

Abstract
During infection and inflammation, circulating monocytes leave the bloodstream and migrate into tissues, where they differentiate into macrophages. Macrophages express surface Toll-like receptors (TLRs), which recognize molecular patterns conserved through evolution in a wide range of microorganisms. TLRs play a central role in macrophage activation which is usually associated with gene expression alteration. Macrophages are critical in many diseases and have emerged as attractive targets for therapy. In the following protocol, we describe a procedure to isolate murine peritoneal macrophages using Brewer’s thioglycollate medium. The latter will boost monocyte migration into the peritoneum, accordingly this will raise macrophage yield by 10-fold. Several studies have been carried out using bone marrow, spleen or peritoneal derived macrophages. However, peritoneal macrophages were shown to be more mature upon isolation and are more stable in their functionality and phenotype. Thus, macrophages isolated from murine peritoneal cavity present an important cell population that can serve in different immunological and metabolic studies. Once isolated, macrophages were stimulated with different TLR ligands and consequently gene expression was evaluated.
Keywords: Immunology, Issue 98, Peritoneal cavity, macrophages, thioglycollate, infection, inflammation, TLRs, RNA extraction
Introduction
The reticuloendothelial phagocytic system is composed of cells in various tissues and organs such as bone marrow, blood, liver and spleen. Macrophages are extensively distributed around the body, where they notably participate in innate and adaptive immune responses to control and clear infections. In addition to their role in host defense, macrophages also play an important role in wound healing and in maintaining tissue homeostasis1,2. Furthermore, macrophages are not only important to immune function but also actively participate in iron homeostasis3. In the body, approximately 80% of iron is present in hemoglobin within erythrocytes, which when senescent are phagocytosed by macrophages4. Daily, these macrophages recycle 25 mg of erythrocyte-derived iron and provide its transport into the plasma5. Moreover, during infection and inflammation, pro-inflammatory macrophages sequester serum iron to reduce iron availability to pathogens, at both the systemic and local levels6-8. As well, studies have shown that macrophages and mainly hepatocytes produce an antimicrobial peptide named hepcidin that is considered the master regulator of iron metabolism9,10. Hepcidin is mainly increased by inflammatory stimuli and is partially responsible for iron sequestration in macrophage upon chronic inflammation11-13. As hepcidin expression in macrophages is not very well understood, we studied the possible role of Toll-like receptors (TLRs) in this regulation. The TLRs are primarily found on macrophages and play a central role in their activation. In addition, LPS induced hepcidin expression in the liver is dependent on TLR413. Therefore, to execute our study, we used a method based on the isolation of murine peritoneal macrophages.
Macrophage cell lines are broadly used in macrophage studies; nonetheless extended culture can provoke gene loss and decreased immune functions in these cell lines. Thus, isolation of macrophages from peritoneal cavity is crucial.
The mouse peritoneal cavity presents an ideal site to harvest macrophages13-15. Isolated murine peritoneal macrophages are convenient for several studies regarding their immunological function. However, the number of macrophages in the peritoneum is insufficient for extensive studies and is estimated around 1 x 106 macrophages per mouse. Thus, to raise macrophage output, a sterile eliciting agent such as thioglycollate was injected into the peritoneal cavity preceding the cell harvest. After thioglycollate injection, the yield of macrophages per mouse was increased by 10-fold. Despite the increase in macrophages yield, Brewer’s thioglycollate medium acts as an irritant that induces an inflammatory response, resulting in the recruitment of macrophages, which may but not necessary affect gene expression. Hence, a control group consisting of non-treated macrophages must be included in each experiment. In our hands, hepcidin expression which is highly stimulated by inflammation was not detected in non-treated thioglycollate elicited peritoneal macrophages. Moreover, studies have shown that Brewer’s thioglycollate recruits numerous macrophages, but does not activate them16. On the other hand, Brewer’s thioglycollate elicited macrophages showed an increase in lysosomal enzyme but a decrease in killing ingested microorganisms17. However, the phagocytic capacity was not affected when compared with non-elicited macrophages16.
Once cultured in dishes, the peritoneal macrophages become adherent, therefore allowing their separation from other type of cells isolated from the peritoneal cavity. Subsequently, the isolated macrophages were challenged with different TLRs agonists. Finally, mRNA was extracted from the cultured cells and gene expression was analysed using quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR).
Protocol
All procedures were performed in accordance with the Canadian Council on Animal Care guidelines after approval by the institutional Animal Care Committee of the Centre de recherche du Centre Hospitalier de l’Université de Montréal (CRCHUM).
1. Isolation, Identification, and Culture of Murine Peritoneal Macrophages
Prepare 3.8% brewer’s thioglycollate medium. To do so, suspend 38 g of thioglycollate medium in 1,000 ml of distilled water. Bring solution to boil to dissolve the medium completely. Sterilize by autoclaving at 121 °C for 15 min. Store up to 3 months in the dark, at RT18. NOTE: Discard if turbidity develops which indicates a bacterial contamination. The solution can be kept for up to one year in the dark if kept sterile.
Using 1 ml syringes attached to 23 G needles, inject 1 ml of 3.8% Brewer thioglycollate medium into the peritoneal cavity of each mouse and wait for 3 days. Use new syringe and needle for each mouse.
Anesthetize mice by intraperitoneal injection of sodium pentobarbital (100 mg/kg) and euthanize mice by cervical dislocation. Confirm proper anesthetization by checking the respiratory rate. Usually rapid respirations indicate that the mouse is not deeply anaesthetized.
Wash the abdomen of each mouse with 70% ethanol. Using a scissor, perform a lateral incision along the bottom midline of the peritoneum.
Using a forceps, pull back the abdominal skin to expose the transparent peritoneal skin. Using 5 ml syringes attached to 20 G needles, inject 5 ml of cold DPBS into the peritoneal cavity of each mouse.
Perform a gentle massage on the peritoneal cavity and then aspirate the fluid carefully without puncturing any organ. Remove the needle and dispense the peritoneal fluid into 50 ml conical centrifuge tubes.
Centrifuge for 10 min at 400 x g in a refrigerated centrifuge. Cells must stay cold during the whole procedure. Discard supernatant and resuspend cell pellet in RPMI medium 1640.
Using a hemocytometer, count cells and adjust to cell density to 1 x 106 cells/ml.
Characterize the phenotype of isolated cells by flow cytometry using 1 x 106 cells per mouse and antibodie against F4/80 (a surface antigen expressed on macrophages).
2. Cell Treatments
Directly, after isolation, add 1 x 106 cells into each well. Leave the murine peritoneal macrophages to adhere into the 6-well plates by culturing them for 1 to 2 hr at 37 °C. Remove non-adherent cells by gently washing 3 times with warm PBS.
Subsequently, culture cells in 900 µl serum-free DMEM for 24 hr in the presence of the following TLR ligands: (Pam3CSK4-TLR1/2 (0.5 mg/ml); Poly(I:C)-TLR3 (10 mg/ml); LPS-TLR4 (100 ng/ml); Flagellin-TLR5 (100 ng/ml); FSL1-TLR6/2 (100 ng/ml); ssRNA40-TLR7 (1 µg/ml); ODN1826-TLR9 (1 µM). NOTE: Prepare for each ligand a 10x stock solution. Add 100 µl of the latter to each well, thus performing a 10-fold dilution.
3. RNA Isolation
Remove all medium and lyse cells directly in the six-well plates by adding 1 ml TRIzol to each well and passing the cell lysate several times through a pipette. Incubate the homogenised samples for 5 to 10 min at RT, to allow the complete dissociation of nucleoprotein complexes.
Transfer lysate into 1.5 ml RNase- and DNase-free microcentrifuge tubes. Add 0.2 ml chloroform per 1ml TRIzol. Shake tubes vigorously by hand for 15 sec and incubate them at RT for 5 to 10 min. Centrifuge at 12,000 x g for 15 min at 4 °C. Observe the mixture separating into a lower red phase (phenol-chloroform), an interphase and a colorless upper aqueous phase. RNA remains exclusively in the aqueous phase.
Transfer carefully the upper aqueous phase to a fresh tube without disturbing the interphase. Precipitate the RNA from it by mixing with 0.5 ml of isopropyl alcohol. Incubate samples at RT for 10 min and centrifuge at 12,000 x g for 10 min at 4 °C. The RNA precipitates forming a white pellet on the side and bottom of the tube.
Remove supernatant. Wash the RNA pellet once with 1 ml of 75% ethanol. Mix the samples by vortexing and centrifuge at 7,500 x g for 5 min at 4 °C. Briefly dry the RNA (air dry for 10 min). Dissolve RNA in 0.1 ml DEPC (RNAse free water) and incubate for 10 min at 55 °C.
Quantify RNA using a spectrophotometer. To do so, dilute samples 100 times in DEPC water and read at wavelengths of 260 nm. The RNA concentration will then be: OD260 x 40 ng/ul x dilution factor.
Equalize RNA concentrations and synthesize cDNA as per manufacturer’s instructions using RT-PCR System for First-Strand cDNA Synthesis Kit. As per manufacturer’s instructions, measure gene mRNA levels by real-time PCR (45 cycles) in a Real-Time DNA detection system. Normalize gene expression levels with two housekeeping gene for example β-actin and GAPDH. NOTE: Normalize RNA concentration to 500 µg/ml and use 0.5 µg for cDNA synthesis.
Representative Results
We first characterized the isolated murine peritoneal macrophages by flow cytometry. To do so, we used (F4/80) antibodies that specifically recognize markers only expressed by macrophages. This characterization is required to determine the percentage of isolated macrophage and to distinguish them among cells obtained during the isolation process. As shown in (Figure 1), the percentage of cells expressing the antigen F4/80 was consistently found to be above 95%. Next, to study gene expression in macrophages, the isolated cells were treated with several TLR ligands: Pam3CSK4 (TLR1/2), LPS (TLR4) and FSL1 (TLR6/2). Subsequently, mRNA levels of Hepcidin (Hamp), our gene of interest, were measured by RT-PCR. As shown in (Figure 2), TLR1/2, TLR4 and TLR6/2 ligands were capable of stimulating hepcidin mRNA in murine macrophages19. Together, these results demonstrate the usefulness of this protocol to successfully isolate murine peritoneal macrophages and to investigate precisely the molecular regulation of gene expression.
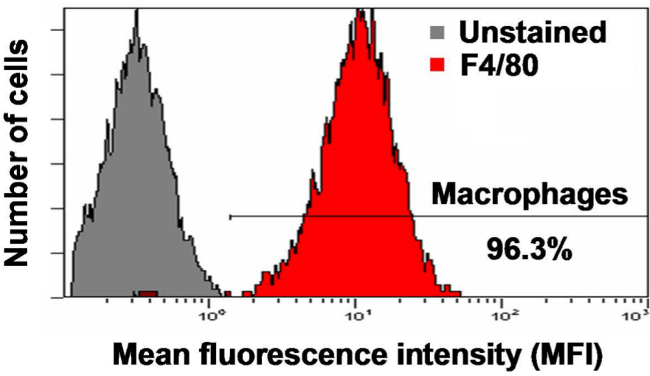 Figure 1. Characterisation of cells isolated from the peritoneal cavity. Enrichment of the recovered macrophages was confirmed by flow cytometric analysis using the F4/80 antibody after blocking nonspecific staining with CD16/CD32 antibodies and was consistently found to be above 95%.
Figure 1. Characterisation of cells isolated from the peritoneal cavity. Enrichment of the recovered macrophages was confirmed by flow cytometric analysis using the F4/80 antibody after blocking nonspecific staining with CD16/CD32 antibodies and was consistently found to be above 95%.
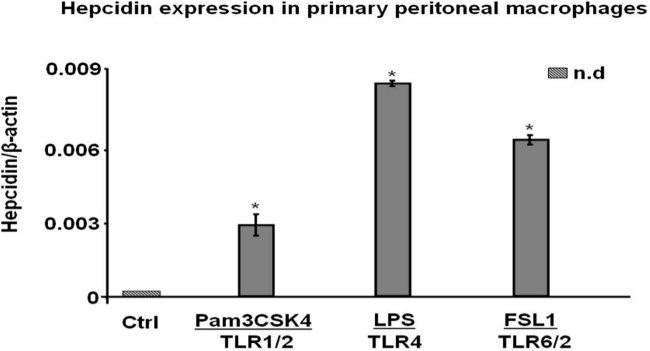 Figure 2. TLR ligands induce hepcidin expression in murine peritoneal macrophages. After murine peritoneal macrophages isolation and stimulation with TLR1/2, TLR4 and TLR6/2 ligands, hepcidin mRNA levels were studied by quantitative reverse transcriptase-polymerase chain reaction. Data are presented as mean ± SEM; n.d. (not detectable); *P < 0.05 versus control (Ctrl). Results are representative of 3 similar experiments performed independently.
Figure 2. TLR ligands induce hepcidin expression in murine peritoneal macrophages. After murine peritoneal macrophages isolation and stimulation with TLR1/2, TLR4 and TLR6/2 ligands, hepcidin mRNA levels were studied by quantitative reverse transcriptase-polymerase chain reaction. Data are presented as mean ± SEM; n.d. (not detectable); *P < 0.05 versus control (Ctrl). Results are representative of 3 similar experiments performed independently.
Discussion
Macrophages are crucial for survival and provide a tempting target to manipulate the host for immunological objectives. The discovery of TLRs and other recognition molecules have conducted the macrophages to the centre of immunological debate. Macrophages respond to a variety of stimuli, including cytokines, damage-associated molecular pattern molecules (DAMPs)20 and molecules associated with groups of pathogens (PAMPs)21. These different stimuli responses represent the course of macrophages activation, and are usually associated with sudden alterations in gene expression22.
In non-inflammatory conditions, the majority of macrophages reside in strategic locations in the body. They can be found in all tissues and as circulating monocytes in the blood. Therefore, macrophages are present in the most susceptible sites for microbial invasion.
The host defense is described as an inflammatory response to the elimination of intracellular pathogens associated with classical macrophage activation. The classical activation of macrophages is induced by microbial products such as lipopolysaccharides in a Th1 cytokine environment which will lead to pro-inflammatory M1-macrophages polarization. The persistence of inflammation results in tissue damage and the development of anti-inflammatory mechanisms necessary to the survival of the host. Therefore, Th2 cytokines allow then the introduction of anti-inflammatory M2-macrophages polarization which inhibits and regulates the M1 response, and also promotes tissue repair. In the protocol described in this paper, thioglycollate injection in the peritoneal cavity stimulates the classical inflammatory cascade and leads to the recruitment of M1 macrophages.
To date, several studies have been carried out using bone marrow, spleen or peritoneal derived macrophages. These macrophages represent heterogeneous populations with different activities. Based on their morphology and surface molecular characteristics, studies have established that peritoneal macrophages are more mature than bone marrow and spleen derived macrophages23. Unlike spleen and peritoneum derived macrophages, bone marrow derived macrophages present a remarkable ability in pahgocytosis and proliferation and can be completely differentiated from macrophage progenitor cells20,24,25. In addition, the isolation of bone marrow macrophages presents a homogenous yield with long lifespan. On the other hand, these macrophages are not fully characterized and their use in experimental studies presents complications due to the inconstancy of their phenotype and functions26 . Unlike bone marrow derived macrophages, spleen and peritoneum macrophages seem to be more functionally and phenotypically stable23 .
Hence, the isolation of murine peritoneal macrophages can serve different immunological studies and gene expression analysis. In addition, the mouse peritoneal cavity affords an ideal site for harvesting resident macrophages27. However, the elicited number is moderate and estimated around 1 x 106 macrophages per mouse. Thus, to increase the harvest of macrophages, eliciting agents such as thioglycollate was injected in the peritoneal cavity 3 days before cell isolation28. This agent will induce an inflammatory response and accordingly increase macrophage crop.
It’s imperative to perform a gentle massage on the peritoneal cavity before withdrawing slowly the peritoneal fluid without puncturing any organ. Pulling out the maximal possible volume is required to collect the largest cell number. In case of blood contamination, a lysis buffer can be used at the end of the procedure to discard red blood cells. The elicited macrophages can then be characterized by flow cytometry using antibodies against antigens that are unique to macrophages as F4/80.
Throughout the procedure, make sure that all reagents are endotoxin-free and all animals were permanently housed under specific pathogen-free conditions. Performing this procedure under pathogen-free conditions is critical because macrophage stimulation prior to impending experiments will alter significantly the results analysis.
Once isolated, the peritoneal macrophages can be used in several studies, including production of inflammatory cytokines, phagocytosis, cell signaling, gene expression, chemotaxis and toxicology29. For example, after stimulation with different TLR ligands, we investigated in the elicited macrophages the molecular regulation of a key regulator of iron metabolism named hepcidin. 24 hr after cell treatments, total RNA was isolated with TRIzol reagent, and gene expression was analysed using qRT-PCR.
As we are studying TLRs activation and gene expression, the use of serum free DMEM is recommended. Serum free medium reduces the degree of contaminants and eliminate any potential source of infectious agents.
Albeit RNA isolation seems a simple process, RNase and DNA contamination must be avoided to prevent RNA degradation and achieve accurate qRT-PCR results. The best way to detect DNA contamination is to include a 'minus-RT' control for each RNA sample in an RT-PCR experiment. If a PCR product is generated from an RNA sample that was not reverse transcribed then the product was amplified from contaminating DNA. In case of DNA contamination, the use of DNase treatment is possible. However, DNase must be completely inactivated prior to RT-PCR so that it doesn't degrade newly synthesized DNA.
Disclosures
The authors have no competing financial interests.
Acknowledgments
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC, grant no 298515-2011). AL is the recipient of a Ph.D. scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC), and MS was supported from a grant from the Canadian Institutes of Health Research (CIHR, grant no. MOP123246).
References
- Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Koury MJ, Ponka P. New insights into erythropoiesis: the roles of folate, vitamin B12, and iron. Annu Rev Nutr. 2004;24:105–131. doi: 10.1146/annurev.nutr.24.012003.132306. [DOI] [PubMed] [Google Scholar]
- Sheftel AD, Kim SF, Ponka P. Non-heme induction of heme oxygenase-1 does not alter cellular iron metabolism. J Biol Chem. 2007;282(14):10480–10486. doi: 10.1074/jbc.M700240200. [DOI] [PubMed] [Google Scholar]
- Hershko C. Storage iron regulation. Prog Hematol. 1977;10:105–148. [PubMed] [Google Scholar]
- Collins HL. Withholding iron as a cellular defence mechanism--friend or foe. Eur J Immunol. 2008;38(7):1803–1806. doi: 10.1002/eji.200838505. [DOI] [PubMed] [Google Scholar]
- Noyes WD, Bothwell TH, Finch CA. The role of the reticulo-endothelial cell in iron metabolism. Br J Haematol. 1960;6:43–55. doi: 10.1111/j.1365-2141.1960.tb06216.x. [DOI] [PubMed] [Google Scholar]
- Layoun A, Huang H, Calve A, Santos MM. Toll-like receptor signal adaptor protein MyD88 is required for sustained endotoxin-induced acute hypoferremic response in mice. Am J Pathol. 2012;180(6):2340–2350. doi: 10.1016/j.ajpath.2012.01.046. [DOI] [PubMed] [Google Scholar]
- Zumerle S, et al. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood. 2014;123(23):3646–3650. doi: 10.1182/blood-2014-01-550467. [DOI] [PubMed] [Google Scholar]
- Nguyen NB, Callaghan KD, Ghio AJ, Haile DJ, Yang F. Hepcidin expression and iron transport in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L417–L425. doi: 10.1152/ajplung.00484.2005. [DOI] [PubMed] [Google Scholar]
- Nemeth E, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Constante M, et al. Distinct requirements for Hfe in basal and induced hepcidin levels in iron overload and inflammation. Am J Physiol Gastrointest Liver Physiol. 2006;291(2):G229–G237. doi: 10.1152/ajpgi.00092.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14 11) doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Leijh PC, van Zwet TL, ter Kuile MN, van Furth R. Effect of thioglycolate on phagocytic and microbicidal activities of peritoneal macrophages. Infect Immun. 1984;46(2):448–452. doi: 10.1128/iai.46.2.448-452.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy M, Debray-Sachs M, Kamoun P, Hamburger J. Effect of thioglycollate on macrophage lysosomal enzymes. Biomedicine. 1978;29(5):167–170. [PubMed] [Google Scholar]
- Li YM, Baviello G, Vlassara H, Mitsuhashi T. Glycation products in aged thioglycollate medium enhance the elicitation of peritoneal macrophages. J Immunol Methods. 1997;201(2):183–188. doi: 10.1016/s0022-1759(96)00224-4. [DOI] [PubMed] [Google Scholar]
- Layoun A, Santos MM. Bacterial cell wall constituents induce hepcidin expression in macrophages through MyD88 signaling. Inflammation. 2012;35(4):1500–1506. doi: 10.1007/s10753-012-9463-4. [DOI] [PubMed] [Google Scholar]
- Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17(4):359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169(5):2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Wang C, et al. Characterization of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol. 2013;14(6) doi: 10.1186/1471-2172-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin PE, McCulloch EA, Till JE. Characterization of the factor in L-cell conditioned medium capable of stimulating colony formation by mouse marrow cells in culture. J Cell Physiol. 1971;77(2):121–134. doi: 10.1002/jcp.1040770202. [DOI] [PubMed] [Google Scholar]
- Cannon GJ, Swanson JA. The macrophage capacity for phagocytosis. J Cell Sci. 1992;101(Pt 4):907–913. doi: 10.1242/jcs.101.4.907. [DOI] [PubMed] [Google Scholar]
- Wang Y, Harris DC. Macrophages in renal disease. J Am Soc Nephrol. 2011;22(1):21–27. doi: 10.1681/ASN.2010030269. [DOI] [PubMed] [Google Scholar]
- Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80(6):1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover DL, Nacy CA. Macrophage activation to kill Leishmania tropica: defective intracellular killing of amastigotes by macrophages elicited with sterile inflammatory agents. J Immunol. 1984;132(3):1487–1493. [PubMed] [Google Scholar]
- Schleicher U, Bogdan C. Generation culture and flow-cytometric characterization of primary mouse macrophages. Methods Mol Biol. 2009;531:203–224. doi: 10.1007/978-1-59745-396-7_14. [DOI] [PubMed] [Google Scholar]