

Abstract
Gene expression and its regulation are very important to understand the behavior of cells under different conditions. Various techniques are used nowadays to study gene expression, but most are limited in terms of providing an overall picture of the expression of the whole transcriptome. DNA microarrays offer a fast and economic research technology, which gives a full overview of global gene expression and have a vast number of applications including identification of novel genes and transcription factor binding sites, characterization of transcriptional activity of the cells and also help in analyzing thousands of genes (in a single experiment). In the present study, the conditions for bacterial transcriptome analysis from cell harvest to DNA microarray analysis have been optimized. Taking into account the time, costs and accuracy of the experiments, this technology platform proves to be very useful and universally applicabale for studying bacterial transcriptomes. Here, we perform DNA microarray analysis with Streptococcus pneumoniae as a case-study by comparing the transcriptional responses of S. pneumoniae grown in the presence of varying L-serine concentrations in the medium. Total RNA was isolated by using a Macaloid method using an RNA isolation kit and the quality of RNA was checked by using an RNA quality check kit. cDNA was prepared using reverse transcriptase and the cDNA samples were labelled using one of two amine-reactive fluorescent dyes. Homemade DNA microarray slides were used for hybridization of the labelled cDNA samples and microarray data were analyzed by using a cDNA microarray data pre-processing framework (Microprep). Finally, Cyber-T was used to analyze the data generated using Microprep for the identification of statistically significant differentially expressed genes. Furthermore, in-house built software packages (PePPER, FIVA, DISCLOSE, PROSECUTOR, Genome2D) were used to analyze data.
Keywords: Molecular Biology, Issue 98, DNA microarrays, gene expression, transcriptomics, bioinformatics, data analysis, pneumococcus, L-serine
Introduction
The study of the whole set of mRNA abundance (transcriptome) encoded by the genome of a unicellular organism or a eukaryotic cell at a specific time or under a specific condition, including gene overexpression or knock-out, is called transcriptomics. Transcriptomics allows us to observe to what extent genes are expressed under a particular condition at a time point X and gives us information about how strongly the genes are expressed relative to a reference.
A microarray is a two-dimensional array on a solid substrate (usually a glass slide or silicon thin-film cell) that can be used to assay large quantities of biological material using high-throughput screening, and miniaturized, multiplexed and parallel processing and detection methods. Microarrays come in various types, including DNA-microarrays, protein microarrays, peptide microarrays, tissue microarrays, antibody microarrays, cellular microarrays and others. A DNA microarray is basically an assembly of microscopic DNA spots fixed to a solid surface, usually glass. DNA microarrays are used to measure the expression levels of a gene or a set of genes simultaneously or to genotype multiple regions of a genome2,3. Picomoles (10-12 moles) of a probe are present within each DNA spot that represents a specific DNA sequence, also known as a reporter. The labelled mRNA molecules from the samples are called ‘targets’. Fluorophores are used to measure probe-target hybridization and detection of fluorophore-labelled targets determines the relative abundance of nucleic acid sequences in the target. A microarray experiment can accomplish multiple genetic tests in parallel because an array may contain tens of thousands of probes. The layout of a simple microarray experiment is shown in Figure 1. Recently, it was established in our and other labs that these arrays are reusable, which makes this technique quite cost-effective.
Different RNA isolation and purification techniques have been developed over the years including C-TAB, SDS and GT methods 4–8. Furthermore, several commercial kits are also available. For gene expression high quality RNA is very important. Therefore, the RNA isolation methods are modified to get a maximum quantity of RNA. Similarly, the steps for cDNA preparation and labelling of cDNA are minimized. Normalization of data after scanning is also performed efficiently by using in-house built software packages and tools9.
Streptococcus pneumoniae is a Gram-positive human pathogen that colonizes the nasopharynx and is the cause of multiple infections such as pneumonia, sepsis, otitis media and meningitis10. The bacterium can utilize a wide variety of the nutrients required for growth and survival 11,12. A number of studies have been carried out on the pneumococcal nitrogen metabolism and regulation emphasizing the importance of amino acids and their role in virulence13,14. In this study, the transcriptomic response of S. pneumoniae to changing concentrations of L-serine, an amino acid abundantly present in the human blood plasma, is reported using DNA microarrays. The transcriptomic response of S. pneumoniae grown in a minimum concentration of L-serine (150 µM) was compared to that grown in a maximum concentration (10 mM) of serine. Chemically defined medium (CDM or minimal medium)15 was used for this study to control the concentration of serine. The focus of this study is to make this technique user-friendly and to provide different tools for data normalization and analysis. Therefore, a number of tools were developed for analysis and data interpretation. FIVA (Functional Information Viewer and Analyzer) provides a platform for processing information contained in clusters of genes having similar gene expression patterns and for constructing functional profiles16. PROSECUTOR is another software package that facilitates the identification of putative functions and annotations of genes 17. By making use of clustering methods, DISCLOSE provides a DNA binding site detection algorithm. Cis-regulatory motifs of genes can be projected by using this algorithm 18. Genome2D offers a Windows-based platform for visualization and analysis of transcriptome data by offering different color ranges to characterize the changes in gene expression levels on a genome map19. The PePPER webserver offers, in addition to the all-in-one analysis method, a toolbox for mining for regulons, promoters and transcription factor binding sites 20. Full annotation of intergenic regions in a bacterial genome can be achieved by using this package. Biologists can greatly benefit from PePPER as it offers them a platform for designing experiments so that the hypothesized information can be confirmed in vitro20. These software packages contribute significantly to the microarray analysis as most of them are freely available and make data normalization and analysis very reliable.
Protocol
1. Preparation of Media, and Cell Culture
Grow S. pneumoniae D39 wild-type strain21 as described previously11. Inoculate cells stored at -80 oC in 10% glycerol (with 1/100 ratio in 50 ml sterile tubes) in 50 ml Chemically Defined Medium (CDM) with a final pH of 6.4 15, but omit L-serine from the amino acid mixture. Note: two different CDMs were used; one containing a minimum concentration of L-serine (150 µM) and the other containing a maximum concentration of L-serine (10 mM). This minimum concentration is basically the concentration of L-serine in human blood plasma and the aim is to compare the gene expression of S. pneumoniae D39 strain under these two conditions.
2. Isolation of Total RNA
- Materials
- Use acid phenol, RNA grade for the isolation of total RNA.
- Macaloid:
- Suspend 2 grams of macaloid in 100 ml TE and boil for 5 min. Cool to RT and sonicate until macaloid gels.Centrifuge the solution at 2,000 × g for 5 min at RT, resuspend in 50 ml TE pH 8 and store at 4 °C.
- Solutions:
- Treat all solutions with diethyl pyrocarbonate (DEPC). Add 100 µl DEPC/100 ml solution, incubate O/N at 37 °C and autoclave for 15 min.
- Methodology
- Grow 50 ml of S. pneumoniae D39 cell culture in 50 ml tubes at 37 oC (no shaking) until mid-exponential phase (OD ~0.3). Centrifuge cultures at 4 °C on 10,000 × g for 2 min. Discard media and immediately freeze the cell pellets in liquid nitrogen.
- Premix 300 µl chloroform:IAA (24:1) and 300 µl phenol (acid phenol, RNA grade). Use 500 µl of the organic phase in step 2.2.3.
- Prepare the following mixture in advance in RNA-free screw-cap tubes: 0.5 g glass beads, 50 µl 10% SDS, 500 µl phenol/chloroform:IAA (as prepared in step 2.2.2), macaloid layer (150-175 µl, not exact as it is very viscous). Resuspend the cell pellets in 400 µl TE (DEPC) and add the resuspended cells into the screw-cap tubes.
- To break the cells, place the screw-cap tubes in a bead beater for 2x 60” pulses (‘homogenize’) with 1 min interval on ice. Centrifuge the samples for 10 min at 10,000 × g (4 °C).
- Transfer the upper phase to a fresh tube, add 500 µl chloroform:IAA (24:1) and centrifuge for 5 min at 10,000 × g (4 °C).
- Transfer 500 µl of the upper phase to fresh tubes, add 2 volumes (1 ml) of lysis/binding buffer and mix by pipetting up and down. Isolate total RNA using the RNA isolation kit and follow the manufacturer recommended protocol.
3. RNA Cleanup
To remove the contamination of DNA from total RNA, add 100 µl DNAseI mix (90 µl DNAse buffer and 10µl DNAseI) and incubate for 20-30 min at 15-25 ºC.
Wash cleaned RNA using the RNAse kit. Obtain 50 µl of eluted volume.
4. Analysis of RNA
- Determine the concentration of RNA on a spectrophotometer. Determine the quality of RNA by using an assay as follows (Figure 2).
- Dilute 1 µl of sample with 50 µl of DEPC water to get a concentration of 20-200 ng/µl.
- Use 1 µl diluted RNA sample to check quality on the Bioanalyser according to the manufacturer’s instructions.
Note: a ratio of 23S:16S of around 2.0 is considered good.1 A260 unit RNA corresponds to 40 µg/ml. A recommended amount for labeling is 10-20 µg.
5. cDNA Preparation and Labelling
NOTE: The following protocol was followed for cDNA preparation and labelling.
- Annealing
- Perform annealing reaction in 300 µl PCR tubes, keeping the concentration of total RNA 10-15 µg for homemade slides or 5 µg for company-made slides. Mix RNA with 2 µl Random nonamers (1.6 µg/µl) and add nuclease-free water if necessary to keep the final volume of the annealing mixture to 18 µl.
- Keep the annealing mixture at 70 °C for 5 min. After that, cool down the mixture to RT for 10 min (annealing) and if needed, spin down the reactions to the bottom of the tube. Place the reaction tubes on ice for at least 1 min.
- Reverse Transcription
- Prepare the reverse transcriptase mix as follows: to 18 µl annealing mix, add 12 µl Master mix (consisting of 6 µl 5x First Strand buffer, 3 µl 0.1 M DDT, 1.2 µl 25x AA-dUTP / nucleotide mix and 1.8 µl reverse transcriptase (1 µl for company-made slides). Keep the reaction mix for 2-16 hr at 42 °C.
6. Degradation of mRNA and Purification of cDNA
To degrade the mRNA from the reaction mixture, add 3 µl of 2.5 M NaOH and place at 37 °C for about 15 min. Subsequently, add 15 µl of 2M HEPES free acid to neutralize the NaOH.
Purify the cDNA mixture by using PCR clean-up columns and follow the manufacturer’s protocol.
7. Measurement of cDNA Concentration
Measure cDNA concentrations on a spectrophotometer. To continue with labelling, check that the concentration of cDNA is at least 60 ng/µl (homemade slides) or 20 ng/µl (company-made slides).
8. Labelling of cDNA with Amine-reactive Dye and Purification
Use amine-reactive dye to label the cDNA. Directly mix the cDNA with one aliquot (5 µl) of amine-reactive dye.
Incubate the mixture at RT, in the dark, for 60 to 90 min and proceed directly to purification of dye-labelled cDNA. Purify dye-labelled cDNA by using PCR clean-up columns and following the manufacturer’s protocol. Elute cDNA in 50 µl of elution buffer.
9. Measurement of Labelled cDNA
Use a spectrophotometer to measure the incorporation of amine-reactive dyes into the cDNA. Check that the concentration of amine-reactive dyes is at least 0.5 pmol/µl in a total volume of 50 µl.
10. Mixing of Labelled cDNA Samples
For homemade slides, use all the labelled cDNA for hybridization with not more than 30% difference in cDNA concentration. For company-made slides, use cDNA with not more than 2-fold difference in cDNA concentration. Note: normally, about 300 ng of cDNA is needed for company-made slides, which is very little as compared to the required amount of cDNA for homemade slides.
11. Hybridization and Washing
Use the following reagents: demi water, ethanol 99%, SHY Buffer (homemade; with 40 µl yeast RNA). Prepare maximum 1 ml of SHY.
- Apparatus and solutions preparation
- Switch on vacuum concentrators and heater at least 1 hr before drying. Switch on hybridization oven, with set point adjusted to the correct hybridization temperature (for S. pneumoniae DNA microarrays, use 45 °C). Similarly, preheat hybridization cassette and oven for 30-60 min.
- Preheat SHY buffer at 68 ˚C for at least 30 min.
- Sample preparation (labelled cDNA)
- Combine equal quantities of labelled cDNAs (max 30% difference). Dry the sample using the vacuum concentrators at high temp (approx. 40 min) until the volume is smaller than 7 µl.
- Lifter-slip
- Use clean lifter-slips. Note: ± 30 µl can be loaded on the slide.
- Clean the lifter-slips with soap, plenty of tap water and 100% ethanol. Note: Dirty lifter slips give high background.
- Air dry the lifter-slips with air pistol to blow away dust particles. Place a clean lifter-slip on the slide with the white teflon lining at the sides facing down.
- Adding hybridization buffer (to labelled cDNA)
- Dissolve the dried dye samples in 7 µl H2O and incubate at 94 °C for 2 min.
- Immediately, add 35 µl preheated SHY buffer (68 °C), mix gently and spin at maximum speed for 1 min to get rid of precipitates. Preheat the probe at 68 ˚C for approximately 5 min until loading.
- Slide-lifter-slip assembly and preheating.
- Place the hybridization slide holder on a heat-block at 50 ˚C. Place DNA microarray slides with the lifter-slip on the heated hybridization slide holder and preheat the slide with lifter-slip for a minute. Perform the next steps as quickly as possible.
- Add 40 µl of the sample target to the end of the slide. Allow the fluid to flow between the glass surfaces by capillary force. Perform all pipetting slowly and carefully.
- Keep the slides with lifter-slip horizontal at all times and move slowly to make sure that the lifter-slip did not move.
- Take the pre-warmed hybridization cassette out of the hybridization oven and close the machine. Place filter-paper soaked with 3 ml 2x SSC (standard saline citrate) in the hybridization cassette.
- Gently place the hybridization slide holder with slides in the hybridization cassette. Close the hybridization cassette and put in the hybridization oven again (for about 16-18 hr).
- Slide washing
- Prepare fresh wash-buffers I, II and III (750 ml per wash step). For 500 ml wash-buffer I, use 2 x SSC / 0.5 % SDS. For 500 ml wash-buffer II, use 1 x SSC / 0.25 % SDS. For 500 ml wash-buffer III, use 1 x SSC / 0.1 % SDS (optional)
- Place the wash-buffers at 30 °C (to be sure SDS is dissolved).
- Submerge the slides as quickly as possible, but very gently, in a falcon tube filled with 50 ml wash buffer I until the glass rests on the conical bottom of the tube.
- After a few seconds, when the lifter-slip sinks to the bottom of the tube, take out the slide with tweezers without scratching the array and put in the rack of the wash station. Continue with the washing with no time gap.
- Wash the slides for 5 min in 500 ml wash-buffer I (in a washing station). Give it a second wash for 20-30 min in 500 ml wash-buffer II (in a washing station). Similarly, wash the slides for 5 min in 500 ml wash-buffer III (optional) (in a washing station).
- Dry the slides for 2 min at 2,000 rpm.
12. Microarray Analysis
Scan images with respective wavelengths in scanner and save in a folder “Jove Project”.
Use software to analyze the scanned files initially as described previously 22. After running this software, select the “Open Image” tab to load the red image file (-.550) as “Red” and green image file (-.635) as “Green”. Upload the gal file (.gal) having S. pneumoniae array list on image file by selecting “Load Array List” tab 22. Note: this array list consisted of 48 grids, on each grid there were 16 rows and 15 columns. Each spot on the grid represents a single gene and spot information including gene name is added through a spot description file. The spot numbers are given from left to right and top to bottom.
After carefully spotting the grids, select tab “Find Array, Find Blocks, Align Features” to align the spots. After aligning all the features, analyze the image by selecting the “Analyzing” tab. Create a new file having results, histogram and scatter plot. Save this file from tab “Save results as” as a .gpr file for further analysis.
Perform further normalization and processing of data with in-house developed Microprep software package as described9.
Use independent biological replicates for DNA microarray data which are dye-swapped. Perform CyberT implementation of a variant of t-test1 and calculate false discovery rates (FDRs) as described9.
For differentially expressed genes, take p < 0.001 and FDR < 0.05 as a standard.
Upload DNA microarray data on NCBI submission page to get a GEO (Gene Expression Omnibus) accession number.
Representative Results
RNA, cDNA isolations and analysis
L-serine is one of the essential amino acids and its concentration in human blood plasma varies from 60-150 µM in children and adults. Its role in the biosynthesis of purines and pyrimidines highlights its importance in metabolism and it is a precursor to several amino acids (glycine, cysteine and tryptophan). To study the impact of L-serine on the whole transcriptome of S. pneumoniae D39 wild-type strain, microarray analysis of the D39 strain grown in CDM with a minimum concentration (150 µM) of L-serine against that grown in a maximum concentration (10 mM) in the same medium was performed. First of all, total RNA from cells grown under both concentrations was isolated. The concentrations of the RNA samples are given in Table 1. The quality of total RNA was examined using the quality check assay. RNA was treated with DNase I before performing this assay to remove the possible genomic DNA. Figure 2 shows the quality of RNA; lane L represents the ladder, lanes 1 and 2 represent RNA from 150µM serine and lanes 3 and 4 represent the RNA from 10 mM serine. The presence of two clear bands corresponding to the two RNA subunits indicated the good quality of RNA and the next step of the experiment could be performed.
After measuring the quality of RNA, cDNA synthesis was done. cDNA was formed by using random nanomers and reverse transcriptase enzyme. The concentrations of the cDNA samples are given in Table 1. This cDNA was labelled with amine-reactive dyes and the concentrations of the labelled cDNA and dye labels are given in Table 1. After labelling, samples were mixed accordingly and then hybridized. After washing, slides were scanned using the scanner, and analysis was performed using Gene Pix Pro software. Figure 3 shows the scatter plot analysis of the amine-reactive dyes ratio. After initial analysis, data was further analyzed using the PicroPrep software package (PrePrep, Prep, PostPrep)9 to reduce noise and Cyber-T was used for final analysis. Table 2 summarizes the results of the microarray studies after applying the criteria of ≥ 2.0 fold difference and p-value < 0.001. A number of genes were differentially expressed in the presence of minimum L-serine as compared to the maximum (Table 2).
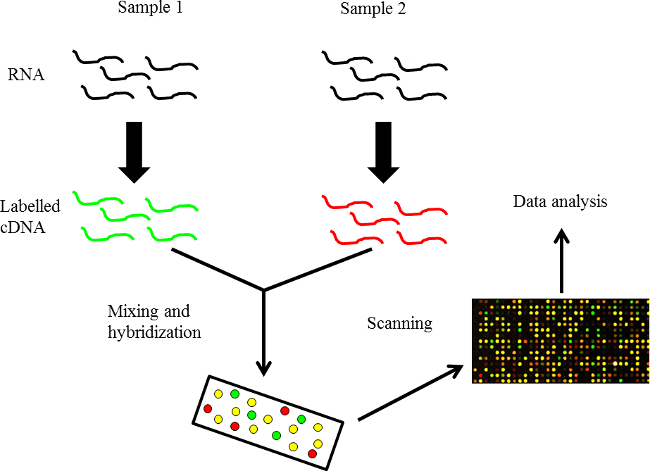 Figure 1. An overview of DNA microarray technology. RNA is isolated from the control and the target samples and labelled cDNA is then hybridized.
Figure 1. An overview of DNA microarray technology. RNA is isolated from the control and the target samples and labelled cDNA is then hybridized.
 Figure 2. Quality check of RNA isolated from S. pneumoniae D39 cells grown in the presence of minimum (150 µM) and maximum (10 mM) serine concentrations. Lane L represents the size-ladder whereas lane 1 and 2 represent RNA samples isolated from cells grown in the presence of minimum serine concentration. Similarly, lane 3 and 4 represent RNA samples isolated from cells grown in the presence of maximum serine concentration. The bands represent the 23S and 16S rRNA. The presence of only two bands shows that there is no gDNA contamination and RNA is of good quality.
Figure 2. Quality check of RNA isolated from S. pneumoniae D39 cells grown in the presence of minimum (150 µM) and maximum (10 mM) serine concentrations. Lane L represents the size-ladder whereas lane 1 and 2 represent RNA samples isolated from cells grown in the presence of minimum serine concentration. Similarly, lane 3 and 4 represent RNA samples isolated from cells grown in the presence of maximum serine concentration. The bands represent the 23S and 16S rRNA. The presence of only two bands shows that there is no gDNA contamination and RNA is of good quality.
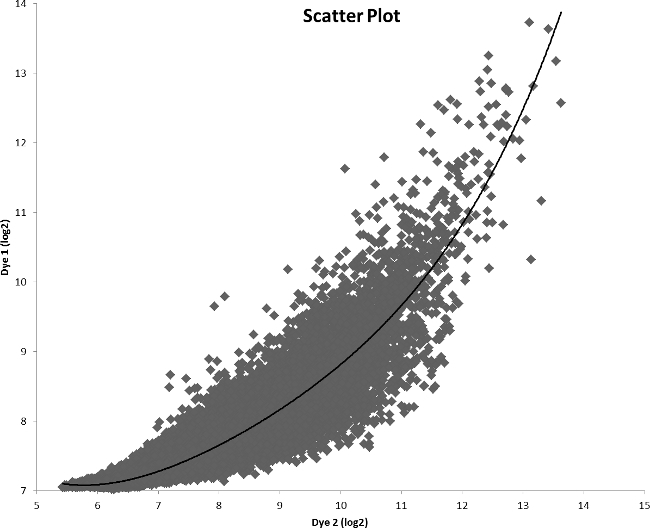 Figure 3. Array comparison scatter plot of a sample mixture. Each spot in the plot represents the mean expression value (log2) of a gene in an experiment with dye 1 on the y-axis and dye 2 on the x-axis.
Figure 3. Array comparison scatter plot of a sample mixture. Each spot in the plot represents the mean expression value (log2) of a gene in an experiment with dye 1 on the y-axis and dye 2 on the x-axis.
| Sample | RNA Sample | Description | RNA concentration (ng/µl) | cDNA concentration (ng/µl) | Labelled cDNA (ng/µl) | DyLight-550 (pmol/µl) | DyLight-650 (pmol/µl) | Hybridization scheme |
| S1 | R1 | D39 wild-type grown in CDM + Minimum L-serine | 2057 | 255 | 225 | 0.8 | R1+R3 | |
| R2 | D39 wild-type grown in CDM + Minimum L-serine | 2566 | 201 | 179 | 1.2 | R2+R4 | ||
| S2 | R3 | D39 wild-type grown in CDM + Maximum L-serine | 2831 | 292 | 276 | 2.3 | ||
| R4 | D39 wild-type grown in CDM + Maximum L-serine | 1867 | 172 | 150 | 1 |
Table 1. Hybridization scheme of the samples used in the microarray analysis.
| Genea | Functionb | Ratioc |
| SPD0600 | Cell division protein DivIB | 4 |
| SPD0445 | Phosphoglycerate kinase | 3.5 |
| SPD0646 | Hypothetical protein | 3.4 |
| SPD0873 | Hypothetical protein | 3.3 |
| SPD1223 | Hypothetical protein | 3.3 |
| SPD0980 | Ribose-phosphate pyrophosphokinase | 2.8 |
| SPD1628 | Xanthine phosphoribosyltransferase | 2.7 |
| SPD1011 | Glycerate kinase | 2.4 |
| SPD0645 | Hypothetical protein | 2.3 |
| SPD0564 | Hypothetical protein | 2.2 |
| SPD0641 | Mannose-6-phosphate isomerase, class I, ManA | 2.1 |
| SPD1333 | Hypothetical protein | 2.1 |
| SPD1384 | Cation efflux family protein | 2.1 |
| SPD1432 | UDP-glucose 4-epimerase, GalE-1 | 2.1 |
| SPD1866 | N-acetylglucosamine-6-phosphate deacetylase, NagA | 2.1 |
| SPD0104 | LysM domain protein | 2 |
| SPD0140 | ABC transporter, ATP-binding protein | 2 |
| SPD0261 | Aminopeptidase C, PepC | 2 |
| SPD1350 | Hypothetical protein | 2 |
| SPD1822 | Ribosomal large subunit pseudouridine synthase, RluD subfamily protein | 2 |
| SPD0453 | Type I restriction-modification system, S subunit | -2 |
| SPD0459 | Heat shock protein GrpE | -2 |
| SPD1006 | Glucose-1-phosphate adenylyltransferase | -2 |
| SPD1799 | Sensor histidine kinase, putative | -2.1 |
| SPD0387 | Beta-hydroxyacyl-(acyl-carrier-protein) dehydratase FabZ | -2.2 |
| SPD1494 | Sugar ABC transporter, permease protein | -2.2 |
| SPD0974 | Class I glutamine amidotransferase, putative | -2.4 |
| SPD1600 | Anthranilate phosphoribosyltransferase | -2.5 |
| SPD1472 | Isoleucyl-tRNA synthetase | -2.6 |
| SPD0681 | Hypothetical protein | -2.7 |
| SPD0501 | Transcription antiterminator, Lict | -3.4 |
Table 2. List of genes regulated in the transcriptome comparison of S. pneumoniae strain D39wild-type grown in CDM15 with minimum concentration of L-serine and CDM15 with maximum concentration of L-serine. aGene numbers refer to D39 locus tags. bD39 annotation/TIGR4 annotation21. cRatio represents the fold increase/decrease in the expression of genes in CDM-maximum as compared to CDM-minimum (minus sign indicates downregulation).
Discussion
We describe a user-friendly protocol that can be applied to perform whole transcriptome analysis of bacteria. The key point about this particular technique is that the condition under which the cells are harvested will vary. After harvesting the cells and RNA isolation, this technique becomes equal for all types of bacterial samples and follows exactly identical steps and therefore, can be applied to any type of bacterial culture. The protocol is very simple and convenient and starts from RNA isolation. Our RNA isolation protocol (using a Macaloid and RNA isolation kit) is time-effective as compared to conventional phenol-chloroform and Trizol methods. In the next step, the preparation of cDNA with transcriptase III enzyme is performed. Next, labelling of cDNA is done by first mixing of cDNA with amine-reactive dyes, and then purifying the labelled cDNA. The labelling is also simple and does not take a lot of time as the samples need to be incubated for about 1 hr in the dark. All these steps can be performed in any standard laboratory as it does not demand any specialized equipment. A microarray scanner is needed to scan slides. For slide scanning and analysis, the GenePix Pro program is used, which is a very simple and user-friendly program22.
After doing all the experiments, analysis was performed using MicroPrep and CyberT. The MicroPrep package, consists of three modules, i.e., PrePreP, PreP and PostPreP9. This data pre-processing framework reduces the time for normalization of data and also reduces the amount of discarded data. The ease with which the software can be used makes it possible for the researcher to have an understanding of the DNA microarray data in minimum time. It takes only a couple of minutes to convert the raw signal data into high-quality data for further processing after slide image analysis is done. Further analysis on the pool of genes regulated in the microarray can be done using different in-house software packages. They include PePPER 20, FIVA 16, DISCLOSE 18, PROSECUTOR 17 and Genome2D 19. These Windows-based tools and software packages are user-friendly and provide deep insight into the data during further investigation. These software packages make it very easy and handy for the researchers to utilize this technology as the data becomes much more meaningful and relevant.
The dyes used in microarrays are known to be susceptible to an ozone-effect, where the dyes become unstable in the presence of ozone and signal strength is so low that it cannot be recognized by the scanner. Amine-reactive dyes which are less sensitive to the ozone-effect are used to label cDNA. The solution to ozone-effect will make this technology even better.
A list was created of genes that were up- or downregulated in the presence of the minimal L-serine concentration as compared to the maximum (Table 2). The upregulated genes can be categorized according to their product’s function. Seven of them encode hypothetical proteins, four are involved in carbohydrate transport and metabolism, a cell division protein DivIB, and certain amino acid transport and metabolism genes. Similarly, there are also certain genes that are downregulated in our tested condition. A BglG-family transcriptional regulator LicT, some carbohydrate genes and some amino acid-specific genes are among the downregulated genes. A heat-shock protein GrpE is also among the down-regulated ones. Therefore, this study provides a complete overview of the genes differentially expressed under the tested conditions. After analysis of the results, sometimes verification of the results is necessary. This can be either done by qPCR or β-galactosidase assays using lacZ promoter fusions.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Anne de Jong and Siger Holsappel for help with the DNA microarrays slide production. Anne de Jong’s support for bioinformatics analysis is also appreciated. We also thank Jelle Slager for reviewing the paper. Muhammad Afzal and Irfan Manzoor are supported by the GC University, Faisalabad, Pakistan under the faculty development program of HEC Pakistan.
References
- Kayala MA, Baldi P. Cyber-T web server: differential analysis of high-throughput data. Nucleic Acids Res. 2012;40:W553–W559. doi: 10.1093/nar/gks420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TW. Binding of cells to matrixes of distinct antibodies coated on solid surface. J. Immunol. Methods. 1983;65:217–223. doi: 10.1016/0022-1759(83)90318-6. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Galau Levi A, A G, Wetzstein HY. A rapid procedure for the isolation of RNA from high-phenolic-containing tissues of pecan. 1992;27(12):1316–1318. [Google Scholar]
- Lopez-Gomez R, Gomez-Lim MA. A method for extraction of intact RNA from fruits rich in polysaccharides using ripe mango mesocarp. 1992. pp. 440–442.
- Salzman RA, Fujita T, Salzman Z, Hasegawa PM, Bressan R. A improved RNA isolation method for plant tissues containing high levels of phenolic compounds or carbohydrates. Plant Molecular Biology Report. 1999;17:11–17. [Google Scholar]
- Kiefer E, Heller W, Ernst D. A simple and efficient protocol for isolation of functional RNA from plant tissues rich in secondary metabolites. Plant Mol. Biol. Report. 2000;18:33–39. [Google Scholar]
- Tattersall EAR, Ergul A, Alkayal F, Deluc L, Cushman JC, Cramer GR. Comparison of methods for isolating high-quality RNA from leaves of grapevine. Am. J. Enol. Vitic. 2005;56:400–406. [Google Scholar]
- Van Hijum SAFT, Garcia de la Nava J, Trelles O, Kok J, Kuipers OP. MicroPreP: a cDNA microarray data pre-processing framework. Appl.Bioinformatics. 2003;2:241–244. [PubMed] [Google Scholar]
- Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat.Rev.Microbiol. 2008;6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- Afzal M, Shafeeq S, Kuipers OP. LacR is a repressor of lacABCD and LacT is an activator of lacTFEG, constituting the lac gene cluster in Streptococcus pneumoniae. Appl. Environ. Microbiol. 2014;80:5349–5358. doi: 10.1128/AEM.01370-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzal M, Shafeeq S, Henriques-Normark B, Kuipers OP. UlaR activates the expression of the ula operon in Streptococcus pneumoniae in the presence of ascorbic acid. Microbiol. Read. Engl. 2014. [DOI] [PubMed]
- Hendriksen WT, et al. Site-specific contributions of glutamine-dependent regulator GlnR and GlnR-regulated genes to virulence of Streptococcus pneumoniae. Infect. Immun. 2008;76:1230–1238. doi: 10.1128/IAI.01004-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman TG, Kuipers OP. Regulation of arginine acquisition and virulence gene expression in the human pathogen Streptococcus pneumoniae by transcription regulators ArgR1 and AhrC. J. Biol. Chem. 2011;286:44594–44605. doi: 10.1074/jbc.M111.295832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman TG, Bijlsma JJE, Kok J, Kuipers OP. To have neighbour's fare: extending the molecular toolbox for Streptococcus pneumoniae. Microbiol. Read. Engl. 2006;152:351–359. doi: 10.1099/mic.0.28521-0. [DOI] [PubMed] [Google Scholar]
- Blom EJ, et al. FIVA: Functional Information Viewer and Analyzer extracting biological knowledge from transcriptome data of prokaryotes. Bioinforma. Oxf. Engl. 2007;23:1161–1163. doi: 10.1093/bioinformatics/btl658. [DOI] [PubMed] [Google Scholar]
- Blom EJ, et al. Prosecutor: parameter-free inference of gene function for prokaryotes using DNA microarray data, genomic context and multiple gene annotation sources. BMC Genomics. 2008;9:495. doi: 10.1186/1471-2164-9-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom EJ, et al. DISCLOSE : DISsection of CLusters Obtained by SEries of transcriptome data using functional annotations and putative transcription factor binding sites. BMC Bioinformatics. 2008;9:535. doi: 10.1186/1471-2105-9-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerends RJS, et al. Genome2D: a visualization tool for the rapid analysis of bacterial transcriptome data. Genome Biol. 2004;5(5):R37. doi: 10.1186/gb-2004-5-5-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong A, Pietersma H, Cordes M, Kuipers OP, Kok J. PePPER, a webserver for prediction of prokaryote promoter elements and regulons. BMC Genomics. 2012;13:229. doi: 10.1186/1471-2164-13-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanie JA, et al. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 2007;189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Devices, Corp. GenePix Pro 6.0 Microarray Acquisition and Analysis Software for GenePix Microarray Scanners-Use's Guide and Tutorial. Molecular Devices, Corp; 2005. [Google Scholar]