

Abstract
GABAergic cortical interneurons, derived from the embryonic medial and caudal ganglionic eminences (MGE and CGE), are functionally and morphologically diverse. Inroads have been made in understanding the roles of distinct cortical interneuron subgroups, however, there are still many mechanisms to be worked out that may contribute to the development and maturation of different types of GABAergic cells. Moreover, altered GABAergic signaling may contribute to phenotypes of autism, schizophrenia and epilepsy. Specific Cre-driver lines have begun to parcel out the functions of unique interneuron subgroups. Despite the advances in mouse models, it is often difficult to efficiently study GABAergic cortical interneuron progenitors with molecular approaches in vivo. One important technique used to study the cell autonomous programming of these cells is transplantation of MGE cells into host cortices. These transplanted cells migrate extensively, differentiate, and functionally integrate. In addition, MGE cells can be efficiently transduced with lentivirus immediately prior to transplantation, allowing for a multitude of molecular approaches. Here we detail a protocol to efficiently transduce MGE cells before transplantation for in vivo analysis, using available Cre-driver lines and Cre-dependent expression vectors. This approach is advantageous because it combines precise genetic manipulation with the ability of these cells to disperse after transplantation, permitting greater cell-type specific resolution in vivo.
Keywords: Developmental Biology, Issue 98, MGE, interneuron, transplantation, lentivirus, cell labeling, somatostatin, Cre
Introduction
GABAergic cortical interneurons comprise ~20-30% of neurons in the mammalian neocortex, while the rest correspond to excitatory, glutamatergic principal neurons. Interneurons are highly diverse in electrophysiological properties, axon and dendrite morphology and synaptic targeting 1, and imbalances in excitatory/inhibitory tone are hypothesized to underlie some phenotypes of neurological/neuropsychiatric disorders including autism, schizophrenia and epilepsy 2. The overall goal of the protocol described herein is to provide a means to efficiently genetically modify GABAergic cortical interneuron progenitors before transplantation for in vivo analyses.
Cortical GABAergic interneurons are born in the medial and caudal ganglionic eminences (MGE and CGE, respectively) 3,4 as well as the preoptic area 5. Cortical interneuron progenitors undergo long-distance tangential migration followed by radial migration to reach their final targets. Upon arrival at their destinations, these cortical interneurons must correctly integrate into the existing neuronal network, and each unique interneuron subgroup will contribute to cortical circuitry in specific ways. Four main subgroups can be distinguished by molecular markers: MGE-derived somatostatin (SST)+ and parvalbumin (PV)+ subgroups, and CGE-derived vasoactive intestinal peptide (VIP)+ and Reelin+;SST- subgroups 6. Different cortical interneuron subgroups are born over different times during embryonic development in the MGE and CGE 7, 8. These and other cortical GABAergic interneuron markers have been used to generate specific Cre-driver lines for many of these subgroups 9-11.
The transplantation of MGE progenitors has emerged as a potential cell-based therapy to treat disorders that may be caused by imbalances in excitation/inhibition 12–24. These therapeutic benefits may be due in part to the unique ability of MGE progenitors (to disperse, differentiate and integrate into a host brain), or potentially because many peri-somatic inhibitory PV+ cells are derived from the MGE. MGE cells can also be quickly and efficiently transduced with lentiviruses before transplantation 15, allowing cells that are genetically modified in vitro to be studied in vivo. The rationale for developing this approach was to overcome roadblocks in studying GABAergic cortical interneuron development and maturation. In particular, MGE transplantation allowed researchers to study the development of mutant cells in vivo, when the mutant mouse would have otherwise died at an early time point. Moreover, by introducing genes of interest before transplantation, the effects of specific genes on a mutant phenotype could be assessed in an efficient manner.
Here, we provide a detailed protocol to transduce MGE cells with lentiviruses prior to transplantation. In addition, we show how this technique can be adapted to express a gene of interest in specific interneuron subgroups from a heterogeneous group of cortical interneuron precursors, using a combination of Cre-dependent expression lentiviruses and available Cre-driver mouse lines. Moreover, this protocol introduces techniques and a platform for researchers to genetically modify GABAergic cortical interneuron precursors for in vivo studies in a unique way. One advantage of this technique over other current approaches is that the transplanted MGE cells will disperse away from the injection site. Also, unlike focal viral injections, after MGE cells disperse their morphology is easier to assess. This approach can be used to study the effect of introducing genes of interest into wild type or mutant cells, introducing a cell type specific reporter to assess morphology, or potentially to study the effect of disease alleles in vivo.
Protocol
Ethics statement: The following procedures have been approved by our institution and animal protocol. Make sure to get approval for all procedures involving survival surgeries before beginning experiments and verify all protocols are up to date.
1. Lentivirus Preparation (Optional Step)
Split three 10 cm plates of HEK293T cells for each lentivirus to be made, in the presence of 10 ml DMEM/10% FBS, and grow to ~60-70% confluence. Grow cells in an incubator at 37 °C with 5% CO2.
Transfect the following DNA plasmids (10 µg total) into each plate using any transfection reagent: 6.4 µg CAG-Flex-GFP lentiviral vector (DNA vector sequence provided in Table 1), 1.2 µg pMD2.G (encodes VSV-g), 1.2 µg pRSV-Rev, and 1.2 µg pMDLg/pRRE. The three lentiviral packaging vectors are commercially available (Table Note: This particular approach utilizes 3rd generation lentiviral vectors but 2nd generation vectors and associated plasmids will also work.
Prepare a waste container containing a 10% bleach solution within a BSL2 certified hood to inactivate any solutions or devices that contacted or contain lentivirus. Next, completely remove the media 4-6 hr after transfection into the bleach solution, replace with the same volume of new media, and allow the cells to grow.
After 4 days, collect media into 50 ml conical tubes and centrifuge at 1,000 x g for 15 min to pellet any cell debris. Next, filter the supernatant through a 0.45 µm membrane. Any low protein binding filter, like PVDF, works well. Caution: place both solid and liquid viral waste into the bleach solution.
Load 30 ml filtered supernatant into ultracentrifuge tubes and into an ultracentrifuge. Centrifuge at 100,000 x g for 2.5 hr at 4 °C. Open the centrifuge tubes in a BSL2 hood and remove supernatant into 10% Bleach. Add ~100 µl of PBS to the pellet, which yields titers of 1x10^7-8 infectious units/ml. Aliquot the following day and store at -80 ºC. Lentivirus aliquots are stable at -80 °C for at least six months.
Important consideration: Many institutions now have viral cores. Acquire concentrated virus with a minimum titer of 1x107 infectious units/ml, for optimal results.
2. Donor Mice for MGE Dissection
First, set up a timed mating between a Cre-expressing line of interest and either a wild type (WT) mouse or a mouse that will express a reporter after Cre-mediated recombination. Note: Many Cre-driver lines are transgenics and are maintained and bred in the hemizygous state. Thus, only half of the embryos will contain the Cre transgene. DNA can be quickly prepared from the embryos for a short PCR to detect the Cre allele. The Cre+ embryos can then be harvested so that only the MGE tissue of the desired genotype is used. Alternatively, a Cre-dependent reporter may be used, to select the embryos for MGE dissection and transplantation.
Calculate which day will correspond to E13.5. If animals were paired the evening before, the day of the plug is 0.5. Order timed-pregnant mice or a WT female with P1 pups in order to time having postnatal (P)1 mice on the day the donor tissue will be E13.5. Alternatively, set up these matings in house one week prior to pairing the donor animals. Note: The former strategy of generating both E13.5 and P1 embryos/pups for the same day is often difficult to do reliably.
3. Preparation of Media, Tools and Equipment.
- Prepare the reagents and tools for MGE cell preparation (Table 2).
- Prepare a clean surface and place clean forceps, scissors and either a microsurgical knife or fine forceps (for precise tissue dissection) on the surface.
- Prepare Hank's balanced salt solution (HBSS), for dissection, and Dulbeco's modified eagle's medium (DMEM) with 10% fetal bovine surem (FBS), for transduction.
- Preincubate 20-30 ml DMEM/10% FBS in a 37 °C tissue culture incubator for about 1 hr. The lid should be loosened to allow for gas exchange and pH equilibration of the media. Note: The media preincubation can be prepared just before starting the dissection.
4. MGE Cell Preparation
Sacrifice the pregnant mouse according to an approved animal protocol and remove the embryos into a 10 cm petri dish containing ice-cold HBSS.
- Remove the brain from each embryo using a dissecting scope (do one at a time, leaving the rest of the embryos in ice-cold HBSS) and then proceed to cut out the MGE tissue. Note: Detailed below, and in Figure 1, are key steps showing how to remove the MGE. In addition, Figure 2 is a movie showing the entire dissection procedure.
- Position the brain with ventral side facing up (Figures 1A, A'). Note: it is also okay to have the dorsal aspect facing up. Cut the brain in half along the saggital plane to generate two pieces. An example hemisected brain is shown (Figures 1B, B'), notice that dorsal (the overlying cortex) and ventral aspects of the brain cover and surround the ganglionic eminence (GE) tissue.
- Next, with a dominant hand, hold either very fine forceps or a stabknife (Table 2), while immobilizing the hemisphere with forceps held by a non-dominant hand. (The hemisphere's medial aspect should be facing up). Unfold the dorsal and ventral tissue with forceps and a stab knife to reveal the underlying GE tissue (Figure 1C and diagrammed in Figure 1C'). Note: Removing some media from the dissection petri dish can make it easier to stabilize the tissue while performing the MGE dissection.
- Make straight cuts with the stabknife or fine forceps to separate the CGE, LGE and septal tissues (see example cuts denoted by red dashed lines, Figures 1D and D'). These cuts can be made in any order. Note: Imagine the MGE as a 'box' that is cut out of the surrounding tissue. Doing so helps reproducibly make similar cuts for each dissection, thus reducing variability in tissue dissections.
- Finally, turn the MGE on its side and trim off the bottom (what was the lateral aspect of the brain). This removes the mantle zone of the MGE (discard in Figures 1E and 1E') from the rest of the MGE. The remaining aspect of MGE contains primarily ventricular zone and subventricular zone portions of the MGE, which contains nearly all the MGE progenitor cells. Proceed with this tissue.
Important consideration: The addition of silicone gel to the bottom of a petri dish can serve as an excellent dissection surface as it protects the delicate tips of forceps and provides a soft surface for pinning tissue with forceps.
4.3 Additional strategies for MGE Dissection.
Cut the skin of the embryo using forceps like scissors to quickly remove the brain. As the embryo lays on its side, hold the tissue at the base of the head with the forceps used to anchor the tissue. First, cut anterior ventral to the brain and posterior ventral to the brain, and then connect the two cuts along the side of the embryo. Then grab the flap of skin and pull it over the top of the brain. The brain can then be quickly dissected away from the remaining tissue.
Alternatively, anchor the posterior aspect of the whole brain behind the cortex. Meanwhile, grasp each hemisphere at the most dorsal caudal aspect of the cortex and tear the cortex away from the anchored tissue in an anterior direction. In this way, both cortices can be removed and the bilateral ganglionic eminences are revealed, still attached to the rest of the tissue preserving the medial-lateral anatomy.
Switch to the stab-knife to make precise cuts around the MGE on each hemisphere. Use either a separate clean set of forceps to collect the MGE or a pipette with a large tip (i.e., P1000). Note: Keep the collected tissue away from all other dissected material, as some media is unavoidably transferred when the MGE is collected.
Collect the MGE and put the tissue into a 1.5 ml collection tube containing DMEM/10% FBS, (a volume of 500 µl is sufficient to collect the MGEs). Repeat for the other hemisphere and place in the same collection tube. Keep samples on ice until all tissue is collected.
5. Lentiviral labeling and Transplantation
Move the tubes of MGE tissue into a BSL2 certified hood and remove the media. Note: The MGE tissue is big enough to be visible to the eye and will settle to the bottom of the tube allowing for the cold media to be easily and gently removed.
Add ~500 ul of DMEM/10% FBS media, that was preincubated in a 37 °C tissue culture incubator. Next, add polybrene to facilitate transduction to each tube at a final concentration of 8 µg/ml. Triturate the MGE tissue to create a single cell suspension, using a P1000 pipette tip. Finally, add ~15-20 µl of concentrated lentivirus to each tube.
Important consideration: A trituration of 10-12 times is sufficient to break up MGE tissue from a single embryo, but more triturations may be required if multiple MGEs are pooled together. Try out different trituration conditions to determine what works best.
Securely close each tube, invert to mix, than place all the tubes in a 37 °C incubator. Incubate the cells in lentivirus for at least 30 min and up to 1 hr. Longer times have resulted in decreased cell viability. Invert the tubes every 10 min until the time is up.
After incubation with lentivirus, remove the tubes from the incubator and centrifuge at ~700 x g for 3 min to pellet the cells. In the BSL2 hood, remove the supernatant and discard into 10% bleach. Next, add 1 ml DMEM/10% FBS and triturate the pellet 2-3 times to wash, then centrifuge again at the same speed to pellet the cells. Repeat this wash-step 2-3 more times to remove excess virus. Note : Perform centrifugation steps at 4 °C, however, reduced viability has not been observed when short spins are performed at RT.
After the final wash, remove as much media as possible and put each tube on ice. Due to residual media on the sides of the tube, ~2-3 µl of media will end up covering the cell pellet by the time the transplantation procedure has begun.
6. Transplantation and Validation
Acquire host P1 pups and prepare the procedure area by spraying down with 70% ethanol. To maintain sterility during the procedure it is recommended to perform these procedures in a dedicated clean procedure room. In addition, use either autoclaved surgical tools sterilize surfaces that the pups will be in contact with using 70% ethanol. An example of representative equipment needed to perform the transplantations and a representative procedure area is shown in Figure 3.
Note: While this procedure is an injection of small volumes, and not a surgical procedure that would require an incision, open wound or sutures, it is still recommended that a new micropipette is used for each mouse to be injected. The micropipettes are heat sterilized when pulled and beveled on a surface that was sprayed with 70% ethanol being stored in a sealed container.
Generate micropipettes to have a diameter at the tip between 40-80 µm. Following these dimensions will yield optimal results. Heat sterilize the micropipettes when pulled and beveled on a surface and spray with 70% ethanol before storing the micropipettes in a sealed container. Note: Alternatively, if access to the devices needed to create these needles (Table 2) is limiting, use any other injection device. However, these may not be as well suited as the micropipettes due to different diameters.
- Draw up mineral oil into a 1 ml syringe, and with a 30 1/2 G needle, fill the glass pipette completely with mineral oil. Next, mount the plunger onto the stereotaxic device, then attach the glass pipette to the stereotaxic device and load onto the plunger. Using the hydraulic drive, move the plunger about halfway into the glass pipette, removing mineral oil that spills out with a clean a clean paper towel.
- Alternatively to using a syringe, submerge the back end of the pipette in mineral oil and filled by capillary action. To prevent air bubbles in the pipette, maintain positive pressure on the syringe until completely out of the glass pipette. Note: Other devices can be used to successfully transplant MGE cells 14. Any device that can deliver a volume less than or near 100 nl works well for this procedure.
Next, use a sterile paper towel, or any absorbent material, with the corner twisted into a fine point to delicately remove excess media above the MGE cell pellet. This step concentrates the cell density so that smaller injection volumes can be achieved. Next, use a low volume (P2 is preferred) pipette to slowly draw up the cell pellet and triturate 1-2 times before moving the cell suspension onto a hydrophobic surface. The volume should be just under 1 µl. Move the tip of the needle into the cell suspension and using the hydraulic drive, draw up the cell suspension into the pipette.
Anaesthetize a P1 pup via hypothermia (wrap in a latex glove and put on ice for ~ 2-4 min). Check for effectiveness of anesthesia with a noxious stimulus. Pinch the skin between the toes with fine forceps: the pup should be unresponsive if pinched. Next, place the pup onto a mold that is under the injection device, then make the skin taut by pulling back the skin on the head and securing with standard lab tape. Note: While hypothermia is very effective on neonatal mice and results in low mortality rate, procedures must be done quickly, as pups will begin to recover in under 10 min. In addition, 4 min on ice is an upper limit of what can be tolerated. Try to only induce hypothermia once if a longer time is required for injections. However, if a pup recovers early, first allow the pup to fully recover before inducing hypothermia again. Moreover, only induce hypothermia one more time to perform injections. Pups will recover within 5-10 min of hypothermia. This can be facilitated by placing the pups with litter and mom or by placing the pup on a warm surface to recover.
Using a stereotaxic device, position the tip of the glass pipette on the surface on the pup's head, perpendicular to the surface of the head. When the pipette is in contact with the head, consider this as '0'. Next, push the pipette through the surface of the skin and skull into the cortex, then insert and retract the pipette ~ 2 times before stopping. For optimal targeting of cells into the neocortex, injections are performed when the micropipette is at a depth of 0.1 mm. However, this should be optimized by the user (see note below). Note: While this depth reliably targets deeper neocortical layers with the devices described above, a few depths should be tested determined empirically. In addition, a range of depths at different rostro-caudal and medio-lateral position should be tested to determine what depth is optimal at each site. Also, the initial puncture should be rapid, as slow penetration into the neocortex diminishes the ability to cleanly penetrate the tissue. Try different speeds when first starting this procedure to find what works best. In addition, for testing coordinates, there is no real substitute for using labeled cells, as dyes have been unreliable to denote position.
Once the needle is in position, rotate the hydraulic device to advance the plunger into the glass pipette, pushing a set volume of cell suspension into the cortex. Inject 50-70 nls per site. Repeat this procedure at 3-6 different sites at different rostral-caudal levels if the goal is to obtain a wide distribution of cells throughout the neocortex. Note: Volumes of 100 nl or greater can cause lesions and lead to large clumps of injected cells that fail to efficiently migrate out of the injection site. Thus, smaller injection volumes (~70 nl or less) are optimal, over more sites if time permits.
When injections are complete, remove the pup from the stage and mark it (toe clipping is a reliable way to identify the pups later). Once the pup has recovered and is able to move on its own (this occurs within minutes of removing them from the stage), put it back with the mom and litter. Note: Since this is a minimally invasive procedure there are no post-surgical procedures once the pup is freely moving and warmed. However, pups should be checked not only after the procedure but also the following day to assure they have no signs of deteriorating health.
Before starting the procedure on the next pup, spray down the area with 70% ethanol to maintain a sterile working area. Allow the injected pups to develop and then assess the tissue at the appropriate developmental stage(s).
Representative Results
Since MGE cells have the unique ability to migrate and integrate when transplanted into a host neocortex 16, they provide an excellent model system for genetic manipulation before in vivo studies. Herein, we show how one can isolate MGE tissue from E13.5 embryos (Figures 1 and 2), which can then be transduced with lentiviruses either in vitro or in a rapid manner before transplantation for in vivo studies. Labeling MGE via lentiviruses has been performed before using an enhancer driving GFP in GABAergic neurons 15. However, the ability to express genes in specific subgroups from a heterogeneous population before transplantation may greatly aid studies of these cells' dispersal and integration. Herein, we sought to develop a means to target expression of a fluorescent protein to a specific interneuron subgroup, those that express somatostatin (SST)+.
We first subcloned a commercially available Cre-dependent GFP reporter (vector information, Table 2) into a lentiviral backbone. The vector was generated by first subcloning the cassette containing GFP flanked by loxP sites from the AAV vector and ligating it into a lentiviral vector backbone 15 using XbaI and PflMI restriction sites, (this cassette contained only some of the CAG promoter). Next, the remainder of the CAG promoter was excised from a pCAGGs mammalian expression vector with SpeI and XbaI sites and ligated into the XbaI site of the lentiviral vector to generate pLenti-CAG-Flex-GFP. The 5' SpeI site of the insert is complimentary to XbaI and thus destroyed the 5' site, while the 3' XbaI site was conserved. The resulting vector (schema Figure 4A, vector sequence Table 1) was first tested in vitro in the presence or absence of Cre (schema of in vitro assay, Figure 4B). MGE primary neurons from Ai14Flox/+E13.5 embryos were cultured and transduced with a combination of lentiviruses. Few to no GFP+ or tdTomato+ cells were observed with only the transduction of the CAG-Flex-GFP lentivirus (Figures 4C, 4F and 4I). TdTomato fluorescence was present in the MGE cells transduced with a CMV-Cre lentivirus, indicative of Cre expression (Figures 4D, 4G and 4J). Finally, co-transduction of the MGE cells resulted in many tdTomato+ cells that also expressed GFP, indicating that the CAG-Flex-GFP lentivirus was working as expected (Figures 4E, 4H and 4K). Some rare GFP+ cells were observed that were not tdTomato+, potentially due to the transduced reporter incorporating next to a strong promoter, which accounted for < 2% of all the GFP+ cells.
MGE cells transplanted into a wild type host neocortex disperse, mature and integrate into the host neocortex 16. E13.5 SST-IRES-Cre+;Ai14Flox/+MGE cells were transplanted into WT neocortices at P1 (schema, Figure 5A) and their distribution examined at 7 and 35 days post transplant (DPT). At both ages, tdTomato+ cells could be seen throughout the neocortex (Figures 5B and 5C). The images in Figures 5B and 5C are representative transplants utilizing the MGE cells from a single embryo. To test how efficiently MGE cells could be labeled with the protocol described herein, MGE cells from SST-IRES-Cre+;Ai14Flox/+ embryos were transduced with the CAG-Flex-GFP lentivirus and transplanted in the same manner. For these experiments, ~15 ul of concentrated lentivirus (~1x107 infectious units/ml) was utilized with the combined MGEs from one embryo for each transplant. The cells were then transplanted into a P1 WT host cortex and analyzed at 7 DPT (Figures 5D-5F). Of the transplanted cells (tdTomato+), approximately 20% were GFP+, indicating transduction with the CAG-Flex-GFP lentivirus (Figure 5G). These data suggest that with the amount and titer of lentivirus described above, ~20% of transplanted MGE cells can be labeled. The amount of virus can either be decreased or increased, or potentially other variables like duration of viral incubation can be altered, to achieve sparser or denser cell labeling.
Using MGE tissue from a Cre-dependent reporter mouse line in conjunction with the CAG-Flex-GFP lentivirus can limit the number of additional markers that can be probed for by immunofluorescence. Therefore, SST-IRES-Cre+ E13.5 MGE tissue was collected and transduced with CAG-Flex-GFP lentivirus in order to visualize those transplanted MGE cells that expressed Cre (will be GFP+). These cells were transplanted into P1 WT cortex and assessed at 35 DPT (see experimental design, Figure 6A), a time point when mature interneuron markers are expressed. At 35 DPT, ~85% of GFP+ cells co-expressed SST, but only ~4% expressed PV (Figures 6B-6H). These numbers are consistent with the lineage analysis of the SST-IRES-Cre mouse line, which also fate maps to ~5-10% of PV+ cells 9 (Vogt and Rubenstein, unpublished results). These data suggest that viral labeling of MGE is efficient and can be an applicable approach to introducing a reporter or potentially a gene of interest before in vivo analysis.
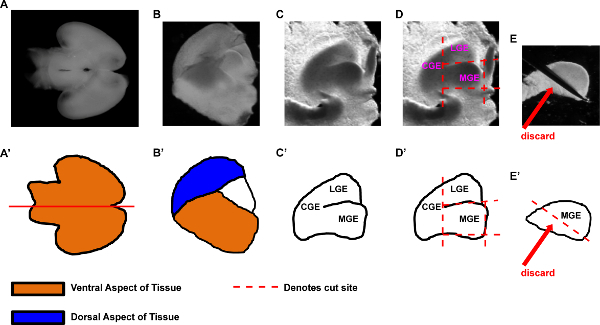 Figure 1. Representative procedure for the dissection of E13.5 MGE tissue. Example dissection of MGE tissue for either primary cultures or transplantations. (A-E) Images of E13.5 brain dissections, and (A'-E') schematics to highlight structures and important steps in the procedure. (A, A') Representative E13.5 brain viewed from the ventral aspect. Red line denotes the first cut made, which will hemisect the brain into two pieces. (B, B') View of one brain hemisection. The medial aspect is visible, with lateral aspect down. Tissue from the dorsal cortex (blue color) overlies the ganglionic eminences. In addition, ventral tissue that will be removed is shown in orange. (C, C') View and illustration of the exposed ganglionic eminences after overlying tissue has been peeled away. (D, D') Same view of exposed ganglionic eminences as in (C, C') but with detailed cut sites shown as red dashed lines. After MGE is cut out according to the lines denoted in (D, D'), the tissue is turned on its side (E, E') and the lateral aspect is trimmed off and discarded. Representative steps in the dissection and isolation of MGE tissue. Abbreviations: (CGE) caudal ganglionic eminence, (LGE) lateral ganglionic eminence, (MGE) medial ganglionice eminence. Please click here to view a larger version of this figure.
Figure 1. Representative procedure for the dissection of E13.5 MGE tissue. Example dissection of MGE tissue for either primary cultures or transplantations. (A-E) Images of E13.5 brain dissections, and (A'-E') schematics to highlight structures and important steps in the procedure. (A, A') Representative E13.5 brain viewed from the ventral aspect. Red line denotes the first cut made, which will hemisect the brain into two pieces. (B, B') View of one brain hemisection. The medial aspect is visible, with lateral aspect down. Tissue from the dorsal cortex (blue color) overlies the ganglionic eminences. In addition, ventral tissue that will be removed is shown in orange. (C, C') View and illustration of the exposed ganglionic eminences after overlying tissue has been peeled away. (D, D') Same view of exposed ganglionic eminences as in (C, C') but with detailed cut sites shown as red dashed lines. After MGE is cut out according to the lines denoted in (D, D'), the tissue is turned on its side (E, E') and the lateral aspect is trimmed off and discarded. Representative steps in the dissection and isolation of MGE tissue. Abbreviations: (CGE) caudal ganglionic eminence, (LGE) lateral ganglionic eminence, (MGE) medial ganglionice eminence. Please click here to view a larger version of this figure.
Figure 2. Movie showing an E13.5 MGE dissection. Movie depicts the removal of an E13.5 brain followed by subsequent unfolding and removal of the overlying tissue. The MGE is labeled and the cuts made to remove surrounding tissue are show in a stepwise order.Please click here to view this video.
 Figure 3. Representative procedure area and example of injection needle for neonatal transplantations.(A, A') Images showing an example setup to perform transplantations into neonatal mice. A microscope (1) is positioned above a stage containing a mold that can hold a neonatal mouse (5) and a stereotaxic device (2). (A') An enlarged image of (A) showing the region of the stereotaxic device that holds the glass injection needle (6) with an inserted plunger (7). The plunger is controlled by a fine hydraulic drive (4) and moves mineral oil within the glass needle that mediates inward and outward flow from the needle. (B) Example of a glass injection needle with a beveled tip. For the ruler in (B), each major demarcation = 100 µm. (C) Inventory list of items shown in (A, A'). Please click here to view a larger version of this figure.
Figure 3. Representative procedure area and example of injection needle for neonatal transplantations.(A, A') Images showing an example setup to perform transplantations into neonatal mice. A microscope (1) is positioned above a stage containing a mold that can hold a neonatal mouse (5) and a stereotaxic device (2). (A') An enlarged image of (A) showing the region of the stereotaxic device that holds the glass injection needle (6) with an inserted plunger (7). The plunger is controlled by a fine hydraulic drive (4) and moves mineral oil within the glass needle that mediates inward and outward flow from the needle. (B) Example of a glass injection needle with a beveled tip. For the ruler in (B), each major demarcation = 100 µm. (C) Inventory list of items shown in (A, A'). Please click here to view a larger version of this figure.
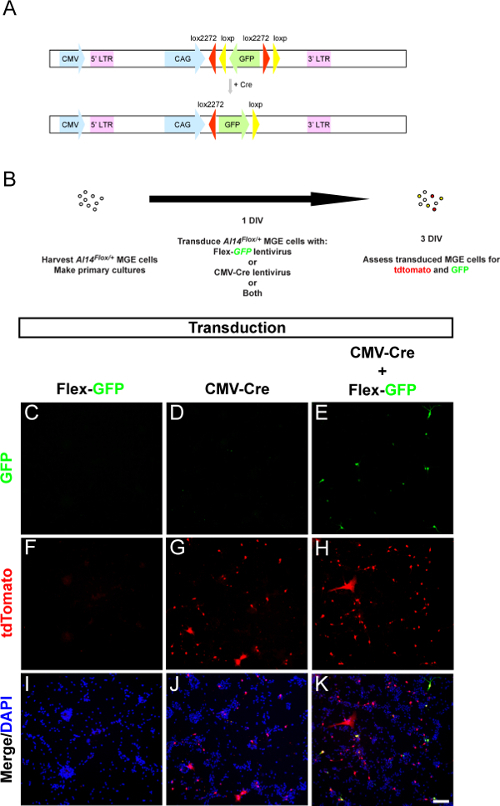 Figure 4. In vitro tests demonstrate the expression of GFP from a CAG-Flex-GFP lentivirus in the presence of Cre.(A) Schema of how the lentiviral CAG-Flex-GFP vector works in the presence of Cre. (B) Schema of primary MGE cell culture experiment to assess if the lentiviral CAG-Flex-GFP would express GFP in those cells with Cre expression. Briefly, MGE cells from a Cre-dependent reporter, Ai14 (expresses tdTomato in the presence of Cre), were grown in vitro and transduced with Cre and/or Flex-GFP lentiviruses. (C-K) Immunofluorescent images of E13.5 MGE primary cultures that were transduced with a CMV-Cre, CAG-Flex-GFP or both. Cultures were imaged for native tdTomato expression, GFP and DAPI. Scale bar in (K) = 100 µm. Please click here to view a larger version of this figure.
Figure 4. In vitro tests demonstrate the expression of GFP from a CAG-Flex-GFP lentivirus in the presence of Cre.(A) Schema of how the lentiviral CAG-Flex-GFP vector works in the presence of Cre. (B) Schema of primary MGE cell culture experiment to assess if the lentiviral CAG-Flex-GFP would express GFP in those cells with Cre expression. Briefly, MGE cells from a Cre-dependent reporter, Ai14 (expresses tdTomato in the presence of Cre), were grown in vitro and transduced with Cre and/or Flex-GFP lentiviruses. (C-K) Immunofluorescent images of E13.5 MGE primary cultures that were transduced with a CMV-Cre, CAG-Flex-GFP or both. Cultures were imaged for native tdTomato expression, GFP and DAPI. Scale bar in (K) = 100 µm. Please click here to view a larger version of this figure.
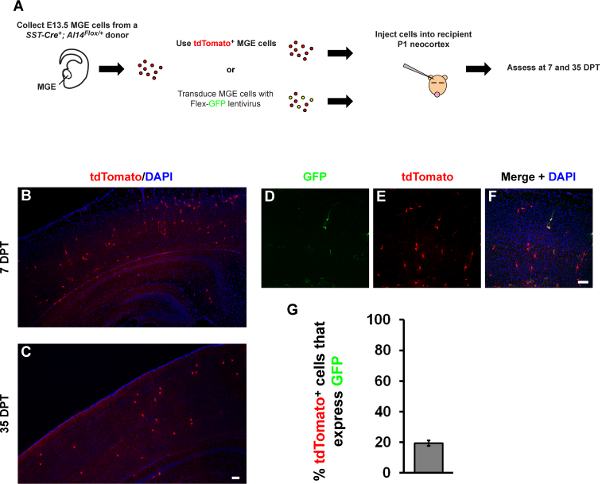 Figure 5. Representative images of transplanted and transduced, SST-IRES-Cre+; Ai14Flox/+ MGE cells.(A) Schema of experimental design to transplant either MGE cells alone or after lentiviral transduction into a wild type (WT) host, (DPT) days post transplant. Briefly, E13.5 SST-IRES-Cre+;Ai14Flox/+ MGE cells were either transplanted or transduced with the CAG-Flex-GFP lentivirus before transplantation into WT neocortices and assessed at 7 DPT. (B, C) Immunofluorescent images of neocortices at 7 or 35 DPT showing representative MGE transplanted cells (tdTomato+). (D-F) Immunofluorescent images of MGE cells transduced with the CAG-Flex-GFP lentivirus assessed at 7 DPT. (G) Quantification of the proportion of tdTomato+ cells that express GFP at 7 DPT. Scale bars in (C) and (F) = 100 µm. Please click here to view a larger version of this figure.
Figure 5. Representative images of transplanted and transduced, SST-IRES-Cre+; Ai14Flox/+ MGE cells.(A) Schema of experimental design to transplant either MGE cells alone or after lentiviral transduction into a wild type (WT) host, (DPT) days post transplant. Briefly, E13.5 SST-IRES-Cre+;Ai14Flox/+ MGE cells were either transplanted or transduced with the CAG-Flex-GFP lentivirus before transplantation into WT neocortices and assessed at 7 DPT. (B, C) Immunofluorescent images of neocortices at 7 or 35 DPT showing representative MGE transplanted cells (tdTomato+). (D-F) Immunofluorescent images of MGE cells transduced with the CAG-Flex-GFP lentivirus assessed at 7 DPT. (G) Quantification of the proportion of tdTomato+ cells that express GFP at 7 DPT. Scale bars in (C) and (F) = 100 µm. Please click here to view a larger version of this figure.
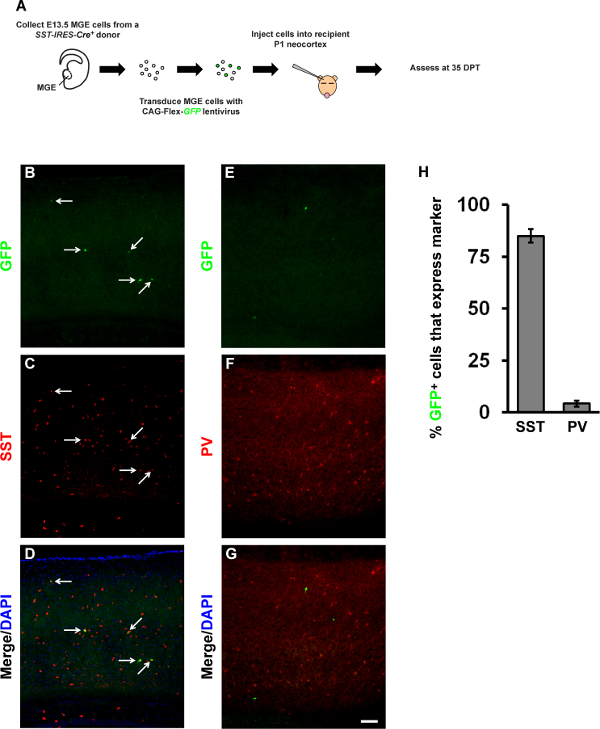 Figure 6. Representative images of CAG-Flex-GFP lentivirus transduced, SST-IRES-Cre+ MGE transplants that co-express MGE-derived interneuron subgroup markers.(A) Schema of E13.5 SST-IRES-Cre+ MGE cell harvest and transduction with a CAG-Flex-GFP lentivirus and transplantation into the neocortex of P1 WT hosts before assessment at 35 DPT. Representative images from the neocortex of transplanted hosts showing expression of GFP, from the Flex vector (B, E), co-stained for either somatostatin (SST) (C) or parvalbumin (PV) (F). (D, G) Merged images co-stained with DAPI. (H) Quantification of the proportion of GFP cells that co-express SST or PV. Data represent ± SEM, (n) = 3. Scale bar in (G) = 100 µm. Please click here to view a larger version of this figure.
Figure 6. Representative images of CAG-Flex-GFP lentivirus transduced, SST-IRES-Cre+ MGE transplants that co-express MGE-derived interneuron subgroup markers.(A) Schema of E13.5 SST-IRES-Cre+ MGE cell harvest and transduction with a CAG-Flex-GFP lentivirus and transplantation into the neocortex of P1 WT hosts before assessment at 35 DPT. Representative images from the neocortex of transplanted hosts showing expression of GFP, from the Flex vector (B, E), co-stained for either somatostatin (SST) (C) or parvalbumin (PV) (F). (D, G) Merged images co-stained with DAPI. (H) Quantification of the proportion of GFP cells that co-express SST or PV. Data represent ± SEM, (n) = 3. Scale bar in (G) = 100 µm. Please click here to view a larger version of this figure.
Table 1. FASTA file of the pLentiviral-CAG-Flex-GFP DNA vector.
| Reagents and equipment for MGE cell preparation and transduction | Catalog # | Company | ||||||
| 1) | Dulbecco's Modified Eagle Medium, with high glucose | 12491-015 | Life Technologies | |||||
| 2) | Heat inactivated Fetal Bovine Serum | 10437-077 | Life Technologies | |||||
| 3) | Hanks balanced salt solution (HBSS), no calcium or magnesium | 14170-112 | Life Technologies | |||||
| 4) | 1.5 ml microcentrifuge tubes (or other collection tubes with lids) | 3810X | Eppendorf International | |||||
| 5) | Polybrene | sc-134220 | SantaCruz Biotechnology | |||||
| 6) | Stab knife straight 22.5 ° (optional) | REF 72-2201 | Surgical specialities corporation | |||||
| 7) | Petri dish (10 cm), for tissue dissections | FB0875713 | Fisher Scientific | |||||
| 8) | 2 fine tip forceps, like Dumont #5 | 11254-20 | Fine Scientific Tools | |||||
| 9) | 37 °C tissue culture incubator with 5% CO2 input | C150 | Binder | |||||
| 10) | Tabletop centrifuge that can spin 1.5 ml microcentrifuge tubes | |||||||
| 11) | Any P1000 pipette that can be used for cell trituration | |||||||
| 12) | Biosafety level 2 hood | |||||||
| Reagents and equipment for in vitro MGE primary cultures | Catalog # | Company | ||||||
| 1) | Lab-TekII chamberslides with cover, 8 well | 154941 | Thermo Fisher | |||||
| 2) | Poly-L-lysine 0.1% weight/volume | P8920 | Sigma Aldrich | |||||
| 3) | Mouse Laminin, 0.5-2 mg/ml | 23017-015 | Life Technologies | |||||
| 4) | Neurobasal medium | 21103-049 | Life Technologies | |||||
| 5) | B27 serum free supplement | 17504044 | Life Technologies | |||||
| 6) | Glutamax, 100x stock | 35050-061 | Life Technologies | |||||
| 7) | Penicillin-Streptomycin, 100x stock | 15070-063 | Life Technologies | |||||
| 8) | Glucose, (prepare a 25% solution in water) | G5400 | Sigma Aldrich | |||||
| Reagents and equipment for MGE cell transplantation | Catalog # | Company | ||||||
| 1) | 1 ml syringe | REF 309602 | BD, Becton Dickinson and company | |||||
| 2) | 30 1/2 G needle | 305106 | BD, Becton Dickinson and company | |||||
| 3) | Precision bore to deliver 5 µl (comes with plunger) | 5-000-1005 | Drummond scientific company | |||||
| 4) | Parafilm | PM-999 | Polysciences, Inc. | |||||
| 5) ** | Stereo microscope with boomstand | MZ6 | Leica | |||||
| 6) ** | Digital just for mice stereotaxic instrument | 51725D | Stoelting company | |||||
| 7) ** | Single-axis oil hydraulic fine micromanipulator | MO-10 | Narishige | |||||
| 8) | Diamond coated rotary beveler | n.a. | made in house | |||||
| 9) | Needle pipette puller | 730 | Kopf instruments | |||||
| 10) | Mineral oil | n.a. | Any drug store or pharmacy | |||||
| 11) | Any pipette and tips that can reliaby measure 1 µl of volume | |||||||
| ** These are the particular models we use, but many other setups should work | ||||||||
| Optional Reagents (for making Lentiviruses) | Catalog # | Company | ||||||
| 1) | 25 mm, 0.45 µm filter | 09-719B | Fisher Scientific | |||||
| 2) | Ultra-clear centrifuge tubes (25 x 89 mm) | 344058 | Beckman Coulter® | |||||
| 3) | Lipofectamine 2000 | (1.5 ml size) | 11668019 | Invitrogen | ||||
| Other commercially available supplies | Catalog # | Company | ||||||
| 1) | pMDLg/pRRE plasmid encoding gag/pol | 12251 | Addgene | |||||
| 2) | pRSV-Rev plasmid encoding Rev | 12253 | Addgene | |||||
| 3) | pMD2.G plasmid encoding VSVG | 12259 | Addgene | |||||
| 4) | pAAV-Flex-GFP | 28304 | Addgene |
Table 2. Inventory list of reagents and tools. An inventory of commercially available reagents including media, dissection tools, representative equipment for transplantation and DNA vectors. (n.a.) Not available or applicable.
Discussion
The use of GABAergic cortical interneuron precursors from the embryonic ganglionic eminences (GEs) for cell based therapies is showing promise for many conditions 12–14. Precise molecular techniques are needed to track, and express genes of interest in specific interneuron subgroups. Here we provide a detailed protocol for labeling embryonic MGE cells with lentiviruses before transplantation and show how this technique can be used to express genes of interest in specific cortical interneuron subgroups for in vivo analysis.
The protocol described herein can be reliably used, however, for optimal reproducibility it is essential to adhere to a few critical steps. First, for viral transduction to be efficient, physiological temperature and pH are necessary. If these parameters are met, MGE cells can be transduced in ~30 minutes. Second, make sure to have a dense MGE pellet before proceeding to do a transplantation. This is important because only a limited volume can be injected into the neonatal mouse brain at each site without incurring damage, and the cells will migrate away from the injection site, so their density at the time of transplant is less of a concern than might be expected. Finally, run some test conditions with MGE cells that express a fluorescent reporter to determine what injection depths are optimal. While this protocol used MGE cells, CGE cells could also be used in the same manner. One drawback is that there are not many Cre-driver lines that restrict expression to the whole CGE or to particular subgroups of CGE-derived interneurons. However, the vasoactive intestinal peptide (VIP) 9 is a reliable Cre-line that could be used to separate out this subgroup of CGE-derived cells. As new lines become available, it will be possible to employ this protocol with more genetic precision.
Previously, we and others have shown the utility of introducing genes into MGE cells before transplantation via electroporation or with lentivirus 15, 17. While these techniques were effective at introducing genes for in vivo analysis, MGE tissue is heterogeneous and gives rise to multiple interneuron subgroups as well as non-neuronal cells 4. Moreover, it has been difficult to express a gene of interest only in a specific interneuron subgroup. Great strides have been made to generate transgenic lines that recapitulate the expression patterns of many cortical interneuron subgroups, including, but not limited to, SST-IRES-Cre, PV-IRES-Cre, PV-2A-Cre and VIP-Cre9-11, and the advent of new lines will help elucidate additional possible populations in the future. Moreover, with the generation of Cre-dependent reporters and expression vectors, including Cre-dependent reporter vectors used herein, a more precise expression of genes of interest can be achieved.
While injection of Cre-dependent reporter viruses into the brains of mice expressing Cre in restricted cells is a powerful method, viruses do not spread far from the site of injection. This may lead to a dense area of overlapping cells making it difficult to study the transduced cells in an individual fashion or away from the injection sites. In contrast, MGE cells have the unique ability to migrate away from the injection site, allowing for a cleaner assessment of the properties of individual cells. A drawback of injecting cells into the neocortex is that they will not traverse the normal tangential migration routes. One option to explore tangential migration using this assay is to prepare the cells in the same manner but inject them into an age-matched MGE of a WT host embryo instead of a neonatal pup. In this experiment, the transplanted cells would undergo the normal tangential routes. By combining MGE transplantation with the spatially-restricted, Cre-dependent expression of a reporter, we show the feasibility of expressing a gene of interest or reporter within a specific subgroup of cells that are themselves well suited for morphological studies.
One limitation to this technique may be the size of the fragment that one can reliably package into a lentiviral vector. In addition, this approach has not yet been tested with AAVs. However, once this technique is optimized by an individual, many variations of this approach may be used. First, the approach described here will allow researchers to express reporters using a Cre-dependent reporter lentivirus in conjunction with MGE donor cells of specific Cre-driver lines. This technique may be used to assess the morphology of individual MGE cells or potentially to express a gene of interest in different subgroups if a second gene is cloned into the vector. Second, it may also be feasible to use a Cre-expressing virus with Cre-dependent reporter MGE donor cells or potentially utilize evolving conserved DNA elements, promoters and enhancers, to drive expression in distinct populations of cells from a heterogeneous population.
A future hurdle to overcome will be labeling human-derived induced pluripotent stem cells or embryonic stem cells to potentially purify and/or study a specific group of cells. Interestingly, conserved DNA enhancers may also be used in viral expression systems to target GABAergic neurons in primary cultures, after injection into the cortex, and before transplantation of MGE cells 15, 18, 19. Enhancers may be an answer to this obstacle and will be of interest as molecular tools, as new techniques are required to study the utility of these cells in therapy. Indeed, inroads to discovering enhancers that could potentially label either GABAergic interneurons 20 or other neuronal subtypes, including glutamatergic neurons 21, are under way. Using the viral labeling method herein, it may be possible to transduce differentiated human-derived stem cells with cell-type specific enhancers to label distinct cells of interest before transplantation and in vivo analysis.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by grants to JLRR from: Autism Speaks, Nina Ireland, Weston Havens Foundation, NIMH R01 MH081880, and NIMH R37 MH049428. PRW was supported by a fellowship from the National Science Council of Taiwan. SFS was supported by F32 (MH103003).
References
- Huang ZJ, Di Cristo G, Ango F. Development of GABA innervation in the cerebral and cerebellar cortices. Nat. Rev. Neurosci. 2007;8(9):673–686. doi: 10.1038/nrn2188. [DOI] [PubMed] [Google Scholar]
- Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, brain, and behavior. 2003;2(5):255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278(5337):474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 2006;7(9):687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Gelman D, et al. A Wide Diversity of Cortical GABAergic Interneurons Derives from the Embryonic Preoptic Area. J. Neurosci. 2011;31(46):16570–16580. doi: 10.1523/JNEUROSCI.4068-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, et al. Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci. 2010;30(5):1582–1594. doi: 10.1523/JNEUROSCI.4515-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Brito R, Fishell G. The developmental integration of cortical interneurons into a functional network. Curr. Top. Dev. Bio. 2009;87:81–118. doi: 10.1016/S0070-2153(09)01203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, Welagen J, Anderson SA. Spatial and Temporal Bias in the Mitotic Origins of Somatostatin. and Parvalbumin-Expressing Interneuron Subgroups and the Chandelier Subtype in the Medial Ganglionic. 2012;22(4):820–827. doi: 10.1093/cercor/bhr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, et al. A Resource of Cre Driver Lines for Genetic Targeting of GABAergic Neurons in Cerebral Cortex. Neuron. 2011;71(6):995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Bio. 2005;3(5):e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell DG, et al. Interneurons from embryonic development to cell-based therapy. Science. 2014;344(6180):1240622. doi: 10.1126/science.1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Girskis KM, Rubenstein JL, Alvarez-Buylla A, Baraban SC. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal. Nature Neuroscience. 2013;16(6):692–697. doi: 10.1038/nn.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JM, et al. Forebrain GABAergic Neuron Precursors Integrate into Adult Spinal Cord and Reduce Injury-Induced Neuropathic Pain. Neuron. 2012;74:663–675. doi: 10.1016/j.neuron.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, et al. Lhx6 Directly Regulates Arx and CXCR7 to Determine Cortical Interneuron Fate and Laminar Position. Neuron. 2014;82(2):350–364. doi: 10.1016/j.neuron.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dolado M, et al. Cortical inhibition modified by embryonic neural precursors grafted into the postnatal brain. J. Neurosci. 2006;26(4):7380–7389. doi: 10.1523/JNEUROSCI.1540-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Xu Q, Ocbina PJ, Anderson SA. NKX2.1 specifies cortical interneuron fate by activating Lhx6. Development. 2008;135(8):1559–1567. doi: 10.1242/dev.015123. [DOI] [PubMed] [Google Scholar]
- Arguello A, et al. Dapper Antagonist of Catenin-1 Cooperates with Dishevelled-1 during Postsynaptic Development in Mouse Forebrain GABAergic Interneurons. PLoS ONE. 2013;8(6):e67679. doi: 10.1371/journal.pone.0067679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, et al. Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron. 2014;81(1):61–68. doi: 10.1016/j.neuron.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJJ, et al. Use of “MGE Enhancers” for Labeling and Selection of Embryonic Stem Cell-Derived Medial Ganglionic Eminence (MGE) Progenitors and Neurons. PLoS ONE. 2013;8(5):e61956. doi: 10.1371/journal.pone.0061956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman K. Transcriptional regulation of enhancers active in protodomains of the developing cerebral cortex. Neuron. 2014;82(5):989–1003. doi: 10.1016/j.neuron.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]