

Abstract
Type 1 diabetes (T1D)–induced osteoporosis is characterized by a predominant suppression of osteoblast number and activity, as well as increased bone marrow adiposity but no change in osteoclast activity. The fundamental mechanisms and alternative anabolic treatments (with few side effects) for T1D bone loss remain undetermined. Recent studies by our laboratory and others indicate that probiotics can benefit bone health. Here, we demonstrate that Lactobacillus reuteri, a probiotic with anti-inflammatory and bone health properties, prevents T1D-induced bone loss and marrow adiposity in mice. We further found that L. reuteri treatment prevented the suppression of Wnt10b in T1D bone. Consistent with a role for attenuated bone Wnt10b expression in T1D osteoporosis, we observed that bone-specific Wnt10b transgenic mice are protected from T1D bone loss. To examine the mechanisms of this protection, we focused on TNF-α, a cytokine up-regulated in T1D that causes suppression of osteoblast Wnt10b expression in vitro. Addition of L. reuteri prevented TNF-α–mediated suppression of Wnt10b and osteoblast maturation markers. Taken together, our findings reveal a mechanism by which T1D causes bone loss and open new avenues for use of probiotics to benefit the bone.
Type 1 diabetes (T1D), the most common form of newly diagnosed diabetes in childhood (1), is characterized by hypoinsulinemia and hyperglycemia. T1D patients are treated with insulin, but attaining perfect glycemic control is difficult, even under therapeutic vigilance. Thus, over time T1D patients are at risk for developing complications such as osteoporosis that can affect their quality of life. T1D increases fracture risk, impairs fracture healing (2, 3), and can negatively affect bone growth/stature in young people (4, 5). Bone loss may further negatively affect systemic metabolism and insulin secretion (6, 7). Understanding the fundamental pathological changes contributing to T1D bone loss is critical for identifying effective therapies and improving the overall health of T1D patients.
T1D rodent models display trabecular and cortical bone loss similar to T1D patients. Bone loss occurs regardless of sex (8), genetic background (9), or model (spontaneous vs pharmacologic) (10, 11) and is characterized by a predominant suppression in bone formation (osteoblast anabolic activity) (12, 13). T1D-associated factors, such as hyperglycemia and inflammation, are known to affect osteoblast maturation and viability (13–17). For example, TNF-α is increased in T1D serum and bone and has been demonstrated to contribute to T1D marrow–induced osteoblast and chondrocyte apoptosis (17–20). Similarly, hyperglycemia and inflammation can affect the proliferation and differentiation of marrow mesenchymal stromal cells that are the precursors for osteoblasts and adipocytes. Bone marrow adiposity is increased in T1D, as shown in streptozotocin (STZ)-induced and spontaneous mouse models (10, 11, 21). This finding suggests that differentiation of mesenchymal stromal cells may be shunted away from the osteoblast lineage and toward the adipocyte lineage under diabetic conditions (21).
One candidate mechanism for T1D suppression of bone formation involves the Wnt signaling pathway. Canonical Wnt/β-catenin signaling through T-cell factor (TCF)/lymphoid enhancer factor (LEF)-induced transcription is a potent regulator of both bone formation and adipocyte differentiation (22, 23). In vitro activation of Wnt signaling inhibits adipogenesis, and similarly inhibition of Wnt signaling promotes adipogenesis (24–27). Among the Wnt proteins, Wnt10b has been shown to be a major enhancer of osteoblast differentiation (28). Similar to the bone phenotype in T1D, Wnt10b knockout mice have low bone density predominantly due to suppressed bone formation (29). It has also been shown that fatty acid binding protein 4 (FABP4)–driven Wnt10b-overexpressing mice have high bone density and low marrow adiposity compared with those of wild-type mice (30). Recently, it was found that TNF-α transgenic (TG) mice display bone loss and decreased Wnt signaling activity, implying a potential interaction between TNF-α and regulation of the Wnt signaling pathway and ultimately bone loss (31). However, the extent to which Wnt10b signaling is involved in T1D bone loss remains unknown.
Probiotics are bacteria that can confer a health benefit to the host. Lactobacillus reuteri is an established and widely used probiotic species that has proven beneficial in clinical trials with few or no side effects (32, 33). L. reuteri (ATCC PTA 6475) stays in the human gastrointestinal tract for about 1 week after ingestion (34 35). What is therapeutically appealing about L. reuteri ATCC PTA 6475 for T1D bone health is that it produces molecules that have potent anti-TNF-α activity in vitro (36), it produces antimicrobial compounds (37), and it has been demonstrated to increase bone volume in male and ovariectomized female mice (38, 39).
It is clear that therapies targeted to increase osteoblast differentiation or activity would benefit T1D bone health. Currently, the only anabolic treatment for osteoporosis approved for use in the United States is intermittent PTH that, although beneficial for T1D bone (40), has been associated with side effects (41, 42) and is not prescribed for long-term use. Therefore, we examined the role of L. reuteri for the treatment of T1D bone loss. Our results indicate that L. reuteri can prevent suppression of osteoblast Wnt10b and Wnt signaling by T1D and TNF-α and prevent suppression of osteoblast activity and bone formation in T1D mice. Taken together, our findings suggest the potential for L. reuteri as therapeutic for treating T1D bone loss.
Materials and Methods
Mice and diabetes induction
Adult (14 weeks old) C57BL/6 male mice were obtained from Harlan Laboratories. Mice were allowed to acclimate to the animal room for 1 week before the start of the experiment. Adult mice were chosen to reduce the contribution of the effects of bone growth on bone density parameters, because adults have a plateaued rate of growth, making bone responses more related to changes in bone remodeling. TG mice expressing Wnt10b in bone (osteocalcin [OC] promoter–driven Wnt10b) (29) were bred and housed at Michigan State University. These mice were genotyped with genomic DNA isolated from an ear punch (DNeasy Blood and Tissue Kit; Qiagen). DNA was amplified by real-time PCR with iQ SYBR Green Supermix (Bio-Rad Laboratories) and primers specific to the transgene synthesized by Integrated DNA Technologies (29). Female TG mice were used because they displayed higher bone density at 14 weeks of age than wild-type mice (whereas males did not), and we have previously shown similar bone loss in both male and female diabetic mice (8).
At 15 weeks of age (unless otherwise noted), mice were injected (intraperitoneally) with either 50 mg/kg STZ (to induce diabetes) or 0.1 M citrate buffer (pH 4.5) vehicle (control) for 5 consecutive days. Diabetes was confirmed 12 days after the first injection (days post injection [dpi]) with an AccuChek compact glucometer (Roche), and a drop of blood was collected from the saphenous vein. Blood glucose of >300 mg/dL was considered diabetic. All mice were maintained on a 12-hour light/dark cycle at 23°C, were given standard laboratory chow, and had food and water ad libitum. Tissues were collected at 4 weeks (28 dpi) for probiotic treatment studies or at 40 dpi for TG mouse studies. Total body and soft tissue masses were measured, and bones were isolated. Animal procedures were completed with the approval of the Michigan State University Institutional Animal Care and Use Committee.
Probiotic bacterial strain, culture, and treatment
L. reuteri ATCC PTA 6475 was cultured under anaerobic conditions in de Man, Rogosa, Sharpe (MRS) medium (Difco) for 16 to 18 hours at 37°C. On the following day, the overnight culture was subcultured into fresh MRS medium and grown until log phase (OD600 = 0.4). Cells were harvested by centrifugation at 4000 rpm for 10 minutes. Mice were treated with the bacterial suspensions (109 colony-forming units/mL) in 0.3 mL by introducing the bacteria (or MRS broth, vehicle control) directly into the stomach of mice with a 24-gauge ball-tipped gavage needle 3 times per week for 4 weeks. Our dosing regime was based on data from pilot studies in which the bacterial concentrations and the number of gavages per week were varied and the optimum regimen was selected. The bone benefits of L. reuteri were maximal when given at 3 times vs 1 time per week. Daily gavaging did not enhance bone density above what was seen at the 3 times per week dosing.
MEM conditioned medium was generated for cell culture studies by resuspending the bacterial pellet in prewarmed α-MEM and incubating with gentle shaking at 37°C for 3 hours. Bacteria were harvested, and the supernatant was separated from the cell pellet, pH neutralized, and filter sterilized using polyvinylidene difluoride membrane filters (0.22 μm pore size; Millipore). Aliquots of 10 mL of supernatant with OD600 = 0.5 were concentrated using Millipore Amicon ultra centrifugal filters with a 3-kDa cutoff and then lyophilized and stored at −80°C. Before use, each aliquot of lyophilized conditioned medium was diluted in 0.85 mL of cell culture medium, and 50 μL of the reconstituted conditioned medium was added to the MC3T3-E1 cells cultured in a 6-well plate.
In vitro cell assays
Preosteoblast MC3T3-E1 cells were plated at 20 000 cells/well in a 6-well plate. Cells were cultured in α-MEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. After cells reached confluence, the medium was further supplemented with 2 mM inorganic phosphate and 25 μg/mL ascorbic acid. The medium was changed every other day. Cells were treated with TNF-α (Sigma-Aldrich) at a final concentration of 10 ng/mL for 24 hours. For some conditions, cells were pretreated with 50 μL of reconstituted L. reuteri conditioned medium added 1 hour before addition of the TNF-α.
Bone marrow cells and marrow-derived osteoblasts were obtained by flushing the marrow from mouse femurs using a syringe containing α-MEM. Cells were spun down and resuspended in complete medium. For bone marrow–derived osteoblast cultures, marrow cells were plated at 10 000 cells/well of a 24-well plate and treated similar to MC3T3 cells for 21 days. For bone marrow cocultures, marrow cells were obtained either from 15-week-old control or 5 dpi STZ-induced diabetic mice. The marrow cells were seeded at 1 million/transwell, which was placed in wells of a 24-well plate atop osteoblasts cultured in differentiation medium for 21 days. Transwells contain a 10-μm thick membrane with 0.4-μm pores at the bottom of the well to allow diffusion of factors between compartments. Osteoblasts were harvested 24 hours later for RNA analysis.
TCF/LEF luciferase reporter assay
To measure the transcriptional activity of β-catenin/TCF, a luciferase reporter assay was performed using the TCF reporter constructs TOPflash and FOPflash (control) (Millipore). Cells were seeded at 10 000 cells/per well of a 24-well plate and transfected 24-hours later with either TOPflash or FOPflash (100 ng) using Lipofectamine 2000 Reagent (Invitrogen). The TOPflash and FOPflash reporters contain 2 sets of 3 copies of wild-type or mutant β-catenin/TCF binding sites, respectively, as well as the thymidine kinase minimal promoter upstream of the firefly luciferase open reading frame. At 24 hours posttransfection, the culture medium was renewed. Then the cells were challenged with 10 ng/mL TNF-α for 24 hours, and luciferase activity was determined using the luciferase reporter assay system (Promega).
Microcomputed tomography (μCT) analyses
Bones were numbered, and measurements were made blindly (without knowledge of condition). After the bones were fixed in 10% formalin for 24 hours, they were transferred to 70% ethanol and subsequently scanned using a GE Explore Locus μCT system at a voxel resolution of 20 μm obtained from 720 views. The beam angle of increment was 0.5, and beam strength was set at 80 peak kV and 450 μA. Each run included bones from each treatment group and a calibration phantom to standardize grayscale values and maintain consistency. Based on autothreshold and isosurface analyses of multiple bone samples, a fixed threshold (800) was used to separate bone from bone marrow. Femur trabecular bone analyses were performed in a region of trabecular bone defined at 0.17 mm proximal to the growth plate of the distal femur extending 2 mm toward the diaphysis and excluding the outer cortical shell. Tibia trabecular bone analyses were performed in a region of trabecular bone defined at 0.17 mm distal to the growth plate of the proximal tibia extending 2 mm toward the diaphysis and excluding the outer cortical shell. Trabecular bone mineral content, bone mineral density, bone volume fraction, thickness, spacing, and number values were computed by a GE Healthcare MicroView software application for visualization and analysis of volumetric image data. Trabecular isosurface images were taken from a cylindrical region in the tibia or femur where analyses were performed measuring 1.0 mm in length and 1.0 mm in diameter.
RNA analyses
Tibias were cleaned of muscle and connective tissue, snap-frozen in liquid nitrogen, and stored at −80°C. Frozen tibias were crushed under liquid nitrogen conditions with a Bessman Tissue Pulverizer (Spectrum Laboratories, Inc.). RNA was isolated with TriReagent (Molecular Research Center, Inc.), and integrity was assessed by formaldehyde-agarose gel electrophoresis. cDNA was synthesized by reverse transcription with a SuperScript II Reverse Transcriptase Kit and oligo dT(12–18) primers (Invitrogen) and amplified by real-time PCR with iQ SYBR Green Supermix and gene-specific primers synthesized by Integrated DNA Technologies. Hypoxanthine guanine phosphoribosyl transferase (HPRT) mRNA levels do not fluctuate in diabetes or with overexpression of Wnt10b and were used as an internal control. HPRT was amplified using 5′-AAGCCTAAGATGAGCGCAAG-3′ and 5′-TTACTAGGCAGATGGCCA CA-3′ (43). Wnt10b was amplified using 5′-TCTCTTTCAGCCCTTTGCTCGGAT-3′ and 5′-ACAACTGAACGGAAGGAGAAGCCT-3′ (44). OC was amplified using 5′-ACGGTATCACTATTTAGGACCTGT G-3′ and 5′-ACTTTATTTTGGAGCTGCTGTGAC-3′ (45). Runt-related transcription factor 2 (Runx2) was amplified using 5′-GACAGAAGCTTGATGACTCTAAAC C-3′ and 5′-TCTGTAATCTGACTCTGTCCTTGT G-3′ (46). FABP4 was amplified using 5′-GCGTGGAATTCGATGAAATCA-3′ and 5′-CCCGCCATCTAGGGTTAT GA-3′ (47). Peroxisome proliferator-activated receptor-γ2 (PPARγ2) was amplified using 5′-TGAAACTCTGGGAGATTCTCC TG-3′ and 5′-CCATGGTAATTTCTTGTGAAGTGC-3′ (48). Real-time PCR was performed for 40 cycles using the iCycler (Bio-Rad), and data were evaluated using iCycler software. Each cycle consisted of 95°C for 15 seconds, 60°C for 30 seconds (except for OC and Runx2, which had annealing temperatures of 65°C), and 72°C for 30 seconds. cDNA-free samples, a negative control, did not produce amplicons. Melting curve and gel analyses (sizing, isolation, and sequencing) were used to verify single products of the appropriate base pair size.
Serum measurements
Blood was obtained from mice at the time of euthanasia, and blood serum was prepared from each sample by centrifugation for 10 minutes at 4000 rpm. Serum was aliquoted and stored frozen at −80°C and did not go through more than 1 freeze/thaw cycle. Serum OC levels were measured using a Mouse OC EIA Kit (Biomedical Technologies, Inc.), according to the manufacturer's instructions.
Bone histomorphometry, immunohistochemistry, and dynamic measures
Fixed femur samples were processed on an automated Thermo Electron Excelsior tissue processor for dehydration, clearing, and infiltration using a routine overnight processing schedule. Samples were embedded in paraffin, and blocks were sectioned at 5 μm on a microtome. Slides were stained for tartrate-resistant acid phosphatase (TRAP) activity and counterstained with hematoxylin according to the manufacturer's protocol (387A-1KT; Sigma-Aldrich). Histomorphometric measures were made blinded to the mouse condition. Visible adipocytes of >30 μm, adipocyte area/ total marrow area, and average adipocyte size were examined in the distal femur and calculated with Image-Pro Plus software.
For immunohistochemical staining of Wnt10b protein, the femur sections were deparaffinized and hydrated to distilled water before being placed in Tris-buffered saline (TBS) for 5 minutes. Then they were pretreated with enzyme pepsin for 10 minutes at 37°C and incubated overnight at 56°C. Slides were further rinsed in distilled water and soaked in TBS with Tween 20 (TBST) for 5 minutes, and these slides were loaded onto an Autostainer and flooded with TBST for 5 minutes. Samples were blocked with 1:28 normal goat serum for 30 minutes and stained with rabbit anti-Wnt10b antibody at 1:200 dilutions for 60 minutes and biotinylated goat anti-rabbit antibody at 1:136 for 30 minutes (Table 1). They were then stained with Vector R.T.U. peroxidase reagent for 30 minutes and Vector Nova Red for 15 minutes. TBS was used to rinse the slides between each staining step. The slides were counterstained with working methyl green for 30 seconds.
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used | DOI or Publication Data |
|---|---|---|---|---|---|---|
| Wnt10b | Anti-Wnt10b | Sigma Aldrich, catalog no. PRS4619 | Rabbit, polyclonal | 1:200 dilution |
For dynamic histomorphometric measures of bone formation, mice were injected intraperitoneally with 200 μL of 10 mg/mL calcein (Sigma-Aldrich) dissolved in sterile saline at 7 and 2 days before harvest. L3–L4 vertebrae were fixed in formalin at the time of harvest and were transferred to 70% ethanol 48 hours later. Vertebrae were then embedded, sectioned, and examined under UV light. Five images were taken, and the distance between the calcein lines and their length along the bone surface was measured and used to calculate the mineral apposition rate (MAR).
Statistical analyses
Power analyses (80% power) were used to determine the number of animals or in vitro experiments needed to detect a 15% difference at a significance level of .05. The number depended on the outcomes measured. All measurements are presented as means ± SEM. To determine the statistical significance (P ≥ .05) of the changes between groups, particularly diabetic mice vs L. reuteri–treated diabetic mice, we used one-way ANOVA and Fisher post hoc tests with GraphPad Prism Software.
Results
L. reuteri treatment prevents T1D bone loss
Studies from our laboratory and others indicate a role for inflammation in T1D bone loss, and therefore we hypothesized that the probiotic L. reuteri 6475, which has anti-inflammatory properties (49, 50), could benefit T1D bone health. To test this, adult control and STZ-induced diabetic male mice were given L. reuteri orally by gavage 3 times per week starting 2 days after their first STZ injection. Untreated mice were gavaged with vehicle (broth). Mice were harvested 4 weeks later, and their bones were analyzed by μCT. Remarkably, T1D trabecular bone loss was prevented in T1D mice treated with L. reuteri as indicated by the representative trabecular isosurface bone images in Figure 1A. Quantitative analysis of the distal femur bone volume fraction further indicates that T1D mice display a 35% reduction in bone volume fraction, whereas T1D mice treated with the probiotic had bone volumes that were similar to those of controls (Figure 1B). Further analyses of bone parameters (Figure 1B) indicate that lower trabecular number and increased trabecular spacing mark T1D trabecular bone loss; L. reuteri treatment significantly protected mice from these changes. Although T1D did not have a pronounced effect on trabecular thickness, L. reuteri treatment significantly increased it. Cortical bone analyses revealed that the significant decrease in T1D mouse femur cortical area and thickness was also prevented with L. reuteri treatment (Table 2).
Figure 1.

L. reuteri treatment prevents T1D bone loss. A, Representative isosurface images of the distal femur trabecular bone volume. B, Average ± SE (n = 10) bone volume fraction (BVF) (bone volume/total volume) and other trabecular measures of the distal femur from control mice (C, white bars), diabetic mice (D, black bars), L. reuteri–treated control mice (C + LR, light gray bars), and L. reuteri–treated diabetic mice (D + LR, dark gray bars). *, P < .05; ***, P < .001. BMC, bone mineral content; BMD, bone mineral density; T.N., trabecular number; Tb.Sp., trabecular spacing; Tb.Th., trabecular thickness.
Table 2.
Cortical Bone Parameters for L. reuteri Study
| Control | Diabetic | Control + Lr | Diabetic + Lr | |
|---|---|---|---|---|
| Tt.Ar. (mm2) | 2.15 ± 0.08 | 2.06 ± 0.05 | 2.20 ± 0.07 | 2.14 ± 0.06 |
| Ct.Ar. (mm2) | 1.13 ± 0.05 | 0.98 ± 0.03a | 1.20 ± 0.03 | 1.16 ± 0.04b |
| Ma.Ar. (mm2) | 1.02 ± 0.04 | 1.07 ± 0.03 | 1.00 ± 0.05 | 0.98 ± 0.04 |
| Ct.Ar./Tt.Ar. | 0.52 ± 0.01 | 0.48 ± 0.01a | 0.55 ± 0.01 | 0.54 ± 0.01b |
| Thickness (mm) | 0.26 ± 0.01 | 0.23 ± 0.01a | 0.29 ± 0.01 | 0.27 ± 0.01b |
| Inner perimeter (mm) | 3.86 ± 0.06 | 3.94 ± 0.05 | 3.84 ± 0.09 | 3.80 ± 0.08b |
| Outer perimeter (mm) | 5.43 ± 0.10 | 5.32 ± 0.06 | 5.56 ± 0.07 | 5.47 ± 0.09 |
| MOI (mm4) | 0.58 ± 0.05 | 0.50 ± 0.03 | 0.63 ± 0.03 | 0.59 ± 0.04b |
Abbreviations: Ct.Ar, cortical bone area; Ct.Ar./Tt.Ar., cortical area fraction; Lr, L. reuteri; Ma.Ar., marrow area; MOI, the cross-sectional moments of inertia at the z-axis; Tt.Ar., total cross-sectional area. Data are averages ± SE. n = 10 per condition.
P ≤ .05 to control.
P ≤ .05 to diabetic group by one-way ANOVA.
L. reuteri treatment did not prevent T1D-induced muscle and peripheral adipose tissue loss
As expected, T1D increased blood glucose levels and L. reuteri treatment did not influence elevated blood glucose levels (Figure 2A). Consistent with a diabetic phenotype and similar to previous findings (17, 51), T1D mice displayed decreased body mass, muscle (tibialis) mass, and inguinal (subcutaneous) and retroperitoneal (visceral) adipose tissue mass (Figure 2). Measures from probiotic-treated diabetic mice did not significantly differ from those for untreated diabetic mice. However in control mice, the probiotic treatment did reduce visceral adipocyte tissue mass. This result suggests that probiotics may affect systemic metabolism.
Figure 2.

L. reuteri does not block effects of T1D on blood glucose or body composition. Measurements taken 4 weeks after induction of diabetes include random-fed blood glucose levels (A), body mass (B), tibialis mass (C), inguinal fat mass (D), and retroperitoneal fat mass (E). Values are averages ± SEM (n = 10 per group). **, P < .01; ***, P < .001. C, control; D, diabetic
L. reuteri treatment restores osteoblast function in T1D mice
To identify what bone cell activity (formation, resorption, or both) is targeted by L. reuteri treatment to prevent T1D bone loss, we examined serum markers of bone remodeling, bone RNA expression, and static and dynamic histomorphometry in untreated and treated control and diabetic mice 4 weeks after treatment. The greatest changes were seen in T1D mice, which displayed suppressed levels of markers of bone formation including serum OC and MAR compared with those of controls; this result is consistent with reports indicating that T1D suppresses bone formation and remodeling. Corresponding with the prevention of T1D bone loss, L. reuteri treatment raised T1D mouse serum OC, osteoblast surface, and MAR to healthy control mouse levels (Figure 3). Osteoclast surface coverage did not differ across the four conditions, but levels of the bone resorption marker TRAP were significantly elevated in both T1D and probiotic-treated T1D mice, although L. reuteri treatment did not change TRAP levels compared with those of untreated mice. Thus, L. reuteri has a predominant beneficial effect on osteoblast activity in this model. This is similar to the anabolic effect that we have previously reported in healthy male mice (38).
Figure 3.

L. reuteri treatment increases bone anabolic markers in diabetic mice. A, Representative images from calcein-labeled trabecular bone 4 weeks after diabetes onset with or without L. reuteri treatment. B, Quantitation of bone MAR. C, Osteoblast surface/total bone surface in distal femur trabecular bone region. D, Serum levels of OC. E, Osteoclast surface/total bone surface in distal femur trabecular bone region. F, Serum levels of TRAP. Values represent averages ± SE (n = 10 per group). *, P < .05; **, P < .01.
Probiotic L. reuteri treatment prevents T1D bone marrow adiposity
Previously we demonstrated that T1D bone loss is associated with increased marrow adiposity (21, 52), implicating altered lineage selection of bone marrow stromal cells toward the adipocyte over osteoblast lineage. Indeed, analysis of bone histological sections revealed more marrow adipocytes in diabetic bone than in control bone. Consistent with bone health benefits, L. reuteri-treated T1D mice did not display this pathological trend (Figure 4, A and B). The increased size of adipocytes (Figure 4, C and D) in T1D mice was also prevented by probiotic treatment.
Figure 4.
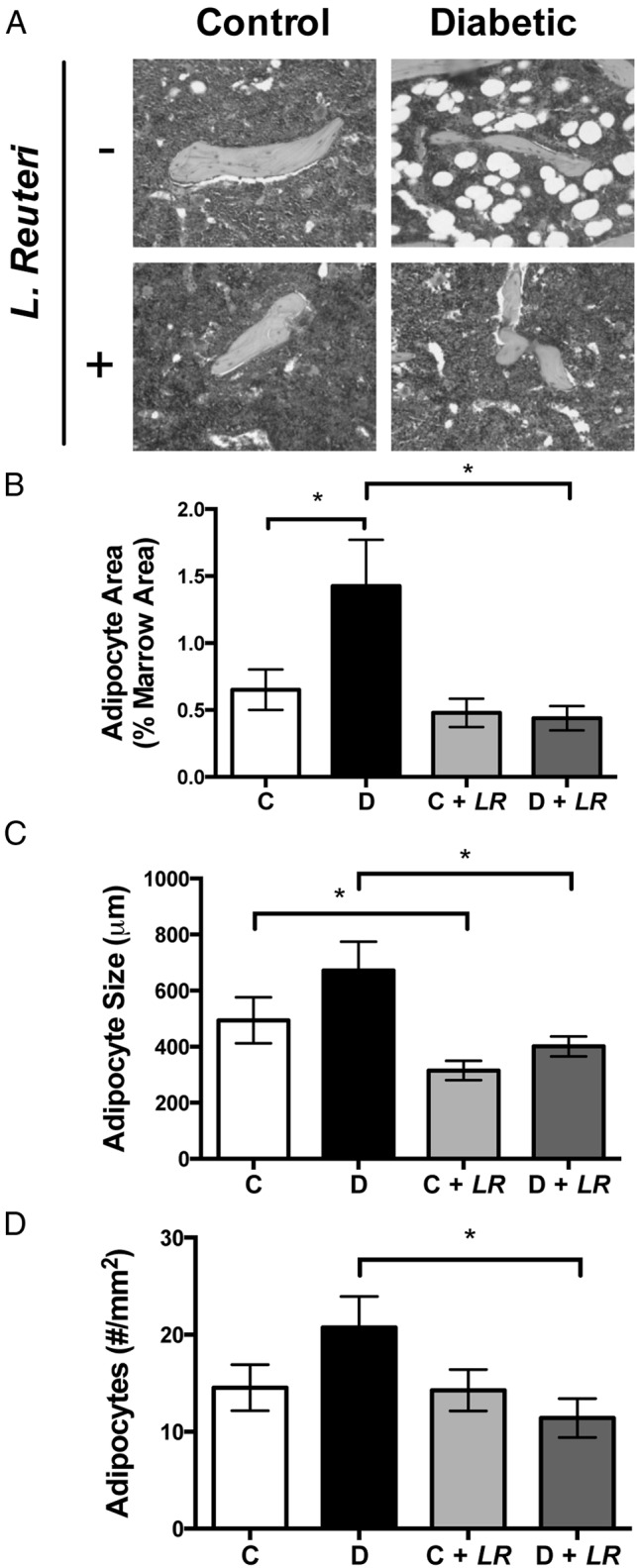
Increased marrow adiposity in bone of T1D mice is not observed with L. reuteri. A, Representative images of marrow adipocytes in the distal femur. Adipocytes are identified by their morphologic appearance (empty oval space) in the hematoxylin and eosin–stained paraffin sections of bone specimens. Image-Pro Plus software was used to calculate the distal femur adipocyte area relative to the total marrow area (B) and the average adipocyte size (C). The total number of adipocytes was counted manually (D). Values represent averages ± SE (n = 10 per group). *, P < .05.
L. reuteri modulates Wnt10b expression in gut and bone
Activation of Wnt10b signaling in mesenchymal precursor cells stimulates osteoblastogenesis and inhibits adipogenesis (30). Previous findings demonstrate that T1D suppresses osteoblast maturation, lineage selection, and viability. Given that Wnt10b intricately controls these processes, we hypothesized Wnt10b involvement in the T1D bone phenotype. Consistent with this, T1D reduces Wnt10b expression in mouse bone (Figure 5A) concurrent with molecular bone phenotype changes (17) that include decreased OC (osteoblast marker) and increased FABP4 (adipocyte marker) RNA levels. We therefore examined whether the probiotic treatment could restore Wnt10b expression in diabetic mice. Remarkably, the suppression in Wnt10b expression in T1D bone was fully returned to normal levels by probiotic treatment (Figure 5, A and B). Immunohistochemical examination of Wnt10b protein levels in the local area of osteoblasts in control and diabetic bone, with and without L. reuteri treatment, demonstrated staining that was consistent with Wnt10b RNA expression levels (Figure 5, B and C). It is notable that although bone Wnt10b expression was up-regulated by probiotic treatment, the expression level of Wnt10b after probiotic treatment is not significantly higher than that of the control untreated mice.
Figure 5.

L. reuteri treatment prevents diabetic suppression of Wnt10b in bone. A, Expression of Wnt10b was measured in the tibia bone in control and diabetic wild-type and Wnt10b TG mice using real-time PCR. HPRT was used as a housekeeping gene to normalize the RNA expression. B, Representative images of Wnt10b immunostaining in the local area of osteoblasts and bone marrow of the femur metaphysis of wild-type control (C) and diabetic (D) mice 4 weeks after STZ injection. Magnification, ×40. Arrows point to Wnt10b-positive cells. C, Bar graph represents the number of Wnt10b-positive cells per square millimeter of bone marrow. Bars represent averages ± SE (n = 7–8 per group). *, P < .05.
Expression of Wnt10b in bone prevents T1D bone loss
Because Wnt10b has been shown by numerous reports to have a role in regulating osteoblast differentiation (29, 30, 53), we hypothesized that decreased Wnt10b expression in T1D bone is one of the underlying mechanisms of T1D bone loss. To test this hypothesis, we induced T1D in wild-type and OC-Wnt10b TG mice (created by fusing the Wnt10b gene with the OC promoter to target Wnt10b expression to osteoblasts) (29). Overexpression of Wnt10b did not affect the levels of blood glucose or fat pat or muscle mass in control mice (Figure 6A). Importantly, T1D OC-Wnt10b TG mice displayed changes in blood glucose and fat and muscle mass similar to those in T1D wild-type mice (21). This indicates that Wnt10b overexpression in bone does not significantly affect systemic glucose and fat metabolism. In contrast, OC-Wnt10b TG mouse tibias were protected from the T1D-induced molecular phenotype changes of decreased OC and increased FABP4 (Figure 6C), thereby indicating the possibility that overexpression of bone-specific Wnt10b could prevent T1D bone loss. To confirm this, bone density was examined in control and diabetic wild-type vs OC-Wnt10b TG mice using μCT (Figure 6B). As expected, T1D decreased the bone density in wild-type mice, whereas no significant difference was observed between control and diabetic OC-Wnt10b TG mice. The group of OC-Wnt10b TG mice showed higher bone density than the wild-type mice, consistent with previous reports (29). These findings indicate that expression of Wnt10b in T1D bone can prevent trabecular bone loss in T1D mice.
Figure 6.

Wnt10b TG mice do not display T1D bone loss and marrow adiposity. A, Random-fed blood glucose levels, fat pad mass, and tibialis mass measured in control (C) and diabetic (D) wild-type (WT) and Wnt10b TG mice. Values are averages ± SE (n = 10 per group). *, P < .05 compared with untreated control mice. B, Representative μCT isosurface images of trabecular bone volume/total volume from distal femurs are shown. Average ± SE (n = 10) bone volume fraction (bone volume/total volume) and other trabecular measures of the distal femur. BMC, bone mineral content; BMD, bone mineral density; T.N., trabecular number; Tb.Sp., Tb.Th., trabecular thickness. C, Expression of OC and FABP4 were measured in the tibia bone using real-time PCR. HPRT was used as a housekeeping gene to normalize the RNA expression. Bars represent averages ± SE (n = 7–8 per group). *, P < .05. C, control (white bars); D, diabetic (black bars); C-TG, TG control (light gray bars); D-TG, diabetic TG mice (dark gray bars).
Diabetic bone marrow decreases osteoblast Wnt10b expression: a negative role for TNF-α and a beneficial role for L. reuteri
Given that T1D is associated with systemic inflammation and bone marrow is the main immune organ where immune cells originate and migrate through, we hypothesized that changes in the T1D bone marrow reduce Wnt10b expression in osteoblasts. To determine the possible causes of T1D suppression of Wnt10b expression, we cocultured bone marrow from the T1D mice with osteoblasts and measured Wnt10b RNA levels. Indeed, Wnt10b expression was attenuated in the osteoblasts cocultured with the diabetic bone marrow compared with that in the control marrow (Figure 7A). TNF-α is a critical proinflammatory factor that promotes bone loss (54) and has been demonstrated to be up-regulated by diabetes (17, 55, 56). In T1D mouse bone, TNF-α RNA levels are demonstrated to increase 5 days after T1D induction (17), which is concurrent with the decrease in Wnt10b and OC and increase in FABP4 RNA levels. Therefore, we investigated whether TNF-α could decrease Wnt10b expression in vitro in osteoblasts at different stages of maturation. Figure 7B indicates that TNF-α treatment consistently decreases osteoblast Wnt10b expression (no matter what stage the osteoblasts are in) and simultaneously decreases OC and increases FABP4 expression. This is analogous to the T1D bone phenotype. Canonical Wnt signaling is one of the mechanisms controlling the development of precursor cells into osteoblasts or adipocytes (30); therefore, we examined whether TNF-α treatment decreases canonical Wnt signaling pathway activity. Using a Wnt downstream transcription factor (TCF) reporter system, we found that TNF-α greatly reduced canonical Wnt signaling activity (Figure 7C). This finding suggests that elevation of TNF-α might be a critical factor leading to the decrease in Wnt10b expression and bone loss. Probiotics have been used as anti-inflammatory agents (49, 50), and, in particular, L. reuteri 6475 secretory products have been shown to have anti-TNF-α activity (50, 57). Thus, we further tested whether L .reuteri 6475 supernatant (secreted products) could prevent the TNF-α–induced decrease in Wnt10b expression in vitro. Indeed, the TNF-α suppression of Wnt10b in cultured osteoblasts was prevented by the addition of L. reuteri 6475 supernatant (Figure 7D). This suggested that the probiotic L. reuteri 6475 could potentially benefit T1D bone loss in vivo.
Figure 7.

L. reuteri rescues the TNF-α–induced down-regulation of Wnt10b in osteoblasts. A, Expression of Wnt10b was measured in MC3T3–E1 osteoblasts cocultured with control (C) and diabetic (D) bone marrow for 24 hours. B, Wnt10b, OC, and FABP4 mRNA levels (relative to HPRT levels) were measured in MC3T3–E1 cells cultured for 0 to 21 days with (C) and without (T) TNF-α treatment. Cells were plated at 10 000 cells/mL at day 0, and TNF-α was added 24 hours before harvest. Gene expression was measured using real-time PCR. C, MC3T3 osteoblasts were transfected with a TCF/LEF reporter. After 24 hours, cells were treated with TNF-α and luciferase activity measured 24 hours later. RLU, relative light unit. D, Confluent MC3T3 osteoblasts were treated with the supernatant/medium of an L. reuteri (LR sup) culture or with media alone (vehicle). After 1 hour of pretreatment, cells were treated with TNF-α at 10 ng/mL. Cells were harvested 24 hours later. Wnt10b expression was examined using real-time PCR. Values represent averages ± SE (n = >6 per group). *, P < .05.
Discussion
The gut-bone signaling axis offers new therapeutic approaches to treating osteoporosis. Ingestion of probiotics has previously been shown to benefit bone health by improving bone density in chickens (58) aging mice (59), male mice (38), and ovariectomized mice (39, 60). However, it was not known whether L. reuteri could rescue bone loss under disease conditions, such as T1D. Our findings demonstrate that L. reuteri ATCC 6475 treatment of T1D mice increases bone density as characterized by increased osteoblast activity and decreased bone marrow adiposity (39). The effects of L. reuteri on adiposity were striking and suggest that L. reuteri can affect mesenchymal stromal cell lineage selection and osteoblast maturation. Consistent with this, L. reuteri treatment restored the expression of the lineage regulator, Wnt10b, and osteoblast activity in T1D bone.
Wnt10b-deficient mouse models demonstrate that reduction/depletion of Wnt10b reduces bone formation by decreasing osteoblast activity without affecting osteoclast parameters (30, 29, 61). In addition, over-expression of Wnt10b regulates mesenchymal stem cell selection, increases osteoblast activity, and reduces adipogenesis (53, 62). These findings are further supported by the observation that FABP4 promoter-driven Wnt10b TG mice have significantly reduced marrow and peripheral fat (30, 63). Whereas other anabolic signaling pathways and other Wnt family members can affect marrow stem cell differentiation, global gene expression analyses indicated that T1D causes a major reduction specifically in Wnt10b levels. Thus, we hypothesized that T1D suppression of Wnt10b expression could contribute to the T1D bone phenotype. This hypothesis is supported in vivo by our finding that T1D OC-Wnt10b TG mice did not display trabecular bone loss and by the finding that L. reuteri treatment prevented T1D suppression of Wnt10b RNA and protein levels as well as bone loss. Whereas Wnt10b overexpression in the TG mouse studies proved that increasing Wnt10b prevents T1D bone loss, it did not prove that Wnt10b is required for the bone-sparing effects of L. reuteri. Alternate anabolic signaling pathways could also be affected by L. reuteri to prevent T1D bone loss; this is an area of ongoing research. Still, our studies demonstrate that a factor secreted by L. reuteri inhibited osteoblast TNF-α signaling and prevented TNF-α suppression of osteoblast Wnt10b expression. TNF-α is known to suppress Wnt10b, osteogenesis, osteoblast maturation, and osteoblast viability in vitro and in various disease models (19, 64, 65, 66); therefore, the suppression of osteoblast responsiveness to TNF-α would probably be of benefit to bone health. In addition, other studies have demonstrated that antibodies to TNF-α can prevent many of the bone pathological changes seen in mice with diabetes as well as in mice or humans with other inflammatory diseases (20, 56, 67–70).
What signaling pathway does TNF-α affect to reduce bone health? Our in vitro data indicated that TNF-α is capable of suppressing Wnt10b downstream signaling and TCF and LEF reporter activity. Previous reports demonstrated that TNF-α suppresses Smad activation (71), leading to reduced Wnt10b expression. Whether TNF-α modulates Wnt10b expression in T1D through the SMAD signaling pathway and whether L. reuteri secreted products can affect this signaling pathway directly are ongoing focuses of our research. Past studies indicated that L. reuteri can prevent TNF-α signaling locally (57) and systematically (49) probably by preventing nuclear factor-κB activation or the MAPK pathway (72), key regulatory pathways of bone formation (64). It is possible that L. reuteri directly regulates Wnt10b through these pathways instead of actively regulating local or systemic TNF-α production.
Our studies demonstrated that the TNF-α effects on osteoblasts are not stage dependent in the context of Wnt10b, OC, and FABP4 responses. Although the baseline levels of Wnt10b and OC were stage dependent, being highest at 7 and 21 days, respectively, TNF-α consistently suppressed Wnt10b and OC expression in osteoblasts at 2, 7, 14, and 21 days after plating. Furthermore, reporter assays demonstrated that not only the level of Wnt10b but also Wnt signaling itself was suppressed by TNF-α. This is probably due to decreased osteoblast secretion of Wnt10b and therefore reduced autocrine and paracrine signaling. In addition, L. reuteri treatment of confluent day 7 osteoblasts that have the highest level of Wnt10b expression demonstrated significant protection from TNF-α suppression of Wnt10b levels.
What specific cells are responsible for making TNF-α and Wnt10b in bone? An important source of marrow TNF-α is what is produced by T cells, and these cells are thought to be involved in mediating the bone loss associated with chronic PTH treatment (73). Regarding Wnt10b, studies also suggest that Wnt10b can be expressed by T cells, specifically CD8+ T cells, and is important for bone anabolic responses to PTH (74). Other cells (such as immune cells, osteoblasts, and osteocytes) are also capable of producing both TNF-α and Wnt10b. Our in vivo gene expression data were obtained from whole bone RNA isolations to reduce isolation variation and error. However, we looked at bone marrow gene expression and found that TNF-α was highly expressed in marrow, whereas Wnt10b mRNA was highly expressed in the whole bone isolations (whereas much lower levels were found in the marrow). Wnt10b protein, on the other hand, was detected throughout the bone including the marrow, indicating that it is secreted and able to interact with a variety of bone cells. Past studies indicate that the components of the Wnt10b receptor, LRP5 and LRP6, are expressed throughout osteoblast maturation, and levels increase with differentiation, suggesting that all stages of osteoblast maturation can respond to Wnt10b (75).
Interestingly, we found that L. reuteri treatment did not significantly affect osteoclast activity in T1D mice, which is consistent with L. reuteri having a predominant anabolic effect in male mice (38) but is in contrast to our finding in ovariectomized mice. One possible reason for the variation in responses is that the L. reuteri effects are bone pathology dependent. In T1D, we hypothesize that inflammation contributes to the suppression of osteoblast activity, whereas the inflammatory effects on osteoclasts are blunted by high glucose conditions. In ovariectomized mice, inflammation contributes predominantly to osteoclast activation with less of an effect on osteoblasts. In this sense, L. reuteri could be an effective regulator that potentially prevents inflammatory pathological changes without overactivating normal functions of the body.
In summary, our studies demonstrate that L. reuteri treatment may be an effective way to suppress T1D bone loss through activation of anabolic pathways potentially as a result of preventing Wnt10b suppression. Although there are still many questions to be answered, our findings demonstrate the potential for targeting the gut-bone signaling axis to treat T1D-induced bone loss.
Acknowledgments
We thank the Investigative Histology Laboratory in the Department of Physiology, Division of Human Pathology, and the Biomedical Imaging Center at Michigan State University for their assistance with histology and imaging, respectively.
Current address for J.Z.: Xi'an Jiaotong University, First Affiliated Hospital of Xi'an Jiaotong University, Frontier Institute of Science and Technology, Bone and Joint Research Center, Xi'an, China 710061.
This work was supported by National Institutes of Health Grants R01 DK101050 (to L.R.M.), R01 AT007695-01 (to L.R.M. and R.A.B.), and R01 DK62876 (to O.A.M.).
Disclosure Summary: R.A.B. and L.R.M. have previously received funding from Biogaia to study L. reuteri (although Biogaia did not support this study). The other authors have nothing to disclose.
Footnotes
- FABP4
- fatty acid binding protein 4
- HPRT
- hypoxanthine guanine phosphoribosyl transferase
- MAR
- mineral apposition rate
- MRS
- de Man, Rogosa, Sharpe
- OC
- osteocalcin
- Runx2
- runt-related transcription factor 2
- STZ
- streptozotocin
- TBS
- Tris-buffered saline
- TBST
- Tris-buffered saline with Tween 20
- TCF/LEF
- T-cell factor/lymphoid enhancer factor
- T1D
- type 1 diabetes, type 1 diabetic
- TG
- transgenic
- TRAP
- tartrate-resistant acid phosphatase.
References
- 1. Sabin MA, Cameron FJ, Werther GA. Type 1 diabetes—still the commonest form of diabetes in children. Aust Fam Physician. 2009;38:695–697. [PubMed] [Google Scholar]
- 2. Kayal RA, Tsatsas D, Bauer MA, et al. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res. 2007;22:560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alblowi J, Kayal RA, Siqueira M, et al. High levels of tumor necrosis factor-α contribute to accelerated loss of cartilage in diabetic fracture healing. Am J Pathol. 2009;175:1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Danne T, Kordonouri O, Enders I, Weber B. Factors influencing height and weight development in children with diabetes. Results of the Berlin Retinopathy Study. Diabetes Care. 1997;20:281–285. [DOI] [PubMed] [Google Scholar]
- 5. Holl RW, Grabert M, Heinze E, Sorgo W, Debatin KM. Age at onset and long-term metabolic control affect height in type-1 diabetes mellitus. Eur J Pediatr. 1998;157:972–977. [DOI] [PubMed] [Google Scholar]
- 6. Hinoi E, Gao N, Jung DY, et al. An osteoblast-dependent mechanism contributes to the leptin regulation of insulin secretion. Ann NY Acad Sci. 2009;1173(suppl 1):E20–E30. [DOI] [PubMed] [Google Scholar]
- 7. Yoshizawa T, Hinoi E, Jung DY, et al. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J Clin Invest. 2009;119:2807–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin LM, McCabe LR. Type I diabetic bone phenotype is location but not gender dependent. Histochem Cell Biol. 2007;128:125–133. [DOI] [PubMed] [Google Scholar]
- 9. Zhang J, Coe L, Motyl KJ, McCabe L. Mouse strain dependent and independent effects of type 1 diabetic bone pathology. Diabetes Metab. 2012;S1–008.22305438 [Google Scholar]
- 10. Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148:198–205. [DOI] [PubMed] [Google Scholar]
- 11. Coe LM, Zhang J, McCabe LR. Both spontaneous Ins2+/− and streptozotocin-induced type I diabetes cause bone loss in young mice. J Cell Physiol. 2013;228:689–695. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz AV, Sellmeyer DE. Diabetes, fracture, and bone fragility. Curr Osteoporos Rep. 2007;5:105–111. [DOI] [PubMed] [Google Scholar]
- 13. McCabe L, Zhang J, Raehtz S. Understanding the skeletal pathology of type 1 and 2 diabetes mellitus. Crit Rev Eukaryot Gene Expr. 21:187–206. [DOI] [PubMed] [Google Scholar]
- 14. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 15. McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem. 2007;102:1343–1357. [DOI] [PubMed] [Google Scholar]
- 16. Botolin S, McCabe LR. Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J Cell Biochem. 2006;99:411–424. [DOI] [PubMed] [Google Scholar]
- 17. Motyl KJ, Botolin S, Irwin R, et al. Bone inflammation and altered gene expression with type I diabetes early onset. J Cell Physiol. 2009;218:575–583. [DOI] [PubMed] [Google Scholar]
- 18. Coe LM, Irwin R, Lippner D, McCabe LR. The bone marrow microenvironment contributes to type I diabetes induced osteoblast death. J Cell Physiol. 2011;226:477–483. [DOI] [PubMed] [Google Scholar]
- 19. Kayal RA, Siqueira M, Alblowi J, et al. TNF-α mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J Bone Miner Res. 2010;25:1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ko KI, Coimbra LS, Tian C, et al. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFα-mediated mechanism. Diabetologia. 2015;58:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-γ2 expression in type I diabetic mice. Endocrinology. 2005;146:3622–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moon RT. Wnt/β-catenin pathway. Sci STKE. 2005;2005:cm1. [DOI] [PubMed] [Google Scholar]
- 23. Baron R, Rawadi G. Targeting the Wnt/β-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635–2643. [DOI] [PubMed] [Google Scholar]
- 24. Bennett CN, Ross SE, Longo KA, et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem. 2002;277:30998–31004. [DOI] [PubMed] [Google Scholar]
- 25. Ross SE, Hemati N, Longo KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. [DOI] [PubMed] [Google Scholar]
- 26. Ross SE, Erickson RL, Gerin I, et al. Microarray analyses during adipogenesis: understanding the effects of Wnt signaling on adipogenesis and the roles of liver X receptor α in adipocyte metabolism. Mol Cell Biol. 2002;22:5989–5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kang S, Bennett CN, Gerin I, et al. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor γ. J Biol Chem. 2007;282:14515–14524. [DOI] [PubMed] [Google Scholar]
- 28. Kubota T, Michigami T, Ozono K. Wnt signaling in bone metabolism. J Bone Miner Metab. 2009;27:265–271. [DOI] [PubMed] [Google Scholar]
- 29. Bennett CN, Ouyang H, Ma YL, et al. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22:1924–1932. [DOI] [PubMed] [Google Scholar]
- 30. Bennett CN, Longo KA, Wright WS, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA. 2005;102:3324–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gilbert LC, Chen H, Lu X, Nanes MS. Chronic low dose tumor necrosis factor-α (TNF) suppresses early bone accrual in young mice by inhibiting osteoblasts without affecting osteoclasts. Bone. 2013;56:174–183. [DOI] [PubMed] [Google Scholar]
- 32. Shornikova AV, Casas IA, Isolauri E, Mykkänen H, Vesikari T. Lactobacillus reuteri as a therapeutic agent in acute diarrhea in young children. J Pediatr Gastroenterol Nutr. 1997;24:399–404. [DOI] [PubMed] [Google Scholar]
- 33. Shornikova AV, Casas IA, Mykkänen H, Salo E, Vesikari T. Bacteriotherapy with Lactobacillus reuteri in rotavirus gastroenteritis. Pediatr Infect Dis J. 1997;16:1103–1107. [DOI] [PubMed] [Google Scholar]
- 34. Frese SA, Hutkins RW, Walter J. Comparison of the Colonization ability of autochthonous and allochthonous strains of lactobacilli in the human gastrointestinal tract. Adv Microbiol. 2012;2:399–409. [Google Scholar]
- 35. Oh PL, Benson AK, Peterson DA, et al. Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J. 2010;4:377–387. [DOI] [PubMed] [Google Scholar]
- 36. Thomas CM, Hong T, van Pijkeren JP, et al. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLOS One. 2012;7:e31951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schaefer L, Auchtung TA, Hermans KE, Whitehead D, Borhan B, Britton RA. The antimicrobial compound reuterin (3-hydroxypropionaldehyde) induces oxidative stress via interaction with thiol groups. Microbiology. 2010;156:1589–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCabe LR, Irwin R, Schaefer L, Britton RA. Probiotic use decreases intestinal inflammation and increases bone density in healthy male but not female mice. J Cell Physiol. 2013;228:1793–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Britton RA, Irwin R, Quach D, et al. Probiotic L. reuteri treatment prevents bone loss in a menopausal ovariectomized mouse model. J Cell Physiol. 2014;229:1822–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Motyl KJ, McCauley LK, McCabe LR. Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. J Cell Physiol. 2012;227:1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vahle JL, Long GG, Sandusky G, Westmore M, Ma YL, Sato M. Bone neoplasms in F344 rats given teriparatide [rhPTH(1–34)] are dependent on duration of treatment and dose. Toxicol Pathol. 2004;32:426–438. [DOI] [PubMed] [Google Scholar]
- 42. Vahle JL, Sato M, Long GG, et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1–34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30:312–321. [DOI] [PubMed] [Google Scholar]
- 43. Vengellur A, LaPres JJ. The role of hypoxia inducible factor 1α in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol Sci. 2004;82:638–646. [DOI] [PubMed] [Google Scholar]
- 44. Fox KE, Colton LA, Erickson PF, et al. Regulation of cyclin D1 and Wnt10b gene expression by cAMP-responsive element-binding protein during early adipogenesis involves differential promoter methylation. J Biol Chem. 2008;283:35096–35105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ontiveros C, McCabe LR. Simulated microgravity suppresses osteoblast phenotype, Runx2 levels and AP-1 transactivation. J Cell Biochem. 2003;88:427–437. [DOI] [PubMed] [Google Scholar]
- 46. Ontiveros C, Irwin R, Wiseman RW, McCabe LR. Hypoxia suppresses runx2 independent of modeled microgravity. J Cell Physiol. 2004;200:169–176. [DOI] [PubMed] [Google Scholar]
- 47. Li J, Takaishi K, Cook W, McCorkle SK, Unger RH. Insig-1 “brakes” lipogenesis in adipocytes and inhibits differentiation of preadipocytes. Proc Natl Acad Sci USA. 2003;100:9476–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kast-Woelbern HR, Dana SL, Cesario RM, et al. Rosiglitazone induction of Insig-1 in white adipose tissue reveals a novel interplay of peroxisome proliferator-activated receptor γ and sterol regulatory element-binding protein in the regulation of adipogenesis. J Biol Chem. 2004;279:23908–23915. [DOI] [PubMed] [Google Scholar]
- 49. Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. [DOI] [PubMed] [Google Scholar]
- 50. Thomas CM, Hong T, van Pijkeren JP, et al. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One. 7:e31951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Motyl K, McCabe LR. Streptozotocin, type I diabetes severity and bone. Biol Proced Online. 2009;11:296–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Motyl KJ, Raetz M, Tekalur SA, Schwartz RC, McCabe LR. CCAAT/enhancer binding protein β-deficiency enhances type 1 diabetic bone phenotype by increasing marrow adiposity and bone resorption. Am J Physiol Regul Integr Comp Physiol. 300:R1250–R1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wright WS, Longo KA, Dolinsky VW, et al. Wnt10b inhibits obesity in ob/ob and agouti mice. Diabetes. 2007;56:295–303. [DOI] [PubMed] [Google Scholar]
- 54. Seriolo B, Paolino S, Sulli A, Ferretti V, Cutolo M. Bone metabolism changes during anti-TNF-α therapy in patients with active rheumatoid arthritis. Ann N Y Acad Sci. 2006;1069:420–427. [DOI] [PubMed] [Google Scholar]
- 55. Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu R, Bal HS, Desta T, Behl Y, Graves DT. Tumor necrosis factor-α mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am J Pathol. 2006;168:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Resta-Lenert S, Barrett KE. Probiotics and commensals reverse TNF-α- and IFN-γ-induced dysfunction in human intestinal epithelial cells. Gastroenterology. 2006;130:731–746. [DOI] [PubMed] [Google Scholar]
- 58. Mutus R, Kocabagli N, Alp M, Acar N, Eren M, Gezen SS. The effect of dietary probiotic supplementation on tibial bone characteristics and strength in broilers. Poult Sci. 2006;85:1621–1625. [DOI] [PubMed] [Google Scholar]
- 59. Kimoto-Nira H, Suzuki C, Kobayashi M, Sasaki K, Kurisaki J, Mizumachi K. Anti-ageing effect of a lactococcal strain: analysis using senescence-accelerated mice. Br J Nutr. 2007;98:1178–1186. [DOI] [PubMed] [Google Scholar]
- 60. Ohlsson C, Engdahl C, Fak F, et al. Probiotics protect mice from ovariectomy-induced cortical bone loss. PLoS One. 2014;9:e92368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stevens JR, Miranda-Carboni GA, Singer MA, Brugger SM, Lyons KM, Lane TF. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res. 25:2138–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kennell JA, MacDougald OA. Wnt signaling inhibits adipogenesis through β-catenin-dependent and -independent mechanisms. J Biol Chem. 2005;280:24004–24010. [DOI] [PubMed] [Google Scholar]
- 63. Longo KA, Wright WS, Kang S, et al. Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem. 2004;279:35503–35509. [DOI] [PubMed] [Google Scholar]
- 64. Zhou FH, Foster BK, Zhou XF, Cowin AJ, Xian CJ. TNF-α mediates p38 MAP kinase activation and negatively regulates bone formation at the injured growth plate in rats. J Bone Miner Res. 2006;21:1075–1088. [DOI] [PubMed] [Google Scholar]
- 65. Boyce BF, Li P, Yao Z, et al. TNF-α and pathologic bone resorption. Keio J Med. 2005;54:127–131. [DOI] [PubMed] [Google Scholar]
- 66. Saidenberg-Kermanac'h N, Corrado A, Lemeiter D, deVernejoul MC, Boissier MC, Cohen-Solal ME. TNF-α antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone. 2004;35:1200–1207. [DOI] [PubMed] [Google Scholar]
- 67. Miheller P, Muzes G, Zagoni T, Toth M, Racz K, Tulassay Z. Infliximab therapy improves the bone metabolism in fistulizing Crohn's disease. Dig Dis. 2006;24:201–206. [DOI] [PubMed] [Google Scholar]
- 68. Pazianas M, Rhim AD, Weinberg AM, Su C, Lichtenstein GR. The effect of anti-TNF-α therapy on spinal bone mineral density in patients with Crohn's disease. Ann NY Acad Sci. 2006;1068:543–556. [DOI] [PubMed] [Google Scholar]
- 69. Uno JK, Kolek OI, Hines ER, et al. The role of tumor necrosis factor α in down-regulation of osteoblast Phex gene expression in experimental murine colitis. Gastroenterology. 2006;131:497–509. [DOI] [PubMed] [Google Scholar]
- 70. Durnez A, Paternotte S, Fechtenbaum J, et al. Increase in bone density in patients with spondyloarthritis during anti-tumor necrosis factor therapy: 6-year followup study. J Rheumatol. 2013;40:1712–1718. [DOI] [PubMed] [Google Scholar]
- 71. Li Y, Li A, Strait K, Zhang H, Nanes MS, Weitzmann MN. Endogenous TNFα lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-κB. J Bone Miner Res. 2007;22:646–655. [DOI] [PubMed] [Google Scholar]
- 72. Iyer C, Kosters A, Sethi G, Kunnumakkara AB, Aggarwal BB, Versalovic J. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-κB and MAPK signalling. Cell Microbiol. 2008;10:1442–1452. [DOI] [PubMed] [Google Scholar]
- 73. Pacifici R. Role of T cells in ovariectomy induced bone loss—revisited. J Bone Miner Res. 2012;27:231–239. [DOI] [PubMed] [Google Scholar]
- 74. Robinson JW, Li JY, Walker LD, et al. T cell-expressed CD40L potentiates the bone anabolic activity of intermittent PTH treatment. J Bone Miner Res. 2015;30:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Riddle RC, Diegel CR, Leslie JM, et al. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS One. 2013;8:e63323. [DOI] [PMC free article] [PubMed] [Google Scholar]