

Abstract
There are many different animal models available for studying the pathogenesis of human inflammatory bowel diseases (IBD), each with its own advantages and disadvantages. We describe here an experimental colitis model that is initiated by adoptive transfer of syngeneic splenic CD4+CD45RBhigh T cells into T and B cell deficient recipient mice. The CD4+CD45RBhigh T cell population that largely consists of naïve effector cells is capable of inducing chronic intestinal inflammation, closely resembling key aspects of human IBD. This method can be manipulated to study aspects of disease onset and progression. Additionally it can be used to study the function of innate, adaptive, and regulatory immune cell populations, and the role of environmental exposures, i.e., the microbiota, in intestinal inflammation. In this article we illustrate the methodology for inducing colitis with a step-by-step protocol. This includes a video demonstration of key technical aspects required to successfully develop this murine model of experimental colitis for research purposes.
Keywords: Immunology, Issue 98, IBD, Colitis, Experimental Models, Adaptive Immunity, T cells, Mucosal Immunity, Inflammation
Introduction
The inflammatory bowel diseases (IBD) Crohn’s disease and ulcerative colitis result from an incompletely defined and complex interaction between host immune responses, genetic susceptibility, environmental factors, and the enteric luminal contents1. Recent genome-wide association studies report associations between immune cell regulatory genes and IBD susceptibility2,3. Both innate and adaptive immune cell intrinsic genes are represented in these studies, indicating a central role for these cell populations in IBD pathogenesis.
There currently exist more than 50 animal models of human IBD. While no one model perfectly phenocopies human IBD, many are useful for studying various aspects of human disease, including disease onset and progression and the wound-healing response. In the method described here, intestinal inflammation is initiated with syngeneic splenic CD4+CD45RBhigh T cell adoptive transfer into T and B cell deficient recipient mice4. The CD4+CD45RBhigh T cell population contains mainly naïve T cells primed for activation that are capable of inducing chronic small bowel and colonic inflammation. This method allows the researcher to modify key experimental variables, including both innate and adaptive immune cell populations, to answer biologically relevant questions relating to disease pathogenesis. Additionally, this method provides precise initiation of disease onset and a well-characterized experimental time course. This permits the kinetic study of clinical features of disease progression in mice. Intestinal inflammation induced by this method shares many features with human IBD, including chronic large and small bowel transmural inflammation, pathogenesis driven by cytokines such as TNF and IL-12, and systemic symptoms such as wasting5. Thus, it is an ideal model system for studying the pathogenesis of human IBD.
The method here describes in detail the protocol for inducing experimental colitis by adoptive transfer of CD4+CD45RBhigh T cells into Rag1-/- mice. We discuss key technical steps, expected results, optimization, and trouble-shooting. We address the required elements for the successful development of this murine model of intestinal inflammation for research purposes.
Protocol
NOTE: Ensure that all animal protocols are approved by and in compliance with Institutional Animal Care and Use Committee (IACUC) regulations and the National Research Council’s Guide for the Care and Use of Laboratory Animals. Donor mice may be either male or female, but recipient mice should be male. If female recipients are to be used, the donor mice must be female5. Maintain colonies using regular, non-sterile bedding and non-acidified water, as these may impact the intestinal microbiota, and, thus, the colitis phenotype of the mice5,6.
1. Experimental Preparation
Use ice-cold media and buffers. Keep cells on ice throughout the experiment.
Perform the experiment in sterile biohazard hood using sterile technique.
2. Isolation of Splenic T cells
Euthanize donor mouse/mice in CO2 chamber followed by cervical dislocation. Spray abdomen with 70% ethanol.
Make a horizontal incision in the abdomen and peel away skin to expose peritoneum. Hold peritoneum away from the internal organs with the forceps and make an incision in the left abdominal peritoneum to expose and excise the spleen.
Place the spleen in 10 ml of Complete Media in a Petri dish. Remove and discard excess tissue from spleen.
Use 2 sterilized glass slides to crush and tease apart spleen into single-cell suspension. Filter cell suspension through a 70 μm strainer into a 50 ml conical tube and rinse strainer with 5 ml of Complete Media. Place up to 5 spleens in one 50 ml conical tube.
Centrifuge cells at 450 x g for 7 min. Discard supernatant by pouring off or by vacuum suction into waste container.
Gently resuspend cells in 5 ml per spleen of Lysis Buffer to lyse red blood cells for 10 min at room temperature. Add an equal volume of Complete Media (5 ml per spleen) to the tube.
Centrifuge cells at 450 x g for 7 min. Discard supernatant by pouring off or by vacuum suction into waste container.
Gently resuspend cells in 10 ml of Labeling Buffer.
- Count cells by trypan blue exclusion.
- Remove 20 μl of cell suspension and add to 180 μl trypan blue to a microfuge tube and mix thoroughly. After 5 min, add 10 μl labeled cells to hemocytometer and count non-blue cells under the microscope. Determine total number of viable cells. Discard cell/trypan blue mix.
Centrifuge cells in 10 ml of Labeling Buffer at 450 x g for 7 min. Discard supernatant by pouring off or by vacuum suction into waste container.
3. Enrichment of CD4+ T cells
NOTE: Follow manufacturer’s instructions for specific products used in this section.
Gently resuspend cells to a single-cell suspension of 20 x 106 cell/ml in cold Labeling Buffer.
Add 5 μl per 1 x 106 cells of biotinylated CD4 T cell enrichment antibodies. Incubate cells on ice for 15 min.
Add 10x volume of Labeling Buffer. Centrifuge cells at 450 × g for 7 min. Carefully aspirate all the supernatant using vacuum suction into waste container.
Thoroughly vortex streptavidin-conjugated magnetic particles. Add 5 μl of particles per 1 x 106 cells.
Mix thoroughly but gently. Keep mix at 6-12 °C for 30 min.
Add Labeling Buffer to a concentration of 20-80 x 106 cells/ml. Transfer up to 1.0 ml labeled cells per 12 x 75 mm round-bottom test tube (referred to as the “positive-fraction tube”).
Place each positive-fraction tube on magnet for 6-8 min.
With the positive-fraction tubes still on the magnet, carefully transfer supernatant from positive-fraction tube with glass Pasteur pipet to a new sterile 50 ml conical tube (referred to as the “enriched fraction”). This enriched fraction contains CD4+ T cells. Be careful not to disrupt the labeled cells attracted to the magnet.
Resuspend cells left in the positive-fraction tubes in the same volume of Labeling Buffer as in step 3.6 by pipetting up and down vigorously. Place positive-fraction tube back on magnet for 6-8 min.
With positive-fraction tubes still on the magnet, carefully transfer supernatant (enriched fraction, CD4+) from positive-fraction tube with glass Pasteur pipet to sterile 50 ml conical tube from step 3.8 without disrupting cells attached to magnet.
Repeat steps 3.9-3.10 to increase the yield of CD4+ T cells obtained. Continue protocol using enriched fraction (CD4+ cells).
4. Labeling and Sorting Cells7
Centrifuge enriched cells at 450 x g for 7 min. Discard supernatant by pouring off or vacuum suction into waste container.
Resuspend cells in 1 ml of Labeling Buffer. Remove aliquot of cells to count and to assess for cell viability by trypan blue exclusion as in step 2.9.
- Add a volume of Labeling Buffer to 10 x 106 cells/ml; if cells are already <10 x 106, centrifuge at 450 x g for 7 min, discard supernatant by pouring off or vacuum suction into waste container, and add volume of Labeling Buffer to 10 x 106 cells/ml.
- Set up separate aliquots of approximately 5-10 × 105 cells each for unstained, isotype-stained, and single-stained control cells in microfuge tubes.
Add 5 μg/ml CD4-FITC and 1 μg/ml CD45RB-PE to cells. Add isotype control stains and single stains at the same concentration to appropriate aliquots in microfuge tubes. Mix well but gently and incubate on ice protected from light for 30 min.
Add 10x volume of Labeling Buffer to cells and controls. Centrifuge 450 x g for 7 min. Discard supernatant by vacuum suction into waste container.
Resuspend in Labeling Buffer to volume in step 4.5. Centrifuge 450 x g for 7 min. Discard supernatant by vacuum suction into waste container.
Resuspend in Labeling Buffer to 10 x 106 cells/ml. Pass cells through 70 μm strainer into FACS tube. Keep on ice protected from light until ready for FACS.
Set up and determine appropriate compensation on the cell sorter with unstained cells and single-stained controls.
Exclude nonviable cells with forward- and side-scatter gating (Figure 1A). Set up gating for CD4+ and CD45RB+ cells with isotype-stained controls. Gate cells on CD4+ population.
Sort CD4+ cells into CD45RBhigh and CD45RBlow populations using a simple histogram for PE-stained cells into tubes with 2 ml of Complete Media. The CD45RBhigh population represents the highest 40% of CD4+CD45RB+ cells (CD45RBhigh), and the CD45RBlow population is the lowest 20% of CD4+CD45RB+ cells (CD45RBlow; Figure 1B).
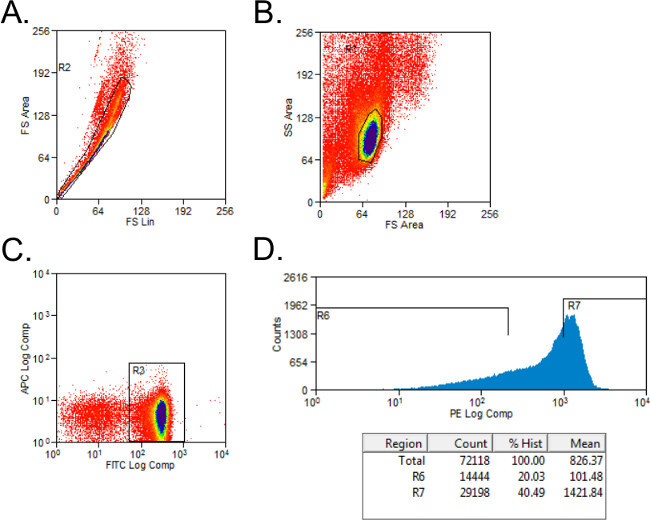 Figure 1: Representative flow cytometry plots of CD4+CD45RB T cell populations during FACS analysis. (A-C) FITC CD4- and PE CD45RB-stained splenocytes from donor C57BL/6 mice were sorted by FACS into CD4+CD45RBhigh and CD4+CD45RBlow T cell populations. (A) Doublet events were excluded on the forward scatter plot. (B) Lymphocytes were gated in the forward and side scatter plot. (C) CD4+ T cells were gated, and (D) CD4+CD45RB+ T cells were plotted on a PE versus Events histogram. CD4+CD45RBlow cells were considered to be the lowest 20% of CD45RB+ cells. CD4+CD45RBhigh cells were defined as the highest 40% of CD45RB+ cells.
Figure 1: Representative flow cytometry plots of CD4+CD45RB T cell populations during FACS analysis. (A-C) FITC CD4- and PE CD45RB-stained splenocytes from donor C57BL/6 mice were sorted by FACS into CD4+CD45RBhigh and CD4+CD45RBlow T cell populations. (A) Doublet events were excluded on the forward scatter plot. (B) Lymphocytes were gated in the forward and side scatter plot. (C) CD4+ T cells were gated, and (D) CD4+CD45RB+ T cells were plotted on a PE versus Events histogram. CD4+CD45RBlow cells were considered to be the lowest 20% of CD45RB+ cells. CD4+CD45RBhigh cells were defined as the highest 40% of CD45RB+ cells.
Run an aliquot of each cell population on the FACS machine to assess purity of populations.
Centrifuge sorted cells at 450 x g for 7 min. Resuspend in 1 ml PBS. Remove aliquot of cells to count and to assess for cell viability by trypan blue exclusion as in step 2.9.
5. Injection of Cells into Recipients
Resuspend sorted cells to 4 x 106 cells/ml (CD45RBhigh) or 2 x 106 cells/ml (CD45RBlow) in PBS.
Transfer 100 μl of CD45RBhigh cells per recipient to new sterile tube. Add 100 μl of PBS per recipient to this tube. Thus, total injection volume per animal is 200 μl, and total amount of cells per recipient is 4 x 105 CD4+CD45RBhigh naïve T cells.
If experimental group receiving T regulatory cells is desired, transfer 100 μl of CD45RBhigh cells per recipient to new sterile tube. Add 100 μl of CD45RBlow cells per recipient to the same tube. Total injection volume per animal is 200 μl; ratio of CD45RBhigh:CD45RBlow cells is 2:1.
Inject 100 μl of CD45RBhigh or CD45RBhigh/CD45RBlow CD4+ cells intraperitoneally into the right and left side of the abdomen (total of 200 μl) of each recipient.
6. Monitoring of Disease Progression in Recipient Animals
- To assess the clinical status of the recipient animals, assign aggregate clinical scores for the following parameters8 on the day of injection, weekly thereafter, and at time of sacrifice:
- Determine wasting by measuring weight loss: 0 – no weight loss; 1 – 0.1-10% loss of initial body weight; 2 – more than 10% loss of initial body weight (Figure 2A).
 Figure 2: Clinical and gross pathological signs of inflammation occur after transfer of wild type CD4+CD45RBhigh T cells into Rag1-/- and NRDKO recipient mice11. (A) NRDKO recipients lost on average 10% of their initial body weights by 5 weeks post-transfer, whereas Rag1-/- recipients did not exhibit clinical signs of disease at this time. Each point represents the mean percentage of initial body weight for the cohort ± SEM. **, p <0.005. (B) Some mice developed severe intestinal inflammation, as demonstrated by the presence of rectal prolapse. This is a representative picture of rectal prolapse in a NRDKO recipient mouse. (C) Grossly, colons from both Rag1-/- and NRDKO recipient mice are thickened and shortened compared to colons from Rag1-/- and NRDKO mice without T cell adoptive transfer. Colons from NRDKO recipient mice show severe inflammation and increased colon weights (data not shown). Please click here to view a larger version of this figure.
Figure 2: Clinical and gross pathological signs of inflammation occur after transfer of wild type CD4+CD45RBhigh T cells into Rag1-/- and NRDKO recipient mice11. (A) NRDKO recipients lost on average 10% of their initial body weights by 5 weeks post-transfer, whereas Rag1-/- recipients did not exhibit clinical signs of disease at this time. Each point represents the mean percentage of initial body weight for the cohort ± SEM. **, p <0.005. (B) Some mice developed severe intestinal inflammation, as demonstrated by the presence of rectal prolapse. This is a representative picture of rectal prolapse in a NRDKO recipient mouse. (C) Grossly, colons from both Rag1-/- and NRDKO recipient mice are thickened and shortened compared to colons from Rag1-/- and NRDKO mice without T cell adoptive transfer. Colons from NRDKO recipient mice show severe inflammation and increased colon weights (data not shown). Please click here to view a larger version of this figure.
- Determine quality of stool by placing animal in clean container until it has passed stool: 0 – none; 1 – soft stool; 2 – watery and/or bloody stool.
- Determine general health of animal by presence of the following signs of disease: 0 – no hunched posture, bristled fur, or skin lesions; 1 – any one of the following present: hunched posture, bristled fur, or skin lesions.
- Determine presence of rectal prolapse: 0 – absent; 1 – present (Figure 2B).
Sacrifice animals by CO2 inhalation followed by cervical dislocation when they have lost 20% of their starting body weight or at desired time point, whichever comes first. Clinical disease is typically apparent starting at week 5 post-repletion.
- Assess for disease severity as previously described5,7.
- Assign clinical scores as in step 6.18.
- Measure colon length and weight (Figure 2C).
- Perform histological analysis of inflammation5.
- Determine spontaneous cytokine expression in intestinal tissue explant cultures9, mesenteric lymph nodes10, and/or serum11.
- Briefly, for explant cultures9, remove colons after sacrifice, open longitudinally, and clean with PBS. Incubate colons on an orbital shaker set at 250 rpm in Complete Media for 30 min at room temperature.
- Chop tissue into small pieces and incubated at 37 °C in Complete Media for 24 hr. Collect the supernatant and use for quantitation of cytokines per 100 mg tissue by ELISA.
- Perform ex vivo characterization of T cell phenotypes and/or function10.
Representative Results
Approximately 10 x 106 CD4+CD45RBhigh T cells from 10 spleens from adult C57BL/6 donor mice are reliably isolated. This number will vary depending on the age and strain of the donor mouse and the proficiency of the researcher. When 4 x 105 C57BL/6 CD4+CD45RBhigh T cells are transferred into C57BL/6 Rag1-/- recipient mice, clinical signs of disease emerge around week 5 post-repletion or sooner if mice are genetically susceptible to more severe disease (Figure 2, 3)11,12. CD3+ T cells are seen accumulating in colons of Rag1-/- recipients as early as 3 weeks (Figure 3A)12.
 Figure 3: Transfer of wild type CD4+CD45RBhigh T cells into Rag1-/- and RKO/δKD recipients induces chronic intestinal inflammation12. (A) (Original magnification 10X, H&E and CD3 immunohistochemistry). H&E staining (top panels) illustrates epithelial hyperplasia, inflammatory cell infiltrates, and crypt abscesses (arrow) present in RKO/δKD recipients but not Rag1-/- recipients. Colons from RKO/δKD recipient mice demonstrated marked accumulation of CD3+ T cells by CD3 IHC (bottom panels) compared to colons from Rag1-/- recipients. (B) Colons from RKO/δKD recipient mice demonstrated more severe inflammation as compared to colons from Rag1-/- recipient mice as determined by histology scoring. (C, D) Colonic explant cultures from RKO/δKD recipient mice secreted less IL-10 (C) and more IL-12p40 (D) than did colonic explant cultures from Rag1-/- recipient mice. Please click here to view a larger version of this figure.
Figure 3: Transfer of wild type CD4+CD45RBhigh T cells into Rag1-/- and RKO/δKD recipients induces chronic intestinal inflammation12. (A) (Original magnification 10X, H&E and CD3 immunohistochemistry). H&E staining (top panels) illustrates epithelial hyperplasia, inflammatory cell infiltrates, and crypt abscesses (arrow) present in RKO/δKD recipients but not Rag1-/- recipients. Colons from RKO/δKD recipient mice demonstrated marked accumulation of CD3+ T cells by CD3 IHC (bottom panels) compared to colons from Rag1-/- recipients. (B) Colons from RKO/δKD recipient mice demonstrated more severe inflammation as compared to colons from Rag1-/- recipient mice as determined by histology scoring. (C, D) Colonic explant cultures from RKO/δKD recipient mice secreted less IL-10 (C) and more IL-12p40 (D) than did colonic explant cultures from Rag1-/- recipient mice. Please click here to view a larger version of this figure.
We previously published that adoptive T cell transfer into Nfil3-/- / Rag1-/- double knockout recipients (NRDKO) develop more severe colitis compared to Rag1-/- recipient mice (Figure 2)11. NFIL3 negatively regulates IL-12p40 in murine colonic macrophages independently of its IL-10-inducing effects13. Dysregulation of IL-12p40 and IL-10 is implicated in the pathogenesis of human IBD1. Thus, unchecked IL-12p40 production in adoptive transfer NDRKO recipient mice results in more rapid disease progression, as shown by weight loss (Figure 2A). Some NRDKO recipient mice developed severe disease resulting in rectal prolapse (Figure 2B). Grossly, colons from NRDKO recipient mice are thickened and shortened, representing a large influx of inflammatory cells into the colon, compared to Rag1-/- recipient mice (Figure 2C).
Comparatively, in Rag1-/- mice with a nonfunctional PI3K catalytic subunit p110δ in non-lymphocyte populations (RKO/δKD), CD4+CD45RBhigh T cell repletion induces a similarly rapid and severe clinical course of disease compared with NRDKO mice (Figure 3)12. Histological analysis at 3 weeks demonstrates colonic tissue hypertrophy, inflammatory cell infiltrates, and crypt abscesses (arrow) in RKO/δKD recipients compared to Rag1-/- recipients (Figure 3A). Recipient mice in this experiment were euthanized at 3 weeks due to a significant number that had lost 20% of their initial body weight. Histological scores, as determined by a pathologist blinded to the experimental groups and based on specific criteria developed in our lab12, were higher in RKO/δKD recipient mice compared to Rag1-/- recipient mice (Figure 3B). Additionally, spontaneous cytokine production from colonic explant cultures demonstrated decreased IL-10 and enhanced IL-12p40 production by RKO/δKD recipient mice compared to Rag1-/- recipient mice (Figure 3C, D)12. Again, increased IL-12p40 and decreased IL-10 production is implicated in the pathogenesis of human IBD1.
Discussion
Here we describe a step-by-step protocol inducing colonic inflammation in mice by adoptive transfer of CD4+CD45RB+ T cells into immunodeficient mice. We used C57BL/6 donor spleens and syngeneic Rag1-/- recipient mice, although other strains (e.g., BALB/c, 129S6/SvEv, non-obese diabetic (NOD)) and genetic models of immunodeficiency (e.g., SCID, Rag2-/-) may also be used4,14-16. It is well established that background strain affects experimental colitis severity in mice1. Furthermore, the enteric microbiota in a murine population varies widely between facilities, even between those within the same institution6,17. While Figures 2 and 3 do not show the development of colitis in Rag1-/- mice at 4 weeks post-transfer, in our and others’ experiences Rag1-/- recipients develop clinical disease between 6 and 9 weeks after transfer of CD4+CD45RBhigh T cells18-23. This variability in clinical course and disease expression and should be taken into account when establishing this model of experimental colitis to minimize intra- and inter-experimental inconsistency.
A critical step for the reproducibility of this experiment is FACS isolation of pure populations of CD4+CD45RBhigh cells without residual activated/memory/regulatory T cells. This involves a proficient understanding of flow cytometry. It is important to first negatively select non-viable and CD4- cells. Then, using a simple histogram for CD45RB-PE cells, select the highest 40% of cells to represent the CD45RBhigh T cells and the lowest 20% as the CD45RBlow T cells. Other markers to consider using for isolation of naïve CD4+ T cells are CD62L and CD25, although in our experience CD45RBhigh and CD45RBlow separation is sufficient and reproducible5. Additionally, it is possible to vary the ratio of CD45RBhigh to CD45RBlow T cells transferred into recipients to study the effects of regulatory T cell populations on disease phenotype. Work with someone who is familiar with flow cytometry initially to set up the FACS protocol and then using that protocol in future experiments. Additionally, a step where significant variation may impact results in this protocol is the intraperitoneal injection of T cells into recipient mice. Here, the intra- and inter-experimenter variability can greatly impact the outcome of the experiment. It is suggested that the person performing this step is comfortable with handling mice and with the injection technique so as to minimize variability in quality and quantity of cells adoptively transferred. Additionally, it is important to change needles between mice, as needle dulling can affect the efficacy of the intraperitoneal injection.
Steps of the protocol that require optimization include Section 3: Enrichment of CD4+ T cells and Section 4: Labeling and Sorting Cells. Optimization of the CD4+ T cell enrichment depends on the magnetic system used and should follow the manufacturers’ instructions. Options for magnetic cell separation systems are listed in the Table of Specific Reagents/Equipment. It is advisable to enrich for CD4+ T cells by negative selection, as directly labeling this population can affect the phenotype of the cells. There are many antibody clones available for labeling cells for FACS. The antibody concentration (step 4.4) should be optimized to ensure specific staining and to obtain an adequate separation of CD4+CD45RB T cell populations during FACS.
Development of this protocol may require trouble-shooting. First, the success of each experiment depends on the proficiency of the researcher and will increase with improved skill in these methods. However, consistently obtaining low numbers of T cells for adoptive transfer may be caused by not keeping cells on ice during isolation, resuspending centrifuged cells too aggressively, or the use of splenocytes from young mice with small spleens. Additionally, optimize the concentration of anti-CD4 and anti-CD45RB labeling antibodies for staining to avoid deficient or non-specific cell staining (see above). If recipients develop widely varying degrees of intestinal inflammation, the most likely cause is imprecise intraperitoneal injection technique. This should improve with practice and can be monitored by immunohistologic staining of CD3+ cells in the intestines of recipient mice. Additional things to consider include the gender of donor and recipient mice and normal variations in disease penetration. When male recipient mice are used, either male or female donor mice are appropriate. However, if female recipient mice are to be used, the donor mice must be female5. In our and others’ experiences, penetration of disease in this model is 85-90%5. Thus it is expected that some mice may not develop intestinal inflammation, given other causes of marked variability in disease severity have been ruled out. Keep in mind there is marked variability in phenotypes between facilities within an organization and outside it with regard to murine gastrointestinal microbiota and practices that influence the microbiota6,17. Thus, the robustness of phenotype or kinetics of disease in Rag1-/- recipient mice may vary widely based on one’s location. Therefore, it is important to determine the best parameters for these experiments based on the timeline and results obtained from each individual facility.
Here we describe the methodology of the well-characterized adaptive transfer murine model of chronic small bowel and colonic inflammation that resembles human IBD5. This step-by-step protocol illustrates key techniques for the successful development of this method for research purposes. This is a particularly useful animal model of human IBD, as it allows for synchronization of disease and manipulation of various cell populations. However, the immunodeficient recipient mice are genetically modified to develop without mature adaptive immune cells, which likely affect downstream disease outcomes. Despite this, the method described here will be very useful in future studies determining the impact of the enteric microbiota, innate immune cells, and adaptive immune regulatory cells in the pathogenesis of human IBD.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by American Gastroenterological Association (AGA) Research Scholars Award and Crohn’s and Colitis Foundation of America (CCFA) Career Development Award (to S.Z.S.), NIH NIDDK F30 DK089692 (to E.C.S.), and University of North Carolina Center for Gastrointestinal Biology and Disease Grant P30 DK34987 (Histology Core). The UNC Flow Cytometry Core Facility is supported in part by an NCI Center Core Support Grant (P30CA016086) to the UNC Lineberger Comprehensive Cancer Center. We thank Luke B. Borst from North Carolina State University College of Veterinary Medicine for his help with histopathological analysis and immunohistochemistry.
References
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140(6):1704–1712. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5(11):1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- Ostanin DV, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):135–146. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma BW, et al. Routine habitat change: a source of unrecognized transient alteration of intestinal microbiota in laboratory mice. PLoS One. 2012;7(10):e47416. doi: 10.1371/journal.pone.0047416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read S, Powrie F. Induction of inflammatory bowel disease in immunodeficient mice by depletion of regulatory T cells. Curr Protoc Immunol. 1999;Chapter 15(Unit 15 13) doi: 10.1002/0471142735.im1513s30. [DOI] [PubMed] [Google Scholar]
- Maillard MH, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J Exp Med. 2007;204(2):381–391. doi: 10.1084/jem.20061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegazi RA, et al. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J Exp Med. 2005;202(12):1703–1713. doi: 10.1084/jem.20051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole A, et al. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J Immunol. 2013;191(5):2771–2779. doi: 10.4049/jimmunol.1301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, et al. NFIL3-deficient mice develop microbiota-dependent, IL-12/23-driven spontaneous colitis. J Immunol. 2014;192(4):1918–1927. doi: 10.4049/jimmunol.1301819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach EC, et al. Innate PI3K p110delta Regulates Th1/Th17 Development and Microbiota-Dependent Colitis. J Immunol. 2014;192(8):3958–3968. doi: 10.4049/jimmunol.1301533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, et al. NFIL3 is a regulator of IL-12 p40 in macrophages and mucosal immunity. J Immunol. 2011;186(8):4649–4655. doi: 10.4049/jimmunol.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach MW, Bean AG, Mauze S, Coffman RL, Powrie F. Inflammatory bowel disease in C.B-17 scid mice reconstituted with the CD45RBhigh subset of CD4+ T cells. Am J Pathol. 1996;148(5):1503–1515. [PMC free article] [PubMed] [Google Scholar]
- Powrie F, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4. T cells. Immunity. 1994;1(7):553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192(2):295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GB, et al. Functional divergence in gastrointestinal microbiota in physically-separated genetically identical mice. Sci Rep. 2014;4:5437. doi: 10.1038/srep05437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, et al. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180(3):1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz CC, et al. Extracellular adenosine regulates colitis through effects on lymphoid and nonlymphoid cells. Am J Physiol Gastrointest Liver Physiol. 2014;307(3):338–346. doi: 10.1152/ajpgi.00404.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma M, et al. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177(5):2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- Ranatunga DC, et al. A protective role for human IL-10-expressing CD4+ T cells in colitis. J Immunol. 2012;189(3):1243–1252. doi: 10.4049/jimmunol.1103421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikrishna G, et al. Carboxylated glycans mediate colitis through activation of NF-kappa. B. J Immunol. 2005;175(8):5412–5422. doi: 10.4049/jimmunol.175.8.5412. [DOI] [PubMed] [Google Scholar]
- Wang F, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131(4):1153–1163. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]