

Abstract
Studying the interaction between proteins is key in understanding their function(s). A very powerful method that is frequently used to study interactions of proteins with other macromolecules in a complex sample is called co-immunoprecipitation. The described co-immunoprecipitation protocol allows to demonstrate and further investigate the interaction between the antiviral myxovirus resistance protein 1 (Mx1) and one of its viral targets, the influenza A virus nucleoprotein (NP). The protocol starts with transfected mammalian cells, but it is also possible to use influenza A virus infected cells as starting material. After cell lysis, the viral NP protein is pulled-down with a specific antibody and the resulting immune-complexes are precipitated with protein G beads. The successful pull-down of NP and the co-immunoprecipitation of the antiviral Mx1 protein are subsequently revealed by western blotting. A prerequisite for successful co-immunoprecipitation of Mx1 with NP is the presence of N-ethylmaleimide (NEM) in the cell lysis buffer. NEM alkylates free thiol groups. Presumably this reaction stabilizes the weak and/or transient NP–Mx1 interaction by preserving a specific conformation of Mx1, its viral target or an unknown third component. An important limitation of co-immunoprecipitation experiments is the inadvertent pull-down of contaminating proteins, caused by nonspecific binding of proteins to the protein G beads or antibodies. Therefore, it is very important to include control settings to exclude false positive results. The described co-immunoprecipitation protocol can be used to study the interaction of Mx proteins from different vertebrate species with viral proteins, any pair of proteins, or of a protein with other macromolecules. The beneficial role of NEM to stabilize weak and/or transient interactions needs to be tested for each interaction pair individually.
Keywords: Immunology, Issue 98, Influenza A virus, nucleoprotein, Mx1, co-immunoprecipitation, N-ethylmaleimide, plasmid transfection, viral infection, antiviral host defense
Introduction
Myxovirus resistance (Mx) proteins are an important part of the innate immune defense against viral pathogens. These proteins are large dynamin-like GTPases that are induced by type I and type III interferons. The corresponding Mx genes are present in nearly all vertebrates in one or multiple copies and their gene products inhibit a wide range of viruses, including Orthomyxoviridae (e.g., influenza virus), Rhabdoviridae (e.g., vesicular stomatitis virus), Bunyaviridae (e.g., la crosse virus) and Retroviridae (e.g., human immunodeficiency virus-1)1-4. It is unclear how these proteins recognize such a broad array of viruses, without any apparent shared primary sequence motifs in these viruses. Analyzing the interaction of Mx proteins with their viral targets, potentially involving higher order complexes with other host cell factors, will help to understand the molecular mechanisms that have evolved in the arms race between viruses and their hosts.
The interaction between mammalian Mx proteins and viral targets has been studied most extensively for human MxA. Human MxA can inhibit the replication of many viruses, including the orthomyxoviruses influenza A and Thogoto virus. MxA binds the Thogoto virus ribonucleoprotein complexes (vRNPs), thereby preventing their nuclear entry, which results in block of infection5. This interaction between MxA and Thogoto virus vRNPs has been demonstrated with co-sedimentation and co-immunoprecipitation experiments6-9. How Mx proteins hinder influenza A viruses is less clear. One major problem is that it is not straightforward to demonstrate an interaction between an Mx protein and an influenza gene product. One report demonstrated an interaction between human MxA and the NP protein in influenza A virus infected cells10. This interaction could only be shown by co-immunoprecipitation if the cells had been treated with the cross-linking reagent dithiobis (succinimidyl propionate) before lysis, suggesting that the interaction is transient and/or weak. More recent studies have shown that the differential Mx sensitivity of different influenza A strains is determined by the origin of the NP protein11,12. In line with this, influenza A viruses can partly escape from Mx control by mutating specific residues in the NP protein13. This suggests that the main target of influenza A viruses for host Mx is the NP protein, most probably NP assembled in vRNP complexes. However, none of these more recent studies demonstrated an interaction between influenza NP or vRNPs and either human MxA or mouse Mx1.
Recently we showed, for the first time, an interaction between the influenza NP and the mouse Mx1 protein with an optimized co-immunoprecipitation protocol14, which is described here in detail. In general, co-immunoprecipitation is one of the most frequently used biochemical approaches to investigate protein-protein interactions. This technique is often preferred over alternative techniques, e.g., yeast two hybrid, since it allows to investigate protein-protein interactions in their natural environment. Co-immunoprecipitation can be carried out on endogenously expressed proteins if antibodies against the proteins of interest are available. Alternatively, the proteins of interest can be expressed in the cell through transfection or infection and an affinity tag may be used. In addition to the above-mentioned advantages, the described co-immunoprecipitation protocol allows the detection of weak and/or transient protein interactions. The main component in this optimized protocol is the addition of N-ethylmaleimide (NEM) in the cell lysis buffer. NEM is an alkylating reagent that reacts with free thiol groups such as present in cysteines, at a pH of 6.5-7.5, to form a stable thio-ester (Figure 1). At higher pH, NEM can also react with amino groups or undergo hydrolysis15. NEM is typically used to block free thiol groups, in order to prevent disulfide bond formation or inhibit enzymatic activity. For example, NEM is often used to block desumoylating enzymes, which are cysteine proteases. In the described co-immunoprecipitation protocol, NEM was initially included in the lysis buffer because it had been reported that the sumoylation of influenza proteins can influence the interaction between viral proteins16. Unexpectedly, the addition of NEM proved to be key to document the interaction between influenza NP and mouse Mx1 by co-immunoprecipitation. It is unclear why the addition of NEM is crucial to detect the NP–Mx1 interaction. Possibly the interaction is too transient and/or weak. NEM could stabilize the interaction, e.g., by preserving a specific conformation of Mx1, a viral protein or even an unknown third component. Such a stabilizing effect of NEM has been observed before, e.g., for the interaction between the ribonucleotide reductase M1 and its inhibitor gemcitabine (F2dC)17. Mx1 and NP both contain multiple cysteine residues which could be modified by NEM. For example, a recent study by Rennie et al. demonstrated that a stalkless MxA-variant contains three solvent exposed cysteine residues which can be modified by iodoacetamide. Mutating these residues to serines did not influence the enzymatic activity of MxA, but prevented disulfide-mediated aggregation18. As these cysteines are conserved in Mx1, this suggests that the analogous cysteines in Mx1 can be modified by NEM and as such influence its conformation or solubility. In addition, NEM might also affect the GTPase activity of Mx1, which is essential for the anti-influenza activity of Mx1, and thereby stabilize the interaction between Mx1 and NP. However, a direct effect of NEM on the GTPase activity of Mx1 is unlikely, as NEM is also required to detect the interaction between influenza NP and GTPase inactive mutants of the Mx1 protein14. Clearly, more research is needed to unravel the effect of NEM on the NP–Mx1 interaction.
In summary, the described co-immunoprecipitation protocol allows to study the interaction between the antiviral Mx1 protein and its viral target, the influenza NP protein. This protocol could also be used to study other weak or transient interactions that depend on the stabilization of specific protein conformations. Protein-protein interaction that depend on specific conformations have been described before, e.g., for calcium-binding proteins such as calmodulin19. Finally, the beneficial role of NEM could also be used in other methods that detect protein-protein interactions, such as co-sedimentation assays.
Protocol
Note: The following transfection and co-immunoprecipitation protocol is established for a 9 cm Petri dish format. Other formats are also possible after scaling the protocol.
1. Seeding the Human Embryonic Kidney (HEK) 293T Cells
Seed the HEK293T cells one day before transfection at 1.2 x 106 cells per 9 cm Petri dish in 12 ml of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 2 mM L-glutamine, 0.4 mM Na-pyruvate, 0.1 mM non-essential amino acids, 100 U/ml penicillin and 0.1 mg/ml streptomycin.
Grow the cells 16 hr at 37 °C and 5% CO2.
Visually inspect the morphology and viability of the cells with an inverted light microscope before transfection. The cells need to be sub-confluent for optimal transfection efficiency.
2. Calcium-phosphate Transfection of HEK293T Cells
Note: Use 0.5-1 µg of pCAXL-NP or empty pCAXL plasmid in combination with 1-3 µg of pCAXL-Mx1 per 9 cm dish. Use an equal amount of total plasmid DNA in all samples; adjust with empty plasmid if necessary.
- Prepare the following transfection buffers:
- Prepare Tris-EDTA (TE) with concentrations of 1.0 mM Tris-HCl pH 8.0 and 0.1 mM EDTA pH 8.0.
- Prepare BS/HEPES with concentrations of 25 mM HEPES (5.96 g/L, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 274 mM NaCl (16 g/L), 10 mM KCl (0.74 g/L), 1.5 mM NaHPO4·12H2O (0.5 g/L) and 11.1 mM dextrose (2 g/L). Adjust pH to 7.05.
- Prepare CaCl2/HEPES with concentrations of 1.25 M CaCl2·2H2O (183.8 g/L) and 125 mM HEPES (29.79 g/L). Adjust pH to 7.05 with NaOH.
Warm the transfection buffers at 37 °C before use.
Prepare the plasmid samples by diluting the plasmid DNA in 600 µl of TE. Prepare these mixtures in wells of a 6-well plate.
Add 150 µl of CaCl2/HEPES in a drop wise manner to the plasmid samples and mix by pipetting 3 times up and down.
Prepare the transfection solution by drop wise adding the plasmid solution (TE + DNA + CaCl2/HEPES; 750 µl) to 750 µl of BS/HEPES buffer provided in a fresh 6-well plate. Distribute the plasmid solution evenly over the complete well containing BS/HEPES buffer.
Shake the transfection solution on a plate shaker for 90 sec at 1,000 RPM.
Incubate the mixture for 5 min at room temperature.
Add the transfection solution (1.5 ml) drop wise to the cells. Use a P1000 micropipet to drip the transfection solution on the cells. Disperse the mixture over the complete 9 cm Petri dish and shake the plate very gently.
Incubate the cells at 37 °C and 5% CO2 for 6 hr. Then remove the medium by aspiration and immediately replace with 12 ml fresh, pre-warmed medium. Gently add the fresh medium to the cells to prevent cell detachment. For this, hold the tip of the pipet against the side of the well and gently push out the medium.
Incubate the cells for an additional 16 hr at 37 °C and 5% CO2.
3. Co-immunoprecipitation
Note: Perform the co-immunoprecipitation 24 hr after transfection.
- Preparation of the low salt lysis buffer and high salt wash buffer.
- Prepare a stock solution of 2 M N-ethylmaleimide (NEM) by weighing the amount of NEM and dissolving it in absolute ethanol. Prepare the NEM stock solution fresh before use. CAUTION: NEM is very toxic, prepare and use this stock solution in a fume hood.
- Prepare low salt lysis buffer at concentrations of 50 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 1% NP40 and a protease inhibitor cocktail (dissolve 1 tablet in 50 ml lysis buffer). Add NEM to a final concentration of 25 mM (i.e., dilute 1:80). Keep on ice after adding the protease inhibitors and NEM. Note: Always add the protease inhibitors and NEM freshly before use.
- Prepare a high salt wash buffer at concentrations of 50 mM Tris-HCl pH 8, 500 mM NaCl, 5 mM EDTA and 1% NP40. Note that the high salt wash buffer does not contain NEM.
- Preparation of cell lysates.
- Remove the medium and wash the cells with 2 ml of ice cold phosphate buffered saline (PBS). Very gently add the wash buffer, as HEK293T cells detach easily.
- Remove the PBS and add 600 µl of ice cold low salt lysis buffer per 9 cm Petri dish.
- Incubate the plates for 20 min on ice. Make sure that the plates are kept horizontal, to assure complete coverage of the plate surface with lysis buffer. Gently shake the plates every 5 min.
- Collect the cell lysate in a 1.5 ml microcentrifuge tube and centrifuge for 3 min at 4 °C and 16,000 x g to pellet the insoluble fraction.
- Transfer the soluble fraction, i.e. the cell lysate to a fresh 1.5 ml microcentrifuge tube and keep on ice. Immediately continue with the co-immunoprecipitation protocol, to prevent dissociation of the interacting proteins. Perform all the following steps as much as possible on ice or at 4 °C to limit proteolytic activity in the lysates.
- Generation of immune-complexes. Note: In this step, the protein of interest is bound by the appropriate antibody. To study the NP–Mx1 interaction, use a mouse anti-NP monoclonal antibody.
- For each sample, mix 135 µl of lysate with 2 µl of anti-NP monoclonal antibody and 113 µl of low salt lysis buffer (total volume of 250 µl). Store the remaining lysate at -20 °C for further analysis such as western blotting, to document the expression levels of the surmised interaction partners in the transfected cells. Note: Alternatively, measure the protein concentration of the lysate (e.g. with Bradford reagent) and use a fixed amount of total protein, e.g., 400 µg, for each lysate.
- Incubate the antibody-lysate mix for 3 hr on a turning wheel at 4 °C. This step can be extended to an overnight incubation.
- Preparation of the protein G beads. Note: The protein G beads are shipped and stored in 20% ethanol for preservation. The bead-slurry normally consists of 50% beads and these beads need to be washed before they are used to immunoprecipitate the immune-complexes.
- Use 50 µl of beads, i.e., 100 µl of bead-slurry, for each sample. Wash the amount of beads needed for all samples in the co-immunoprecipitation assay in one tube. Cut the tip of a 1 ml pipette tip to ease pipetting of the bead-slurry.
- Centrifuge the protein G bead-slurry at 8,000 x g and 4 °C for 30 sec. Remove the ethanol solution and add an equal volume of low salt lysis buffer. Centrifuge the protein G bead-slurry at 8,000 x g and 4 °C for 30 sec and remove the supernatant gently. Repeat this wash step 3 times. Note: The low salt lysis buffer used to wash the beads does not need to contain protease inhibitors or NEM.
- Estimate the volume of protein G beads and add an equal volume of low salt lysis buffer to make a new 50% bead-slurry in low salt lysis buffer.
- For each sample, transfer 100 µl of bead-slurry in a fresh 1.5 ml microcentrifuge tube and store on ice until use. Be careful to resuspend the bead-slurry before dividing, as these beads quickly sediment to the bottom of the tube.
- Immunoprecipitation of the immune-complexes by protein G beads and their elution.
- Before using the protein G beads for immunoprecipitation, centrifuge all tubes 30 sec at 8,000 x g and 4 °C and verify by visual inspection that there is an equal amount of beads present in all samples. If necessary adjust the amount of beads in some of the samples and centrifuge again. Discard the supernatants. Be careful not to disturb the pelleted protein G beads.
- Briefly centrifuge the immune-complexes (i.e., lysates with antibody, 250 µl) for 30 sec at 8,000 x g and 4 °C to collect the complete sample at the bottom of the tube. Transfer the immune-complexes to the protein G beads (50 µl).
- Incubate 60 min on a turning wheel at 4 °C. Do not incubate these immune complexes longer than 75 min with the beads to reduce nonspecific binding of proteins to the protein G beads.
- Centrifuge the protein G beads (with bound immune-complexes) for 30 sec at 8,000 x g and 4 °C and remove the supernatants. Be careful not to disturb the pelleted protein G beads. Optional: store this supernatants at 4 °C or -20 °C for later analysis, e.g., to estimate the amount of unbound protein.
- Wash the protein G beads for approximately 5 min with 900 µl of high salt lysis buffer. Make sure that the beads are completely resuspended in the wash buffer for optimal washing. Centrifuge the protein G beads for 30 sec at 8,000 x g and 4 °C and discard the supernatants. Repeat this wash step 4 times. Be careful not to disturb the pelleted protein G beads to avoid loss of immunoprecipitated material.
- After the last wash step, add 50 µl of 2x Laemmli sample buffer to the beads and heat the suspension for 10 min at 95 °C to elute the (co)immunoprecipitated proteins.
- Prepare 10 ml of 6x Laemmli buffer with 1 g of sodium dodecyl sulfate, 3.5 ml of glycerol, 3.5 ml of 1 M Tris-HCl pH 6.8 and 420 µl of β-mercaptoethanol. Adjust to a total volume of 10 ml by adding distilled water. Dilute 3 times in distilled water to obtain 2x Laemmli buffer. CAUTION: β-mercaptoethanol is toxic, prepare and use Laemmli buffer in a fume hood.
- After heating, centrifuge the protein G beads for 30 sec at 8,000 x g and store the samples at 4 °C (short term) or -20 °C (long term).
4. Analyze the (co-)Immunoprecipitated Proteins
Visualize the proteins present in the cell lysate and eluate of the co-immunoprecipitation by SDS-PAGE20 and western blotting21,22. Typically load half of the Laemmli eluate on gel. Be careful not to disturb the pelleted protein G beads when taking samples for gel loading. Mx1 and NP expression were revealed with an anti-Mx1 and anti-NP antibody, respectively14. The bands were detected with HRP-based chemiluminescence and an X-ray film developer.
Representative Results
N-ethylmaleimide is an organic compound that can be used to irreversibly modify free thiol groups, e.g. to inhibit cysteine proteases (Figure 1).
The antiviral Mx1 protein inhibits influenza A virus replication by interacting with the viral nucleoprotein. The optimized co-immunoprecipitation protocol described here allows to study this NP–Mx1 interaction. HEK293T cells were transfected with expression vectors for the antiviral Mx1 protein in the absence or presence of the influenza NP protein. Next, the NP protein was pulled-down from total cell lysates with an NP-specific monoclonal antibody. Figure 2 shows that the Mx1 protein is only co-immunoprecipitated in the presence of co-expressed NP. Possible nonspecific co-immunoprecipitation of Mx1 in the absence of NP is caused by either nonspecific pull-down of the Mx1 protein by the anti-NP antibody or by nonspecific binding of Mx1 to the protein G beads. Therefore, always include a negative control to assess this nonspecific co-immunoprecipitation. Figure 3 shows that the NP–Mx1 interaction can only be detected in the presence of NEM. In this experiment, nonspecific binding of Mx1 to the protein G beads was assessed with a control co-immunoprecipitation reaction in the absence of anti-NP antibody.
This protocol can also be used to study the interaction between Mx1 and the influenza NP protein isolated from infected cells or from virions. For this application, the above protocol was slightly adapted by combining lysates of Mx1 expressing cells with lysates containing the viral NP protein before starting the co-immunoprecipitation protocol. Figure 4 shows the co-immunoprecipitation of Mx1 with NP isolated from transfected cells, infected cells or virions.
In conclusion, these results show that this co-immunoprecipitation protocol can be used to study the interaction between an antiviral protein and its viral target.
 Figure 1: Irreversible modification of free thiol groups by N-ethylmaleimide.
Please click here to view a larger version of this figure.
Figure 1: Irreversible modification of free thiol groups by N-ethylmaleimide.
Please click here to view a larger version of this figure.
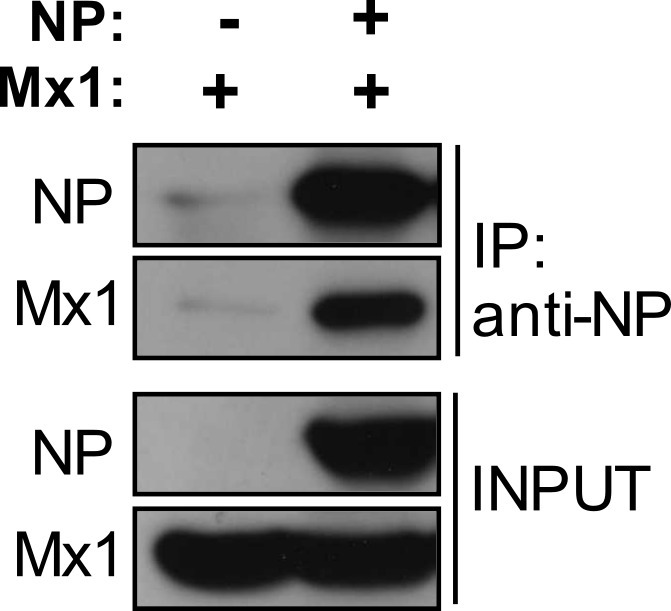 Figure 2: Mx1 interacts with NP. Western blot analysis of a co-immunoprecipitation experiment with two samples: one in which both Mx1 and NP are present and one control setup in which the NP protein is absent. NP was immunoprecipitated with anti-NP and NP and Mx1 were visualized by western blotting. This figure has been modified from14.
Figure 2: Mx1 interacts with NP. Western blot analysis of a co-immunoprecipitation experiment with two samples: one in which both Mx1 and NP are present and one control setup in which the NP protein is absent. NP was immunoprecipitated with anti-NP and NP and Mx1 were visualized by western blotting. This figure has been modified from14.
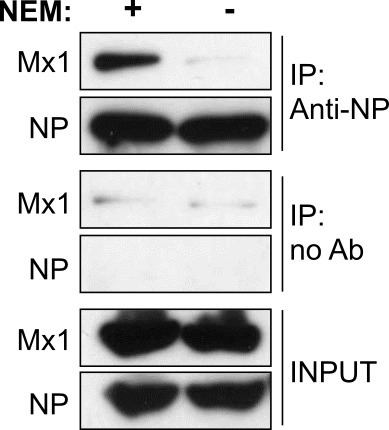 Figure 3: N-ethylmaleimide is important to detect the NP–Mx1 interaction. Western blot analysis of a co-immunoprecipitation experiment performed in the presence or absence of N-ethylmaleimide. NP was immunoprecipitated with anti-NP and Mx1 and NP were visualized by western blotting.
Figure 3: N-ethylmaleimide is important to detect the NP–Mx1 interaction. Western blot analysis of a co-immunoprecipitation experiment performed in the presence or absence of N-ethylmaleimide. NP was immunoprecipitated with anti-NP and Mx1 and NP were visualized by western blotting.
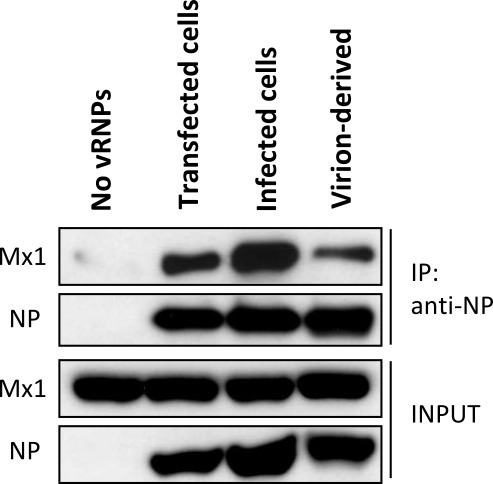 Figure 4: Mx1 interacts with NP isolated from infected cells or from virions. Lysates containing Mx1 were combined with lysates containing NP from different sources: a control lysate (no vRNPs), transfected vRNPs, vRNPs from infected cells or vRNPs isolated from influenza A virions. After mixing the lysates, co-immunoprecipitation with anti-NP was performed and Mx1 and NP were visualized by western blotting.
Figure 4: Mx1 interacts with NP isolated from infected cells or from virions. Lysates containing Mx1 were combined with lysates containing NP from different sources: a control lysate (no vRNPs), transfected vRNPs, vRNPs from infected cells or vRNPs isolated from influenza A virions. After mixing the lysates, co-immunoprecipitation with anti-NP was performed and Mx1 and NP were visualized by western blotting.
Discussion
Studying the interaction between antiviral proteins and their viral targets is very important to understand the details of the antiviral mechanism of these proteins. This can give new insights into how viruses and their hosts co-evolved and be the basis for the development of new antiviral strategies. The optimized co-immunoprecipitation protocol described here allows to study the interaction between the mouse Mx1 protein and its viral target, the influenza NP protein. The most important aspect of this protocol is the addition of NEM in the lysis buffer, as the NP–Mx1 interaction is undetectable in the absence of NEM (Figure 3). To date, it is not known why the presence of NEM is essential to detect this interaction. However, this protocol could be useful to study other weak and/or transient interactions which depend on the stabilization of specific protein conformations, especially if cysteines are involved.
An important limitation of co-immunoprecipitation assays in general is the availability of specific and high quality antibodies that recognize conformational epitopes in one of the interaction partners with high affinity. The NP–Mx1 interaction could not be demonstrated with our in house produced anti-Mx1 polyclonal antiserum. This antiserum also immunoprecipitates the influenza NP protein, even in the absence of Mx1. Furthermore, the anti-NP monoclonal antibody that was used, recognizes the NP protein of the A/Puerto Rico/8/34 influenza strain, but unfortunately it is not suitable to pull-down NP of avian influenza virus strains. Another limitation of this co-immunoprecipitation protocol is the nonspecific binding of Mx1 to the protein G beads. The latter binding can be overcome by increasing the salt concentration in the wash buffer and reducing the contact time between the lysate and the protein G beads. In addition, reducing the amount of beads used for each immunoprecipitation reaction from 50 µl to 25 µl, can further decrease nonspecific binding of Mx1 to these beads. In general, the unintended pull-down of contaminating proteins, caused by nonspecific binding to the protein G beads, can also be reduced by other strategies. For example, these proteins can be removed during a pre-clear step, in which these proteins are removed by incubating the lysate with protein G beads in the absence of antibody. The contaminating proteins are then removed together with the beads and the pre-cleared lysate is used for co-immunoprecipitation. This strategy is only advantageous if the contaminating proteins are different from the protein(s) under study. Alternatively, the unspecific binding sites on the protein G beads could be blocked with BSA. However, this strategy is only recommended if the beads are first coated with the antibody, as BSA can also reduce the precipitation of immune-complexes (i.e., reduced antibody binding by the coated-beads). Taken together, it is very important to include proper controls to exclude nonspecific binding of the proteins of interest to the protein G beads or to the antibodies used.
The described co-immunoprecipitation protocol can be modified to study the interaction of Mx1 with the NP protein present in a different environment, e.g., in infected cells or in purified vRNPs. Mx1 inhibits the expression of viral proteins, including NP, during influenza virus infection23. Therefore, it is technically extremely difficult to study the NP–Mx1 interaction in Mx1 transfected cells that are subsequently infected. Still, this co-immunoprecipitation protocol can also be performed after combining lysates of Mx1 expressing cells and infected cells, allowing successful detection of the NP–Mx1 interaction (Figure 4). As the crucial target of NEM is not known, NEM was added during the lysis of both cell populations. If desired, the pH of the lysis buffer can be changed. The experiments described here are performed at pH 8 (as in Turan et al.10), but the NP-Mx1 co-immunoprecipitation was also successfully performed with a pH 7.2 lysis buffer. Actually a pH of 7.2 is preferred, because at this pH NEM reacts exclusively with free thiol groups and this pH also increases the extraction yield of the Mx1 protein. In addition, the protein G beads could be replaced by protein A beads, depending on the host-species from which the used antibodies are derived. However, cleaner results are obtained with protein G beads. Finally, this protocol can also be used to study the interaction between Mx1 and other influenza proteins, e.g., PB214. In this case a V5 epitope tagged PB2 was used, which could in principle be combined with commercially available anti-V5 agarose affinity gel.
In future experiments, this protocol could be valuable to identify the regions in Mx1 and NP that are important for the NP–Mx1 interaction. One such region in Mx1 could be loop L4, as this loop has been shown to be critical for the interaction between MxA and Thogoto virus NP8,9. If suitable NP-specific antibodies become available, this protocol will also allow to determine if the increased sensitivity of avian influenza A strains for Mx1 correlates with a stronger NP–Mx1 interaction or not, which could help to understand the mechanism of Mx sensitivity. In addition, the interaction between Mx1 and components of viruses belonging to families other than the Orthomyxoviridae, can also be addressed by this adapted co-immunoprecipitation protocol. Finally, further studies to unravel the effect of NEM on the NP–Mx1 will be very valuable to better understand this specific interaction, but also to gain insight in the broader applications of the addition of this compound during cell lysis and co-immunoprecipitation experiments.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by FWO-Vlaanderen, the IOF project IOF10/StarTT/027 and Ghent University Special Research Grant BOF12/GOA/014.
References
- Verhelst J, Hulpiau P, Saelens X. Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol Mol Biol Rev. 2013;77(4):551–566. doi: 10.1128/MMBR.00024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 2013;502(7472):559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013;502(7472):563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, et al. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe. 2013;14(4):398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- Kochs G, Haller O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc Natl Acad Sci U S A. 1999;96(5):2082–2086. doi: 10.1073/pnas.96.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohr F, Schneider-Schaulies S, Haller O, Kochs G. The central interactive region of human MxA GTPase is involved in GTPase activation and interaction with viral target structures. FEBS Lett. 1999;463(1-2):24–28. doi: 10.1016/s0014-5793(99)01598-7. [DOI] [PubMed] [Google Scholar]
- Kochs G, Haller O. GTP-bound human MxA protein interacts with the nucleocapsids of Thogoto virus (Orthomyxoviridae) J Biol Chem. 1999;274(7):4370–4376. doi: 10.1074/jbc.274.7.4370. [DOI] [PubMed] [Google Scholar]
- Mitchell PS, et al. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe. 2012;12(4):598–604. doi: 10.1016/j.chom.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzina C, Haller O, Kochs G. Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J Biol Chem. 2014;289(9):6020–6027. doi: 10.1074/jbc.M113.543892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan K, et al. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004;32(2):643–652. doi: 10.1093/nar/gkh192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmann J, et al. Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J Virol. 2008;82(7):3624–3631. doi: 10.1128/JVI.01753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P, Manz B, Haller O, Schwemmle M, Kochs G. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J Virol. 2011;85(16):8133–8140. doi: 10.1128/JVI.00712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz B, et al. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog. 2013;9(3):e1003279. doi: 10.1371/journal.ppat.1003279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhelst J, Parthoens E, Schepens B, Fiers W, Saelens X. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J Virol. 2012;86(24):13445–13455. doi: 10.1128/JVI.01682-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer CF, Riehm JP. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal Biochem. 1967;18(2):248–255. [Google Scholar]
- Wu CY, Jeng KS, Lai MM. The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J Virol. 2011;85(13):6618–6628. doi: 10.1128/JVI.02401-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhou J, Zhang Y, Bepler G. Modulation of the ribonucleotide reductase M1-gemcitabine interaction in vivo by N-ethylmaleimide. Biochem Biophys Res Commun. 2011;413(2):383–388. doi: 10.1016/j.bbrc.2011.08.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie ML, McKelvie SA, Bulloch EM, Kingston RL. Transient dimerization of human MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure. 2014;22(10):1433–1445. doi: 10.1016/j.str.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405(2):199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- Separating Protein with SDS-PAGE. JoVE; 2014. Available from: http://www.jove.com/science-education/5058/separating-protein-with-sds-page. [Google Scholar]
- Gallagher S, Chakavarti D. Immunoblot analysis. J Vis Exp. 2008. [DOI] [PMC free article] [PubMed]
- Eslami A, Lujan J. Western blotting: sample preparation to detection. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Pavlovic J, Haller O, Staeheli P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol. 1992;66(4):2564–2569. doi: 10.1128/jvi.66.4.2564-2569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]