of the fluorescent protein in cells (Step 2), treatment of permeabilized cells with protease will provide information on whether the fluorophore (and its adjacent protein domain) are in the cytosol and accessible to protease digestion, or are located within an organelle lumen and thus protected from protease digestion (Step 4). LMTK2 is a membrane anchored kinase15, and fluorescence protease protection assays have been used to determine its membrane topology16. The carboxyl tail of LMTK2 was fused to GFP and expressed transiently. Fluorescent LMTK2-GFP signal was lost rapidly following addition of proteinase K, indicating the location of the carboxyl terminus of LMTK2 being located within the cytosol (Figure 3). Caveolin-GFP (Cav1-GFP)17 is a plasma membrane protein with a GFP-moiety attached to the cytosolic domain. As with LMTK2, the signal from caveolin-YFP is digitonin insensitive, but sensitive to the subsequent addition of protease (Figure 3).
Luminal soluble proteins can also be identified using the FPP protocol. In this example, the ER retention signal KDEL is appended to DsRed. In contrast to the signals from LMTK2 and caveolin, the signal from the KDEL-DsRed is insensitive to both digitonin and protease, as digitonin is unable to permeabilize the ER membrane and the fluorescent signal is protected from protease degradation. An example of a membrane domain located within the lumen of an organelle is given by the mitochondrial protein is subunit VIII of human cytochrome c oxidase18,19. pDsRed-Mit, encodes the red fluorescent protein from Discosoma sp. fused to the mitochondrial targeting sequence from cytochrome c oxidase. In cells expressing pDsRed-Mito, fluorescent signal is seen in the cell. Following digitonin permeabilization, the fluorescent signal associated with pDSRed-Mito remains stable. Moreover, following addition of proteinase K, the fluorescence associated with pDsRed-Mito persists, consistent with the fluorophore in the mitochondria and not exposed to the cytosol and thus protected from protease activity (Figure 3).
Extracellular Domains
Protease treatment of non-permeabilized cells should rapidly quench the fluorescence signal when the fluorescent moiety attached to a membrane protein is facing the cell exterior. The glycosylphosphatidylinositol (GPI) anchored protein PrP20 serves as an excellent example. A construct containing the yellow fluorescent protein (YFP) attached to the amino terminus of PrP (YFP-PrP)21,22, a generous gift from Dr. Ehud Cohen, from the Hebrew University of Jerusalem. Prior to protease treatment, the fluorescent signal from the extracellular YFP was bright (Figure 4). Following protease treatment, the extracellular fluorescence signal disappeared rapidly.
Time Considerations
Step 2 requires an hour for cell counting, transfection and plating, plus 6-24 hr to detect protein expression in transfected cells. Steps 4-6 require about 20-30 min per coverslip for image collection of pre-treatment, digitonin and protease additions. Step 7, data analysis, and signal quantitation require a further 10-30 min per sample.
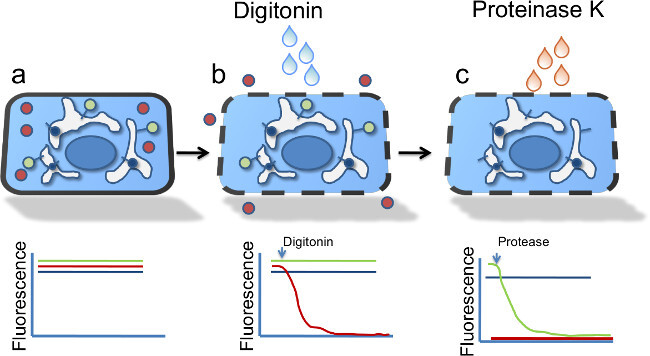 Figure 1:Selective permeabilization of the plasma membrane. (a) Cartoon model of the FPP assay showing a single cell going through the experimental protocol. The red symbol represents red fluorescent protein (RFP) in the cytosol, the green and blue symbols represent transmembrane proteins with the fluorescent tag either facing the organelle lumen (blue) or cytosol (green). (b) Following digitonin permeabilization, free cytosolic proteins diffuse away, reducing the RFP signal. (c) Following application of proteinase K, the cytosolic green signal is degraded, whereas the blue signal is protected from degradation by its presence in the organelle lumen.
Figure 1:Selective permeabilization of the plasma membrane. (a) Cartoon model of the FPP assay showing a single cell going through the experimental protocol. The red symbol represents red fluorescent protein (RFP) in the cytosol, the green and blue symbols represent transmembrane proteins with the fluorescent tag either facing the organelle lumen (blue) or cytosol (green). (b) Following digitonin permeabilization, free cytosolic proteins diffuse away, reducing the RFP signal. (c) Following application of proteinase K, the cytosolic green signal is degraded, whereas the blue signal is protected from degradation by its presence in the organelle lumen.
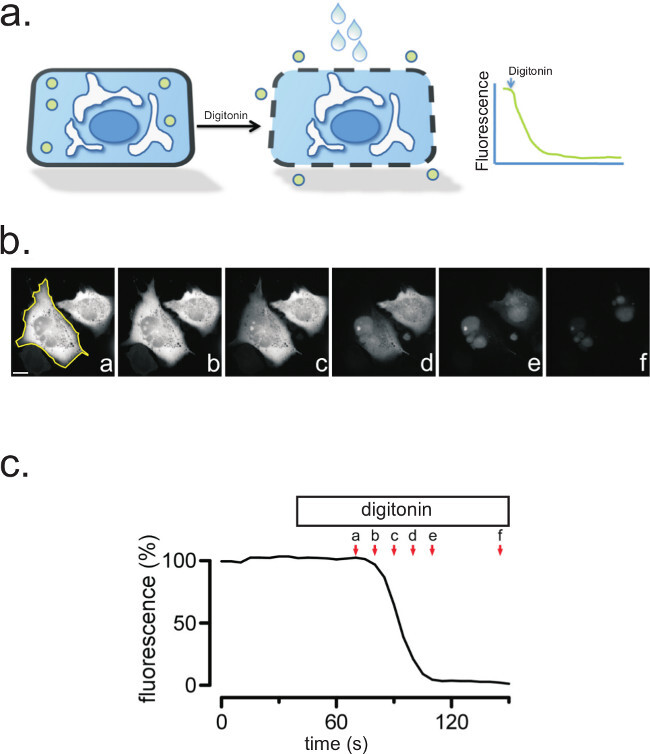 Figure 2: Digitonin treatment permeabilizes the plasma membrane and releases cytosolic GFP. (a) Cartoon of a single cell expressing soluble GFP, before and after digitonin treatment. (b) Digitonin treatment permeabilizes the plasma membrane and releases cytosolic GFP. A region of interest (ROI) is drawn around CHO cells expressing soluble GFP. Following treatment with digitonin (50 µM), images were taken before and after the addition of digitonin, and at the indicated time points. Scale bar, 10 µm. (c) The quantification of the fluorescence intensities of the full time-series (a-f) is shown.
Figure 2: Digitonin treatment permeabilizes the plasma membrane and releases cytosolic GFP. (a) Cartoon of a single cell expressing soluble GFP, before and after digitonin treatment. (b) Digitonin treatment permeabilizes the plasma membrane and releases cytosolic GFP. A region of interest (ROI) is drawn around CHO cells expressing soluble GFP. Following treatment with digitonin (50 µM), images were taken before and after the addition of digitonin, and at the indicated time points. Scale bar, 10 µm. (c) The quantification of the fluorescence intensities of the full time-series (a-f) is shown.
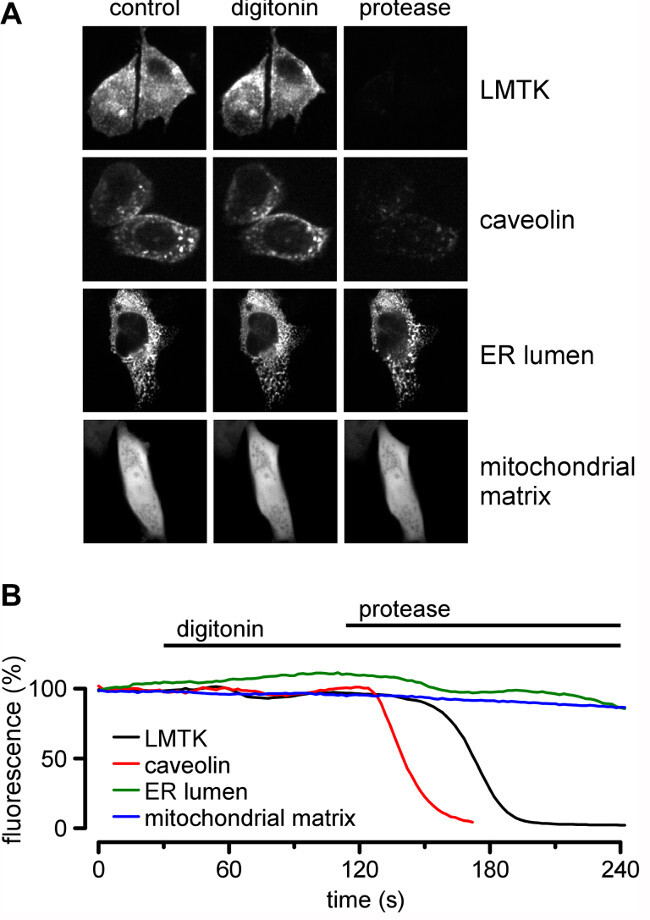 Figure 3: FPP reveals the topology of the endosomal protein LMTK2. (a) Cartoon of FPP showing loss of DsRed (red balls) but retention of LMTK2-GFP (green balls) following digitonin permeabilization, followed by signal loss on application of proteinase K. (b) FPP assay of CHO Cells expressing DsRed and LMTK2-GFP. Images were taken before and after digitonin treatment, and following proteinase K, as indicated. Scale bar: 10 µm.
Figure 3: FPP reveals the topology of the endosomal protein LMTK2. (a) Cartoon of FPP showing loss of DsRed (red balls) but retention of LMTK2-GFP (green balls) following digitonin permeabilization, followed by signal loss on application of proteinase K. (b) FPP assay of CHO Cells expressing DsRed and LMTK2-GFP. Images were taken before and after digitonin treatment, and following proteinase K, as indicated. Scale bar: 10 µm.
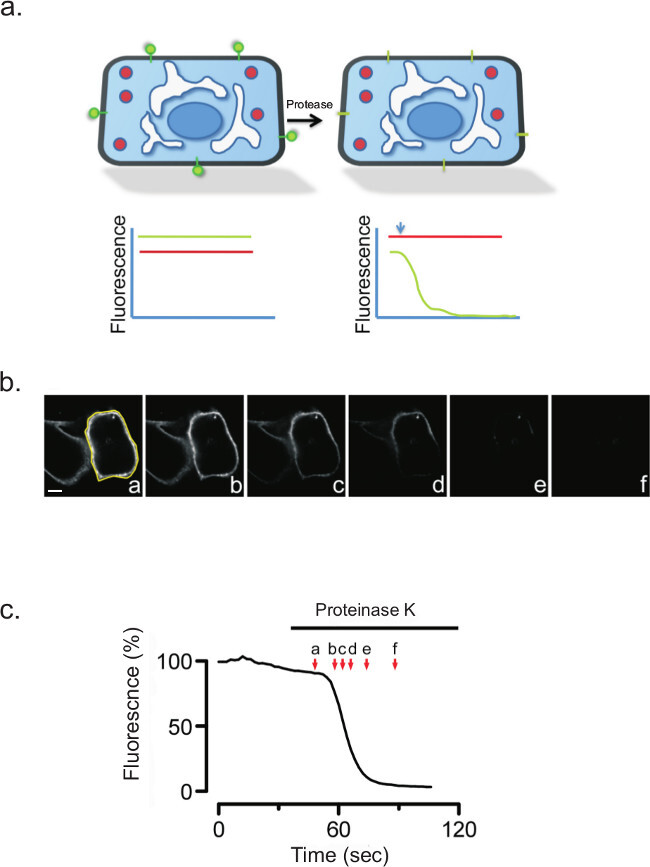 Figure 4: (a) Cartoon model showing the topology and location of YFP-PrP (green) before and after proteinase K treatment. CHO cells expressing YFP-PrP were subjected to proteinase K for 100 sec. (b) Images taken before and after protease treatment are shown at the indicated times. YFP-PrP signal was completely lost following protease in the absence of digitonin. Yellow shows region of interest (ROI) used for quantitation shown in (c).
Figure 4: (a) Cartoon model showing the topology and location of YFP-PrP (green) before and after proteinase K treatment. CHO cells expressing YFP-PrP were subjected to proteinase K for 100 sec. (b) Images taken before and after protease treatment are shown at the indicated times. YFP-PrP signal was completely lost following protease in the absence of digitonin. Yellow shows region of interest (ROI) used for quantitation shown in (c).
Discussion
The correct orientation and topology of membrane proteins is essential for their proper function. Despite the importance of understanding membrane protein topology, there are many proteins for which such data is completely lacking. FPP provides an easy and efficient way of determining membrane protein topology, and one that can be performed by most laboratories. The FPP approach affords significant advantages over previous methods for determining protein topology. For example, there is no need to have a panel of multiple antibodies directed again various domains of the protein of interest31. In many cases such a panel of antibodies is not available; this is especially true for newly cloned proteins. As the assay can be performed on a single cell level, large biochemical quantities of protein are not required for downstream biochemical analyses such as proteolysis or SDS-PAGE. Indeed, relatively few transfected cells are needed to unambiguously assign a topology to the protein of interest. This is important if the investigator is evaluating topology in hard to transfect cells such as epithelial cells or neurons. Simple addition of a GFP moiety to either the carboxyl or amino terminus of the protein can be easily achieved using numerous commercially available GFP containing plasmids, and in most cases, the addition of the GFP moiety does not hamper proper protein folding or subcellular localization. However, the investigator must confirm that this is true for their protein, as there is the potential for GFP to affect the protein of interest23,24. Many proteins are already available as GFP fusion proteins, thus requiring minimal additional effort to initiate topological studies. Although continuous monitoring of fluorescence provides a quick and efficient means of determining protein topology, for investigators who do not have access to live-cell imaging equipment, the assay is readily adapted to a formaldehyde fixation approach using standard epifluorescence of confocal microscopy.
There are however some limitations in the applicability of the FPP assay. Should proteolytic processing, or other post-translational modifications result in slightly different membrane topologies for the protein of interest, the FPP assay will only assess the dominant protein form. In addition, appending a GFP moiety to the termini of some proteins can affect their function, for example interfering with carboxyl terminal PDZ domains. Nevertheless, the FPP assay affords a simple approach to establishing orientation and topology of many membrane proteins.
It is critical that initial experiments be performed to determine the correct concentration of digitonin to release intracellular soluble GFP. If the GFP does not diffuse out of the cell following digitonin application, the concentration of digitonin should be increased gradually, to determine the minimal concentration required to facilitate release. However, too much digitonin will affect the morphology of intracellular vesicles, whose integrity should be confirmed following digitonin application. Organelles in some cells are very sensitive and may require a more stabilizing buffer composition25. If the fluorescence signal from the chimera being evaluated is low, selecting a single clone with a higher expression level, or performing a transient expression, will often increase the signal strength to an acceptable level. Seeking out newer, brighter plasmids should also be considered with low signal intensity. Finally, care should be taken to ensure that the loss of signal seen is due to protease degradation and not to photobleaching of fluorophores. Reducing light intensity and/or acquisition rate will help to reduce photobleaching.
Disclosures
The authors declare no conflict of interest.
Acknowledgments
We wish to thank the Calcium Imaging Core Facility at CMS for their help and guidance in image capture and analysis. This study was supported by the U.S. National Institutes of Health (NIH) HL102208 to N.A.B. This approach was originally pioneered by Holger Lorenz and Jennifer Lippincott-Schwartz at NIH.
References
- Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Current Opinion In Cell Biology. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Ott CM, Lingappa VR. Integral membrane protein biosynthesis: why topology is hard to predict. J Cell Sci. 2002;115:2003–2009. doi: 10.1242/jcs.115.10.2003. [DOI] [PubMed] [Google Scholar]
- Traxler B, Boyd D, Beckwith J. The topological analysis of integral cytoplasmic membrane proteins. The Journal of Membrane Biology. 1993;132:1–11. doi: 10.1007/BF00233047. [DOI] [PubMed] [Google Scholar]
- Cserzo M, Wallin E, Simon I, von Heijne G, Elofsson A. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng. 1997;10:673–676. doi: 10.1093/protein/10.6.673. [DOI] [PubMed] [Google Scholar]
- Geest M, Lolkema JS. Membrane topology and insertion of membrane proteins: search for topogenic signals. Microbiol Mol Biol Rev. 2000;64:13–33. doi: 10.1128/mmbr.64.1.13-33.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Zhang W, Xie J, Dowhan W. Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAM(TM)): application to lipid-specific membrane protein topogenesis. Methods. 2005;36:148–171. doi: 10.1016/j.ymeth.2004.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang XB, Hou YX, Jensen TJ, Riordan JR. Mapping of cystic fibrosis transmembrane conductance regulator membrane topology by glycosylation site insertion. J Biol Chem. 1994;269:18572–18575. [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- Zamyatnin AA, et al. Assessment of the integral membrane protein topology in living cells. Plant J. 2006;46:145–154. doi: 10.1111/j.1365-313X.2006.02674.x. [DOI] [PubMed] [Google Scholar]
- Lorenz H, Hailey DW, Lippincott-Schwartz J. Fluorescence protease protection of GFP chimeras to reveal protein topology and subcellular localization. Nat Methods. 2006;3:205–210. doi: 10.1038/nmeth857. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. a Laboratory Manual. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shah K, McCormack CE, Bradbury NA. Do you know the sex of your cells? American journal of physiology. Cell Physiology. 2014;306:C3–C18. doi: 10.1152/ajpcell.00281.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Brautigan DL. A novel transmembrane Ser/Thr kinase complexes with protein phosphatase-1 and inhibitor-2. J Biol Chem. 2002;277:49605–49612. doi: 10.1074/jbc.M209335200. [DOI] [PubMed] [Google Scholar]
- Nixon A, Jia Y, White C, Bradbury NA. Fluorescence protease protection reveals the topology of the integral membrane protein Lemur Tyrosine Kinase 2 (LMTK2) Am J Physiol. 2012;304(2):C164–C169. doi: 10.1152/ajpcell.00288.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa A, et al. Assembly and trafficking of caveolar domains in the cell: caveolae as stable, cargo-triggered, vesicular transporters. J Cell Biol. 2005;170:769–779. doi: 10.1083/jcb.200506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, et al. A gene specifying subunit VIII of human cytochrome c oxidase is localized to chromosome 11 and is expressed in both muscle and non-muscle tissues. J Biol Chem. 1989;264:10595–10600. [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Pizzo P, Murgia M, Pozzan T. Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr Biol. 1995;5:635–642. doi: 10.1016/s0960-9822(95)00128-x. [DOI] [PubMed] [Google Scholar]
- Lorenz H, Windl O, Kretzschmar HA. Cellular phenotyping of secretory and nuclear prion proteins associated with inherited prion diseases. J Biol Chem. 2002;277:8508–8516. doi: 10.1074/jbc.M110197200. [DOI] [PubMed] [Google Scholar]
- Ben-Gedalya T, et al. Cyclosporin-A-induced prion protein aggresomes are dynamic quality-control cellular compartments. J Cell Sci. 2011;124:1891–1902. doi: 10.1242/jcs.077693. [DOI] [PubMed] [Google Scholar]
- Rogers M, et al. Epitope mapping of the Syrian hamster prion protein utilizing chimeric and mutant genes in a vaccinia virus expression system. J Immunol. 1991;147:3568–3574. [PubMed] [Google Scholar]
- Lisenbee CS, Karnik SK, Trelease RN. Overexpression and mislocalization of a tail-anchored GFP redefines the identity of peroxisomal ER. Traffic. 2003;4:491–501. doi: 10.1034/j.1600-0854.2003.00107.x. [DOI] [PubMed] [Google Scholar]
- Brandee AP, et al. Mislocalization and degradation of human P23H-Rhodopsin-GFP in knockin mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52:9728–9736. doi: 10.1167/iovs.11-8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, et al. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]