

Abstract
RPE65 is essential in the operation of the visual cycle and functions as a chaperone for all-trans-retinyl esters, the substrates for isomerization in the visual cycle. RPE65 stereospecifically binds all-trans-retinyl esters with a KD of 47 nM. It is shown here by using a quantitative fluorescence technique, that Accutane (13-cis-retinoic acid), a drug used in the treatment of acne but that causes night blindness, binds to RPE65 with a KD of 195 nM. All-trans-retinoic acid binds with a KD of 109 nM. The binding of the retinoic acids to RPE65 is competitive with all-trans-retinyl ester binding, and this competition inhibits visual cycle function. A retinoic acid analog that binds weakly to RPE65 is not inhibitory. These data suggest that RPE65 function is rate-limiting in visual cycle function. They also reveal the target through which the retinoic acids induce night blindness. Finally, certain forms of retinal and macular degeneration are caused by the accumulation of vitamin A-based retinotoxic products, called the retinyl pigment epithelium–lipofuscin. These retinotoxic products accumulate during the normal course of rhodopsin bleaching and regeneration after the operation of the visual cycle. Drugs such as Accutane may represent an important approach to reducing the accumulation of the retinotoxic lipofuscin by inhibiting visual cycle function. The identification of RPE65 as the visual cycle target for the retinoic acids makes it feasible to develop useful drugs to treat retinal and macular degeneration while avoiding the substantial side effects of the retinoic acids.
Keywords: visual cycle, rate-limiting step, retinylester binding
The vertebrate visual cycle is comprised of the enzymatic reactions responsible for the biosynthesis of 11-cis-retinal, the visual chromophore (Scheme 1) (1). A three-component system comprised of lecithin retinol acyl transferase (LRAT), RPE65, and isomerohydrolase (IMH) is involved in the actual processing of all-trans-retinol (vitamin A) into 11-cis-retinol (1). LRAT catalyzes the esterification of vitamin A by using lecithin as the acyl donor to generate the all-trans-retinyl esters (1). LRAT is an essential component in the pathway because longchain all-trans-retinyl esters serve as substrates for the isomerization reaction (2–5). The high degree of hydrophobicity of these esters requires the availability of a chaperone to mobilize and deliver them to the appropriate enzyme, in this case the IMH. RPE65 serves this role in the visual cycle (6–8). This protein, which is essential for vertebrate vision (9–11), acts as a chaperone to render the highly hydrophobic all-trans-retinyl esters available for IMH processing into 11-cis-retinol (7, 8). The membrane-bound form of RPE65 was shown by a quantitative fluorescence technique to bind all-trans-retinyl palmitate stereospecifically (7). Finally, in the last enzymatic step in the pathway, 11-cis-retinol is oxidized by an 11-cis-retinol dehydrogenase to produce 11-cis-retinal (12).
Scheme 1.

The mammalian visual cycle.
The normal operation of the visual cycle produces chemically reactive intermediates that are toxic to the visual system. For example, all-trans-retinal, the product of rhodopsin bleaching (Scheme 1), is a chemically reactive species, and can readily engage in Schiff base formation with proteins and lipids, in addition to its being reduced to vitamin A in the visual cycle. Of particular consequence is the reaction of all-trans-retinal with the membrane lipid phosphatidyl ethanolamine, which initiates the formation of the highly retinotoxic molecule(s) in the lipofuscin series (A2E) (13, 14) (Scheme 2). The retinyl pigment epithelium (RPE)–lipofuscin are also readily photooxidized into reactive oxirane derivatives, which cause DNA damage and apoptosis (15). In a general way, the formation of these kinds of adducts is akin to the formation of the toxic advanced glycosylation end products (AGE) formed from aldoses, such as glucose, and the amino moieties of proteins (16).
Scheme 2.

Formation of retinotoxic A2E.
The accumulation of A2E is the cause of Stargardt's disease, a macular degenerative disease of genetic origins (17–19). The elevated levels of A2E found in Stargardt's disease are caused by mutations in the photoreceptor disk ATP-binding cassette retina (ABCR) transporter, which pumps all-trans-retinal and early Schiff base reaction products with phosphatidyl ethanolamine out of the photoreceptor disk membranes before they are irreversibly committed to A2E formation (17–19). A2E formation occurs even in the absence of mutations in the ABCR pump because all-trans-retinal generation occurs as a consequence of the normal operation of the visual cycle (Scheme 1) and the reaction with phosphatidyl ethanolamine always occurs, albeit at a reduced rate compared to that found in Stargardt's disease. Indeed, some forms of age-related retinal and macular degeneration may be caused as a consequence of the age-related accumulation of the RPE–lipofuscin. It has been suggested that retinal and macular degenerative diseases may benefit from limiting the processing of vitamin A in the visual cycle (20). Supporting this view is the observation that 13-cis-retinoic acid (13cRA, trade name Accutane) (Scheme 3), a drug used in the treatment of skin disorders, limits both vitamin A processing in the visual cycle and the formation of A2E in ABCR knockout mice (20). Accutane is known to produce night blindness because it slows the rate of 11-cis-retinal biosynthesis (21).
Scheme 3.

Retinoic acid analogs.
The precise molecular target of 13cRA and congeners in visual processing is of substantial interest, because its identification would facilitate the design of anti-maculopathy drugs that avoid the acute toxic side effects of a retinoic acid but preserve the partial inhibition of the visual cycle. In addition, because mice treated with Accutane are also protected against retinal degeneration, it is possible that molecules of this type may be of broad clinical interest in the treatment of retinal degenerative diseases (20, 22).
One visual cycle target of 13cRA is the 11-cis-retinol dehydrogenase shown in Scheme 1 (21). However, subsequent to the identification of this potential target, it was found that 13cRA strongly inhibits net 11-cis-retinoid formation in treated animals, in addition to inhibiting the 11-cis-retinol → 11-cis-retinal conversion (20, 22). Therefore, there must be a more profound and recondite target for this drug, other than 11-cis-retinol dehydrogenase. Inhibition of the dehydrogenase would simply affect the partitioning between 11-cis-retinol/11-cis-retinal without affecting overall 11-cis-retinoid levels. The identification of this putative target is the central issue of this article. We show here that both all-trans retinoic acid (atRA) and 13cRAs powerfully and specifically bind to and inhibit RPE65 function.
Materials and Methods
Materials. Frozen bovine eyecups devoid of retinas were purchased from W. L. Lawson (Lincoln, NE). Ammonium bicarbonate, BSA, EDTA, DEAE-Sepharose, phenyl-Sepharose CL-4B, all-trans-retinol, all-trans-retinyl palmitate, N-(4-hydroxyphenyl)retinamide (4-HPR), and Trizma base were from Sigma. DTT was from ICN Biomedicals. 11-cis-Retinol, all-trans-retinyl palmitate [15-3H] was synthesized by following the procedures described elsewhere (23). All-trans-retinol [11,12-3H2] (52.0 Ci/mmol; 1 Ci = 37 GBq) was obtained from Perkin–Elmer. Anagrade 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was from Anatrace. HPLC-grade solvents were from Sigma. Anti-RPE65 (NFIT-KVNPETLETIK) antibody was obtained from Genmed. Histagged recombinant cellular retinaldehyde-binding protein (rCRALBP) pET 19b starter culture and anti-CRALBP antibody were generous gifts from John Crabb (Cleveland Clinic, Cleveland, OH). LB medium was obtained from Invitrogen. Nickel-nitrilotriacetic acid (NTA) resin and nickel-NTA spin column were purchased from Qiagen (Valencia, CA). The precast gels (4–20%) for SDS/PAGE, BenchMark prestained, and Magic molecular mass markers were from Invitrogen. DEAE Sepharose was from Amersham Pharmacia. Buffers were changed by dialysis in the request buffer overnight in a slide-a-lyser cassette from Pierce (10-kDa molecular mass cutoff). RPE65 solutions were concentrated with an Amicon Ultra centrifugal filtration device (30-kDa cutoff) from Millipore. All reagents were analytical grade unless specified otherwise.
Purification of RPE65. The purification of the membrane associated form of RPE65 was carried out as described (24). All of the studies performed here are on this form of RPE65. The purity of RPE65 was verified by silver staining or Coomassie staining and Western blot analysis (1:4,000 primary antibody, 1 h at room temperature, and 1:4,000 secondary antibody, 0.5 h at room temperature).
Purification of rCRALBP. Purification was performed as described (25). The purity of these proteins were verified by silver staining or Coomassie staining and Western blot analysis (1:4,000 primary antibody, 1 h at room temperature, and 1:4,000 secondary antibody, 0.5 h at room temperature).
Fluorescence Binding Assays. RPE65 in PBS/1% CHAPS, pH 7.4 was used in the fluorometric titration studies. Protein concentrations were measured by a modified Lowry method (26). All titrations were performed at 25°C. The samples in PBS buffer were excited at 280 nm, and the fluorescence was scanned from 305–500 nm. Fluorescence measurements, using 450-μl quartz cuvettes with a 0.5-cm path length, were made at 25°C on a Jobin Yvon Fluoromax 2 employing the right-angle detection method.
The fluorescence of the protein solution was measured after equilibrating it at 25°C for 10 min. The sample was then titrated with a solution of retinoid dissolved in dimethyl sulfoxide. For each titration, to a 250-μl solution of the protein was added an equal amount of retinoid, typically 0.2 μl, and thoroughly mixed before allowing it to equilibrate for 10 min before recording the fluorescence intensity. The addition of dimethyl sulfoxide (0.1% per addition) did not have any effect on the fluorescence intensity. The binding constant (KD) was calculated from the fluorescence intensity by using the following equation (7):
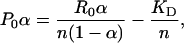 |
[1] |
where P0 is the total protein concentration, α = Fmax - F/Fmax - F0, n is number of independent binding sites, R0 is the total retinoid concentration at each addition, KD is dissociation constant, Fmax is the fluorescence intensity at saturation, and F0 is the initial fluorescence intensity.
Competitive Binding of Retinoic Acid (all-trans and 13-cis) and all-trans-Retinyl Palmitate to RPE65. Buffer exchange experiments were performed to investigate the abilities of the retinoic acids (all-trans and 13-cis) to displace all-trans-retinyl palmitate binding from RPE65. To RPE65 (0.5 μM) (PBS/1% CHAPS, pH 7.4) was added 6 μM retinoic acid (all-trans and 13-cis), and the mixture was incubated at 4°C for 30 min. A control sample of RPE65 was incubated minus retinoic acids at 4°C for 30 min. At the end of this incubation, the samples were incubated for 30 min with [3H]all-trans-retinyl palmitate (0.65 μM, 20.31 Ci/mmol). At the end of this incubation period, the buffer (PBS/1% CHAPS) was exchanged 104-fold with a Centricon 30-kDa molecular mass cutoff filter. The sample retained and the buffer flow through were counted on a liquid scintillation counter to measure the amount of [3H]all-trans-retinyl palmitate retained.
Effect of atRA, 13cRA, and 4-HPR on IMH. To 1 ml of buffered suspension of RPE membranes (100 mM Tris, pH 8.0/76.7 μgof protein) was added 60 μMor6 μM atRA, 13cRA, or 4-HPR, and the mixture was incubated at room temperature for 15 min. A control reaction mixture without any inhibitor was also incubated at room temperature for 15 min. At the end of the 15-min incubation, all-trans-retinol [11,12-3H2] (0.2 μM) was added to the reaction mixtures 0[100 mM Tris, pH 8.0/76.7 μg of RPE protein/0.2% BSA/100 μM l-α-dipalmitoylphosphatidylcholine (DPPC)/1 mM DTT/0.2 μM all-trans-retinol [11,12-3H2]] and incubated at room temperature for 30 min. At the end of this 30-min incubation, aliquots of the reactions were quenched to verify the equal addition of all-trans-retinol [11,12-3H2] and the effect of these inhibitors on LRAT. After this, the control reaction mixture was incubated with atRA (60 and 6 μM), 13cRA (60 and 6 μM), or 4-HPR (60 and 6 μM) for 15 min. Then, all of the reaction mixtures were incubated with 30 μM apo-rCRALBP [100 mM Tris, pH 8.0/7.7 μg of RPE protein/0.2% BSA/100 μM DPPC/1 mM DTT/30 μM apo-rCRALBP/0.2 μM all-trans-retinol [11,12-3H2]] at 37°C for 30 min. At the end of this incubation period, the 200-μl reaction mixture was quenched by the addition of 750 μl of ice-cold methanol, after which 100 μl of 1 M sodium chloride solution was added, and 500 μl of hexane (containing butylated hydroxy toluene at 1 mg/ml) was added to effect extraction of the retinoids. The retinoids were analyzed as described (27). The amount of 11-cis-retinol formed was used as a measurement of IMH activity. All experiments were performed in triplicate, and the average values of these measurements were used for analysis.
Results
Specific Binding of the Retinoic Acids to RPE65. In the current study, bovine RPE65 was purified by standard methods (24), and its interaction with atRA, 13cRA, and 4-HPR was quantitatively measured by fluorescence methods as previously reported to reveal the specific binding of all-trans-retinyl esters to this protein (7). Fig. 1 shows data for the specific binding of atRA to purified RPE65. As shown in Fig. 1 A, the binding of atRA to RPE65 led to an exponential decay in protein fluorescence. This decay follows a saturable binding isotherm (Fig. 1B), and yields an average KD for binding of ≈109 nM (SD = 10 nM, n = 4) (Fig. 1C). Similar data are shown in Fig. 2 for 13cRA, yielding an average KD for binding of ≈195 nM (SD = 20 nM, n = 4) (Fig. 2C). These experiments show that both retinoic acids specifically bind to RPE65 with high affinities. By way of comparison, under the same binding conditions, all-trans-retinyl palmitate binds to RPE65 with a KD of 47 nM (data not shown). To further assess specificity of binding, the binding interactions of an additional retinoid, 4-HPR (Scheme 3) with RPE65 was studied. The binding of 4-HPR to RPE65 is expected to be considerably weaker than for the retinoic acids, because analogs in the retinamide series only weakly induce night blindness (28). In fact, 4-HPR binds rather weakly to RPE65. Data shown in Fig. 3 yield an average KD for binding of ≈3,547 nM (SD = 280 nM, n = 4) (Fig. 3C). Thus, the observed weak binding for 4-HPR is what is predicted by the hypothesis that RPE65 is the night blindness target.
Fig. 1.

Fluorescence titration of RPE65 (0.6 μM) with atRA. The excitation wavelength was at 280 nm and emission was observed through a 0.5-cm layer of solution. The titration solution consisted of 0.6 μM of RPE65 in 100 mM PBS (150 mM), pH 7.4, and 1% CHAPS. (A) The emission spectra of RPE65 with increasing concentrations of atRA. (B) The change in the fluorescence intensity at 338 nM with increasing concentrations of atRA. (C) The linear square fit plots for the titration of RPE65 vs. atRA.
Fig. 2.

Fluorescence titration of RPE65 (0.6 μM) with 13cRA. The excitation wavelength was at 280 nm, and emission was observed through a 0.5-cm layer of solution. The titration solution consisted of 0.6 μM of RPE65 in 100 mM PBS (150 mM), pH 7.4, and 1% CHAPS. (A) The emission spectra of RPE65 with increasing concentrations of 13cRA. (B) The change in the fluorescence intensity at 338 nM with increasing concentrations of 13cRA. (C) The linear square fit plots for the titration of RPE65 vs. 13cRA.
Fig. 3.

Fluorescence titration of RPE65 (0.6 μM) with 4-HPR. The excitation wavelength was at 280 nm, and emission was observed through a 0.5-cm layer of solution. The titration solution consisted of 0.6 μM RPE65 in 100 mM PBS (150 mM), pH 7.4, and 1% CHAPS. (A) The emission spectra of RPE65 with increasing concentrations of 4-HPR. (B) The change in the fluorescence intensity at 338 nM with increasing concentrations of 4-HPR. (C) The linear square fit plots for the titration of RPE65 vs. 4-HPR.
Retinoic Acid Displaces All-trans-Retinyl Palmitate from RPE65. The direct binding studies reported above for the retinoic acids are not determinative as to whether these molecules are competitive with the binding of all-trans-retinyl esters, the physiologically relevant ligands of RPE65 (7). This can readily be shown by prebinding [3H]all-trans-retinyl palmitate to RPE65 and showing that atRA competes with the binding (Fig. 4). This experiment was performed by first incubating RPE65 with atRA or 13cRA and (-) retinoic acid (control), after which the protein was incubated for 30 min with [3H]all-trans-retinyl palmitate. Excess retinoids were removed by buffer exchange, and the flow through and retained solutions were counted by using a liquid scintillation counter. The data show that the retinoic acids and all-trans-retinyl esters apparently compete for the same binding site on RPE65. The next issue to address is whether the retinoic acids interfere with 11-cis-retinol formation in an in vitro system and whether the nature of the putative inhibition is consistent with the inhibition of RPE65.
Fig. 4.

Competitive binding of retinoic acid (all-trans and 13-cis) and all-trans-retinyl palmitate. RPE65 (0.5 μM) in PBS/1% CHAPS was incubated with the retinoic acid (6 μM) for 30 min and then with [3H]all-trans-retinyl palmitate (0.65 μM) for 30 min, and then the buffer (PBS) was exchanged (104-fold, Centricon 30-kDa molecular mass cutoff). Column A is the total radioactivity of all-trans-retinyl palmitate added, column B is the radioactivity of RPE65 retained, and column C is the activity in the flow through. Each experiment was done in triplicate, the average value was used, and SD is displayed as error bars.
Retinoic Acids Inhibit RPE65 Function. RPE65 is the chaperone for all-trans-retinyl esters and, as such, is essential for the mobilization of these hydrophobic molecules for isomerization (7, 8). In the current study, a bovine retinal pigment epithelial membrane system is used to process added all-trans-retinol (vitamin A) to form 11-cis-retinol (27). Because RPE65 is essential for the biosynthesis of 11-cis-retinol (4, 8, 11), RPE65 inhibitors could block this synthesis. In the experiments shown in Fig. 4, the off-rate for the binding of all-trans-retinyl esters to RPE65 is sufficiently slow to allow the complex to survive centrifugation on Centricon spin columns. The same is true for atRA (data not shown). This finding suggests that the order of incubation of inhibitors of RPE65 would be expected to be important to reveal effective inhibition. Preincubation of these membranes with vitamin A rapidly produces all-trans-retinyl esters through rapid esterification mediated by LRAT (2). The synthesized all-trans-retinyl esters are tightly bound to RPE65, and then processed into 11-cis-retinol by IMH (7, 8). This system is not susceptible to inhibition by atRA or 13cRA incubated at 60 μM (data not shown). This is the expected result because the retinoic acids are known not to directly inhibit IMH (21). However, preincubation with the retinoic acids produced a markedly different result, as shown in Fig. 5. In this case, substantial inhibition of 11-cis-retinol formation occurs in the presence of the retinoic acids because they have access to RPE65. Interestingly, the inhibition observed with 4-HPR proved to be substantially weaker (Fig. 5). This is the expected result, given its relatively weak affinity for RPE65.
Fig. 5.

Effect of atRA, 13cRA, and 4-HPR on 11-cis-retinol biosynthesis. These experiments were performed as generally indicated in Materials and Methods. NoI shows the data for the absence of inhibitor, atRA (60 μM) shows the data for the preincubation of RPE membranes with 60 μM atRA, atRA (6 μM) shows the data for the preincubation of RPE membranes with 6 μM of atRA, 13cRA (60 μM) or (6 μM) shows the data for the preincubation of RPE membranes with 60 or 6 μM 13cRA, and 4-HPR (60 μM) or (6 μM) shows the data for the preincubation of RPE membranes with 60 or 6 μM 4-HPR. Each experiment was done in triplicate, the average value was used, and SD is displayed as error bars.
Discussion
The in vitro experiments described here make it highly likely that the retinoic acids induce night blindness by blocking the processing of all-trans-retinyl esters into 11-cis-retinol as a consequence of their interference with RPE65 function. The experiments show that both 13-cis and all-trans-retinoic acid competitively and saturably bind to RPE65 with high affinities. The binding is competitive with all-trans-retinyl palmitate, a natural ligand of RPE65 (7). Moreover, the retinoic acids also interfere with RPE65 function in vitro. Finally, 4-HPR, a relatively weakly binding retinoic acid analog, did not significantly impair 11-cis-retinol formation. The results from these in vitro experiments are also consistent with other data on the effects of 13cRA on the visual cycle, and components thereof (20, 22).
That RPE65 is the primary target of retinoic acid action (because 13cRA and atRA are interconverted in vivo, we will refer to them as retinoic acid; ref. 29) is consistent with the available information on the ocular effects of this drug. Other possible visual cycle targets (Scheme 1) do not seem to be relevant. LRAT is not a target for two reasons. First, in vivo data show that retinoic acid strongly inhibits the resynthesis of 11-cis-retinoids, subsequent to bleaching with light, without inhibiting all-trans-retinyl ester synthesis (20, 22). Secondly, it has been directly shown that the retinoic acids are not inhibitors of LRAT (21). IMH is not a target for retinoic acid either (21). This result is reconfirmed here as well. Thus, neither LRAT nor IMH are inhibited, and the inhibition of 11-cis-retinol dehydrogenase by itself would not prevent overall 11-cis-retinoid biosynthesis, but just affect the partitioning between 11-cis-retinol/11-cis-retinal. In this regard, it is interesting to note that studies on the 11-cis-retinol dehydrogenase knockout in mice do not reveal a critical role for this enzyme in visual pigment regeneration (30). By comparison, 11-cis-retinoids are not synthesized in RPE65 knockout mice, revealing an essential role for this protein in mediating rhodopsin regeneration (11). Therefore, the inhibition of RPE65 by retinoic acid would be expected to interfere with 11-cis-retinoid synthesis. This interference also strongly implies that RPE65 is involved in the rate-limiting step in the visual cycle.
The results reported here may have important medical implications in the treatment of certain retinal and macular degenerative disorders that would benefit from limiting visual cycle turnover because an essential target has been identified. It should be readily possible to design potent and specific antagonists of RPE65 that do not share the toxic side effects associated with the use of the retinoic acids (16). Conversely, the medical utility of the retinoic acids could be expanded by readily screening retinoid analogs for binding to RPE65. Those retinoids that do not bind to RPE65 will not produce night blindness.
Acknowledgments
This work was supported by National Institutes of Health Grant EY-04096.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: LRAT, lecithin retinol acyl transferase; IMH, isomerohydrolase; RPE, retinyl pigment epithelium; 13cRA, 13-cis-retinoic acid; atRA, all-trans-retinoic acid; 4-HPR, N-(4-hydroxyphenyl)retinamide; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; CRALBP, cellular retinaldehyde-binding protein.
References
- 1.Rando, R. R. (2001) Chem. Rev. 101, 1881-1896. [DOI] [PubMed] [Google Scholar]
- 2.Trehan, A., Cañada, F. J. & Rando, R. R. (1990) Biochemistry 29, 309-312. [DOI] [PubMed] [Google Scholar]
- 3.Gollapalli, D. R. & Rando, R. R. (2003) Biochemistry 42, 5809-5818. [DOI] [PubMed] [Google Scholar]
- 4.Moiseyev, G., Crouch, R. K., Goletz, P., Oatis, J., Jr., Redmond, T. M. & Ma, J. X. (2003) Biochemistry 42, 2229-2238. [DOI] [PubMed] [Google Scholar]
- 5.Batten, M. L., Imanishi, Y., Maeda, T., Tu, D. C., Moise, A. R., Bronson, D., Possin, D., Van Gelder, R. N., Baehr, W. & Palczewski, K. (2004) J. Biol. Chem. 279, 10422-10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jahng, W. J., David, C., Nesnas, N., Nakanishi, K. & Rando, R. R. (2003) Biochemistry 42, 6159-6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gollapalli, D. R., Maiti, P. & Rando, R. R. (2003) Biochemistry 42, 11824-11830; and correction 43, 7226. [DOI] [PubMed] [Google Scholar]
- 8.Mata, N. L., Moghrabi, W. N., Lee, J. S., Bui, T. V., Radu, R. A., Horwitz, J. & Travis, G. H. (2004) J. Biol. Chem. 279, 635-643. [DOI] [PubMed] [Google Scholar]
- 9.Hamel, C. P., Tsilou, E., Pfeffer, B. A., Hooks, J. J., Detrick, B. & Redmond, T. M. (1993) J. Biol. Chem. 268, 15751-15757. [PubMed] [Google Scholar]
- 10.Bavik, C. O., Levy, F., Hellman, U., Wernstedt, C. & Eriksson, U. (1993) J. Biol. Chem. 268, 20540-20546. [PubMed] [Google Scholar]
- 11.Redmond, T. M., Yu, S., Lee, E., Bok, D., Hamasaki, D., Chen, N., Goletz, P., Ma, J. X., Crouch, R. K. & Pfeifer, K. (1998) Nat. Genet. 20, 344-351. [DOI] [PubMed] [Google Scholar]
- 12.Simon, A., Hellman, U., Wernstedt, C. & Eriksson, U. (1995) J. Biol. Chem. 270, 1107-1112. [PubMed] [Google Scholar]
- 13.Shaban, H. & Richter, C. (2002) Biol. Chem. 383, 537-545. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Shabat, S., Parish, C. A., Vollmer, H. R., Itagaki, Y., Fishkin, N., Nakanishi, K. & Sparrow, J. R. (2002) J. Biol. Chem. 277, 7183-7190. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Shabat, S., Itagaki, Y., Jockusch, S., Sparrow, J. R., Turro, N. J. & Nakanishi, K. (2002) Angew. Chem. Int. Ed. Engl. 41, 814-817. [DOI] [PubMed] [Google Scholar]
- 16.Asif, M., Egan, J., Vasan, S., Jyothirmayi, G. N., Masurekar, M. R., Lopez, S., Williams, C., Torres, R. L., Wagle, D., Ulrich, P., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 2809-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., Gerrard, B., Baird, L., Stauffer, D., Peiffer, A., et al. (1997) Nat. Genet. 15, 236-246. [DOI] [PubMed] [Google Scholar]
- 18.Sun, H. & Nathans, J. (1997) Nat. Genet. 17, 15-16. [DOI] [PubMed] [Google Scholar]
- 19.Weng, J., Mata, N. L., Azarian, S. M., Tzekov, R. T., Birch, D. G. & Travis, G. H. (1999) Cell 98, 13-23. [DOI] [PubMed] [Google Scholar]
- 20.Radu, R. A., Mata, N. L., Nusinowitz, S., Liu, X., Sieving, P. A. & Travis, G. H. (2003) Proc. Natl. Acad. Sci. USA 100, 4742-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law, W. C. & Rando, R. R. (1989) Biochem. Biophys. Res. Commun. 161, 825-829. [DOI] [PubMed] [Google Scholar]
- 22.Sieving, P. A., Chaudhry, P., Kondo, M., Provenzano, M., Wu, D., Carlson, T. J., Bush, R. A. & Thompson, D. A. (2001) Proc. Natl. Acad. Sci. USA 98, 1835-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi, Y.-Q., Furuyoshi, S., Hubacek, T. & Rando, R. R. (1993) Biochemistry 32, 3077-3080. [DOI] [PubMed] [Google Scholar]
- 24.Ma, J. X., Zhang, J., Othersen, K. L., Moiseyev, G., Ablonczy, Z., Redmond, T. M., Chen, Y. & Crouch, R. K. (2001) Invest. Ophthalmol. Vis. Sci. 42, 1429-1435. [PubMed] [Google Scholar]
- 25.Crabb, J. W., Chen, Y., Goldflam, S., West, K. & Kapron, J. (1998) Methods Mol. Biol. 89, 91-104. [DOI] [PubMed] [Google Scholar]
- 26.Lowry, Rosebrough, N. J., Farr, A. L. & Randall, R. J. (1951) J. Biol. Chem. 193, 265-275. [PubMed] [Google Scholar]
- 27.Winston, A. & Rando, R. R. (1998) Biochemistry 37, 2044-2050. [DOI] [PubMed] [Google Scholar]
- 28.Abou-Issa, H. Curley, R. W., Jr., Alshafie, G. A., Weiss, K. L., Clagett-Dame, M., Chapman, J. S. & Mershon, S. M. (2001) Anticancer Res. 21, 3839-3844. [PubMed] [Google Scholar]
- 29.Tsukada, M. Schroder, M., Roos, T. C., Chandraratna, R. A., Reichert, U., Merk, H. F., Orfanos, C. E. & Zouboulis, C. C. (2000) J. Invest. Dermatol. 115, 321-327. [DOI] [PubMed] [Google Scholar]
- 30.Driessen, C. A. Winkens, H. J., Hoffmann, K., Kuhlmann, L. D., Janssen, B. P., Van Vugt, A. H., Van Hooser, J. P., Wieringa, B. E., Deutman, A. F., Palczewski, K., et al. (2000) Mol. Cell. Biol. 20, 4275-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Norris, A. W., Cheng, L., Giguère, V., Rosenberger, M. & Li, E. (1994) Biochim. Biophys. Acta 1209, 10-18. [DOI] [PubMed] [Google Scholar]
- 32.Kochhar, D. M. & Christian, M. S. (1997) J. Am. Acad. Dermatol. 36, S47-S59. [DOI] [PubMed] [Google Scholar]