

Abstract
LH/human chorionic gonadotropin receptor (LHR) undergoes down-regulation during preovulatory LH surge or in response to exposure to a supraphysiological concentration of its ligands through a posttranscriptional mechanism involving an RNA binding protein designated as LHR mRNA binding protein (LRBP). miR-122, a short noncoding RNA, has been shown to mediate the up-regulation of LRBP. In the present study, we show that inhibition of miR-122 using a locked nucleic acid (LNA)-conjugated antagomir suppressed human chorionic gonadotropin (hCG)-induced up-regulation of LRBP as well as its association with LHR mRNA, as analyzed by RNA EMSA. Most importantly, inhibition of miR-122 resulted in the abolishment of hCG-mediated LHR mRNA down-regulation. We also show that the transcription factor, sterol regulatory element binding protein (SREBP) (SREBP-1a and SREBP-2 isoforms), is an intermediate in miR-122-mediated LHR mRNA regulation. HCG-stimulated increase in the activation of both SREBP-1a and SREBP-2 was inhibited by pretreatment with the miR-122 antagomir. The inhibition of cAMP/protein kinase A (PKA) and ERK pathways, upstream activators of miR-122, abolished SREBP activation after hCG treatment. SREBP-mediated regulation of LRBP expression is mediated by recruitment of LRBP promoter element to SREBP-1a, because chromatin immunoprecipitation assay revealed that association of LRBP promoter to SREBP was increased by hCG treatment. Pretreatment with miR-122 antagomir suppressed this response. Inhibition of SREBP activation by pretreating the rats with a chemical compound, fatostatin abrogated hCG-induced up-regulation of LRBP mRNA and protein. Fatostatin also inhibited LHR-LRBP mRNA-protein complex formation and LHR down-regulation. These results conclusively show that miR-122 plays a regulatory role in LH/hCG-induced LHR mRNA down-regulation by increasing LRBP expression through the activation of SREBP pathway.
Luteinizing hormone/human chorionic gonadotropin receptor (LHR), a member of the glycoprotein subfamily of large G protein-coupled receptor family (1, 2), plays a central role in regulating ovarian function. LHR expression undergoes changes throughout the ovarian cycle in response to changes in the secretion of FSH and LH and other paracrine factors (3–11). Most significant change in LHR expression is seen in response to preovulatory LH surge when the LHR undergoes transient down-regulation. This is followed by a full recovery and then the expression level increases with the development of corpus luteum. LHR expression then falls with the regression of corpus luteum. The ligand-induced down-regulation of LHR is not limited to rodents. Granulosa cells isolated from the follicular aspirates derived from women undergoing ovulation induction for in vitro fertilization showed a sharp decline in the expression of LH receptor mRNA transcripts, in response to treatment with high dose of human chorionic gonadotropin (hCG) before oocyte retrieval (12). We have shown that the rapid changes in LHR expression in human and rodent ovaries under conditions that mimics the preovulatory LH surge occurs through a posttranscriptional mechanism (8, 13, 14). We have identified an RNA binding protein designated as LHR mRNA binding protein (LRBP), which plays a critical role in the regulation of LH receptor (8, 13, 14). Further studies showed that LRBP, which has been identified as mevalonate kinase, regulates LHR expression through a posttranscriptional mechanism (15–20). During LH/hCG-induced down-regulation of LH receptor, the expression of LRBP and its association with LHR mRNA increases significantly leading to the formation of an untranslatable mRNA-protein complex (mRNP) which is then routed for degradation in p bodies (16, 21, 22). We have also shown that activation of cAMP/protein kinase A (PKA)-ERK pathway is upstream of LH/hCG-induced up-regulation of LRBP (23).
microRNAs are small noncoding RNAs that interact with mRNAs inducing their translational suppression and degradation (reviewed in Ref. 24). Nevertheless, several studies have revealed that miRNAs have the capability of activating gene expression directly or indirectly in response to different conditions (25). Our most recent studies have identified a microRNA, miR-122, that plays a regulatory role in LH/hCG-mediated LRBP up-regulation, during LHR down-regulation (26). miR-122 was found to be expressed downstream of cAMP/PKA in this pathway (26). Moreover, there was a temporal relationship in the expression of miR-122, activation of sterol regulatory element binding proteins (SREBPs) and up-regulation of LRBP, after LH/hCG-stimulation. SREBPs belong to a large class of transcription factors whose target genes include enzymes involved in cholesterol biosynthesis such as 3-hydroxy-3-methyl-glutaryl-CoA reductase, and mevalonate kinase (LRBP) (reviewed in in Ref. 27). Of the 3 SREBPs that have been identified, SREBP-1a and SREBP-1c are produced from a single gene through the use of alternate promoters and SREBP-2 from a separate gene (27). SREBP-1c is more selective in activating fatty acid biosynthetic genes and SREBP-2 is more specific for controlling cholesterol biosynthesis, whereas SREBP-1a is involved in both (27, 28). In this study, we have examined the mechanism by which miR-122 regulates LHR expression. By using pharmacological inhibitors and specific antagomir, we show that the LH/hCG-mediated activation of cAMP/PKA-phospho-ERK-miR-122 pathway stimulates the activation of SREBPs, which then leads to the induction of LRBP causing LHR mRNA degradation.
Materials and Methods
Materials
Pregnant mare serum gonadotropin was purchased from Calbiochem. Highly purified hCG (CR 127) was purchased from Dr A. F. Parlow (National Hormone and Peptide Program). EDTA-free protease inhibitor mixture tablets and Quickspin (G-50 Sephadex) columns for radiolabeled RNA purification were purchased from Roche Applied Science. Real-time PCR primers for LHR, LRBP (mevalonate kinase, MVK), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNAs, and 18S rRNA (TaqMan Assay-on-Demand Gene Expression Products), as well as MVK promoter (Custom Plus TaqMan RNA Assays), were from Applied Biosystems/Life Technologies. Because LRBP was identified as MVK, anti-N-terminal mevalonate kinase IgG was raised against the first 15 N-terminal amino acids of MVK (MLSEVLLVSAPGKVI), and this antibody is referred to as the LRBP antibody in the text. SREBP-1a mouse monoclonal antibody (IgG-2A4) was raised against amino acids 301–407 of human SREBP-1a (American Type Culture Collection). Antibodies against β-tubulin and SREBP-2 were from Sigma and Santa Cruz Biotechnology, Inc, respectively. The Super Signal West Femto chemiluminescence kit and antirabbit/antimouse IgG conjugated to horseradish peroxidase were obtained from Pierce. BCA reagent was purchased from GE Healthcare Life Sciences. Locked nucleic acid (LNA)-conjugated miR-122 antagomir was from Exiqon. [α-32P]-uridine triphosphate was obtained from PerkinElmer Life Sciences and Maxiscript T7 kit was from Ambion. Ribonuclease inhibitor (RNasin) was from Promega. The inhibitors U0126 and H-89 were purchased from Calbiochem and fatostatin from Tocris (R&D Systems). EZ-magna Chip kit was from EMD Millipore (Table 1).
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| SREBP-2 | 833–1141 of SREBP-2 of mouse, rat, and human | SREBP-2 antibody (IC6) | Santa Cruz Biotechnology, Inc; catalog SC13552 | Mouse monoclonal | 1:1000 |
| β-Tubulin | Monoclonal anti-β-tubulin (clone 2-28-33) | Sigma; catalog T 5293 | Mouse monoclonal | 1:10 000 | |
| SREBP-1a | Amino acids 301–407 of human SREBP1 | SREBP-1a | ATCC | Mouse monoclonal | 1:1000 |
| MVK | First 15 N-terminal amino acids of MVK (MLSEVLLVSAPGKVI) | LRBP | Pacific immunology | Rabbit polyclonal | 1:500 |
Animals and tissues
Superovulation was induced in 23 day old female Sprague-Dawley rats by sc injection of 50 IU of pregnant mare serum gonadotropin followed by 25 IU of hCG 56 hours later to induce superovulation. Five days later, the animals were treated again with a single dose of hCG (50 IU) to down-regulate LHR mRNA expression. The ovaries were then collected at different time periods after hCG injection and were frozen in liquid nitrogen until further use. To block SREBP activation, the superovulated rats were treated ip with fatostatin (30 mg/kg body weight [b.w.]), based on the results of our preliminary studies using different doses and times to achieve optimum inhibition, 4 hours before the second hCG treatment. To block PKA and ERK1/2, the day-5 superovulated rats were injected sc with H-89 (100 mg/kg b.w.) or U0126 (10 mg/kg b.w.) 1 hour before hCG treatment. To inhibit miR-122, on day 4 of superovulation, the rats were injected with an LNA-conjugated miR-122 antagomir (10 mg/kg b.w.), directly into the bursa of the ovary. The rats were allowed to recover for 24 hours before hCG treatment. Animal handling and treatments were conducted in accordance with the accepted standards of humane animal care, as outlined in the Ethical Guidelines of University of Michigan and were reviewed and approved by University Committee on Use and Care of Animals.
Real-time PCR (quantitative PCR) analysis
Total RNAs were reverse transcribed and subjected to real-time PCR quantitation as per the manufacturer's instructions (Applied Biosystems). The fold change in gene expression was calculated from the threshold cycle (Ct) using the delta delta Ct method with 18S rRNA as the internal control (CTL), as described previously (23).
Western blot analysis
For this, tissue homogenates (S10 fractions) in Radioimmunoprecipitation assay (RIPA) buffer were subjected to 10% SDS-PAGE under reducing conditions followed by Western blot analysis as previously described (23). The presence of immune complexes was detected by chemiluminescence.
RNA EMSA (REMSA)
REMSA was performed by incubating S100 cytosolic fractions from CTL and down-regulated ovaries with a fixed concentration of [α-32P]UTP-labeled LRBP binding sequence (LBS), as described previously (13). The labeled RNA for the binding assay was prepared using the Maxiscript kit and S100 fractions were prepared from the ovaries, as described in detail in our previous manuscripts (13). The RNA-protein complexes were resolved by 5% native polyacrylamide (70:1) gel electrophoresis and analyzed by autoradiography.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the EZ-magna Chip kit (Millipore), as per the manufacturer's instructions with slight modifications (29). Briefly, whole ovaries from CTL and treated rats were placed in 1% formaldehyde solution for cross-linking the DNA and incubated at room temperature for 15 minutes. Cross-linking was stopped by the addition of 2.5M glycine. The ovaries were washed (3×) and then homogenized in ice cold PBS/protease inhibitor mix. Homogenates were centrifuged at 14 000 revolutions per minute for 5 minutes at 4°C. The pellets were then lysed in the cell lysis buffer (from the kit) and, after incubation on ice for 10 minutes, centrifuged, and resuspended in nuclear lysis buffer, after which it was sonicated. One tenth of the resulting samples were removed for input, and remaining samples were immunoprecipitated with SREBP-1a antibody (mouse monoclonal) overnight at 4°C. ChIP using mouse IgG and antibody against RNA polymerase II (Pol II), which binds to the GAPDH promoter, were used as negative and positive CTLs, respectively. The cross-linking was reversed by incubating at 65°C for 2 hours and purified with phenol-chloroform, followed by ethanol precipitation. The purified DNA samples from positive and negative CTLs along with input DNA were amplified by PCR using rat primer sequences for GAPDH gene promoter. The primers designed for the proximal promoter regions of GAPDH are 5′-CGTAGCTCAGGCCTCTGCGCCCTT-3′ (forward) and 5′-CTGGCACTGCACAAGAAGATGCGGCTG-3′ (reverse). The PCR products were separated on 1.2% agarose/ethidium bromide gels and visualized. The DNA from the rest of the samples were analyzed by real-time PCR using the Applied Biosystems 7300 Real-Time PCR System, using primers for MVK promoter, designed from the region flanking a putative sterol response element (SRE) element in the proximal region (product number AJ01XA1, Custom Plus TaqMan RNA Assays; Life Technologies). Rat LRBP (MVK) promoter (∼2019 nucleotides) was deduced using the software https://genome.ucsc.edu (Supplemental Figure 1). The putative SRE element (Bold sequences), 275 nucleotides upstream of the transcription start site, was identified using the MacVector software. The input DNA was also subjected to real-time PCR using primers and probes for GAPDH (product number Rn99999916_s1, detects gDNA; Applied Biosystems). Fold changes of the enriched DNA were calculated using the ΔΔCt method, with GAPDH levels from the input DNA used as the internal CTL. All the real-time PCR assays comprised triplicate data with samples from at least 3 independent experiments.
Statistical analysis
Statistical analysis was carried out using one-way ANOVA followed by the Tukey multiple comparison test. Values were considered statistically significant for P < .05.
Results
miR-122 antagomir inhibited hCG-induced up-regulation of LRBP mRNA
To demonstrate the role of miR-122 in ligand-induced down-regulation of LHR mRNA expression, we examined the effect of miR-122 inhibition on hCG-mediated LRBP up-regulation. miR-122 was inhibited by injecting rats with a specific LNA-conjugated antagomir before hCG treatment. We have previously shown that miR-122 expression was abolished by treatment with this antagomir (26). A 3.7-fold increase was seen in the LRBP mRNA levels 4 hours after hCG treatment, compared with CTL (P < .05 vs hCG) (Figure 1). Pretreatment with the miR-122 antagomir resulted in an inhibition of hCG-mediated increases in LRBP mRNA levels in the ovaries (0.9-fold vs CTL, P < .05 vs hCG). Conversely, treatment with LNA scrambled negative CTL produced no inhibition of hCG-induced increases in LRBP mRNA (2.84-fold vs CTL, nonsignificant vs hCG). Supplemental Figure 2 shows the inhibition of miR-122 expression in the ovaries in rats treated with LNA-conjugated miR-122 antagomir and the absence of inhibition when treated with scrambled negative CTL.
Figure 1.

Inhibition of miR-122 abrogated hCG-induced increases in LRBP gene expression. LNA-conjugated miR-122 antagomir (10 mg/kg b.w.) or scrambled LNA CTL was injected into the bursa of the ovaries of superovulated rats on day 4. Animals were allowed to recover and treated with hCG (50 IU) on day 5. Ovaries were collected 4 hours later and were processed for total RNA isolation. Total RNAs were reverse transcribed, and the resulting cDNAs were subjected to real-time PCR quantitation using predesigned primers and probes for LRBP as described in Materials and Methods. The graphs represent ratio of LRBP levels normalized to 18S rRNA and are expressed as percent change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG; n = 4.
Inhibition of miR-122 decreased hCG-induced association of LRBP protein with LHR mRNA
Because miR-122 inhibition resulted in the reversal of hCG-induced LRBP up-regulation, it would be expected that the decrease in LRBP might lead to an expected decrease in the association of LRBP with LHR mRNA. To examine this possibility, we performed RNA electrophoretic gel mobility shift assay. As previously shown, the LRBP-LHR mRNP complex is observed around 45-kDa region in the gel (13). There was a marked increase in the intensity of the complex formation between LRBP and labeled LHR mRNA probe after hCG treatment (Figure 2, lane 3) compared with the CTL (Figure 2, lane 2). The intensity of this band was significantly reduced with miR-122 antagomir treatment (Figure 2, lane 5), whereas no significant change was seen in the samples from miR-122 antagomir alone pretreated group (Figure 2, lane 4). Quantitative analysis of the autoradiograms using densitometry showed that there was a 4.6-fold increase in the intensity of mRNP complex in the hCG-treated samples compared with CTL, which was reduced to a 2.3-fold increase (vs CTL) when exposed to antagomir before hCG treatment (P < .05 vs hCG).
Figure 2.

Inhibition of miR-122 using a specific antagomir inhibits hCG-induced increase in LRBP binding activity. LNA-conjugated miR-122 antagomir was injected into the bursa of the ovaries of superovulated rats on day 4, followed by hCG on day 5. Ovaries were collected 4 hours later and S100 fractions isolated as described previously (23). Electrophoretic gel mobility shift analysis was performed using S100 fractions isolated from ovaries from different treatment groups, containing equal amounts of total protein, after incubating with [32P]-labeled LBS (1.5 × 105 c.p.m). The autoradiogram shown is representative of 3 independent experiments (biological replicates). The graph represents quantitative analysis of the bands in the autoradiogram. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG; n = 3.
Inhibition of miR-122 abrogated hCG-induced LHR mRNA down-regulation
Analysis of LHR mRNA levels using real-time PCR showed that the inhibition observed for LRBP-LHR mRNA association in the presence of miR-122 antagomir was also reflected in LHR mRNA down-regulation. As shown in Figure 3, the levels of LHR mRNA in the hCG-treated samples were reduced by almost 80% after 12 hours of hCG treatment (P < .05 vs CTL). This inhibition was significantly reversed, bringing the LHR mRNA levels very close to the CTL levels, when the rats were pretreated with miR-122 antagomir before hCG treatment (miR-122 antagomir+hCG, 0.72-fold vs CTL, P < .05 vs hCG), but not for scrambled CTL (0.37-fold vs CTL, nonsignificant vs hCG). These results confirmed our notion that miR-122 mediated up-regulation of LRBP plays a crucial role in hCG-mediated LHR mRNA down-regulation.
Figure 3.

miR-122 antagomir significantly reversed hCG-induced LHR mRNA down-regulation. LNA-conjugated miR-122 antagomir or scrambled LNA CTL was injected into the bursa of the ovaries of superovulated rats on day 4, followed by hCG on day 5. Ovaries were collected 12 hours later and were processed for total RNA isolation. Total RNAs were reverse transcribed, and the resulting cDNAs were subjected to real-time PCR quantitation using specific primers and probes for LHR. The graph represents changes in mRNA levels normalized to 18S rRNA and shown as fold change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG; n = 6.
Inhibition of cAMP/PKA-ERK1/2-miR122 pathway abolished hCG-induced activation of SREBP-1a/SREBP-2
Because LRBP, the target of miR-122, has been characterized as mevalonate kinase, the potential intermediary role of SREBP in mediating the effect of miR-122 action was examined. The rationale for this approach was that it has been well established that SREBPs regulate the expression of oxysterol responsive gene products, such as mevalonate kinase (27). We therefore examined what effect inhibiting miR-122 will have on SREBP levels. The results showed that, as expected hCG increased the activation of both SREBP-1a and SREBP-2, as evidenced by the increase in the intensity of the active (68 kDa) band (Figure 4A, lane 2, panels 1 and 2, respectively). The expression of LRBP was also increased (3.1-fold vs CTL; Figure 4A, lane 2, panel 3). However, when miR-122 was inhibited by pretreating superovulated rats with a specific LNA-conjugated antagomir, the activation of both SREBP-1a (2.7-fold vs CTL) and SREBP-2 (2.0-fold vs CTL) as well as up-regulation of LRBP (3.11-fold vs CTL) were completely blocked as evidenced by the significant decrease in the intensities of the bands in Figure 4A, lane 4 (SREBP-1a, 0.78 ± 0.08-fold; SREBP-2, 0.44 ± 0.15-fold; and LRBP, 0.8 ± 0.11-fold vs CTL; P < .05 vs hCG). This suggested that SREBP activation is an intermediate step in miR-122-mediated up-regulation of LRBP in response to hCG treatment. We have previously shown that miR-122 is induced in response to hCG treatment through the activation of cAMP/PKA and ERK pathways (26). To confirm the role of cAMP/PKA and ERK1/2 pathways, we treated rats with the inhibitors H-89 and U0126 before hCG treatment, as previously shown (26). The results in Figure 4B show that the hCG-mediated activation of both SREBP-1a and SREBP-2 was inhibited by pretreatment with U0126 or H-89 (Figure 4B, lanes 3 and 4, panels 1 and 2). The quantitative analysis showed that there was a 1.78 ± 0.18- and 1.84 ± 0.21-fold (vs CTL) increase in SREBP-1a and SREBP-2, respectively, in the hCG-treated samples. H-89 pretreatment brought the levels of the active SREBP-1a and SREBP-2 significantly down to 0.56 ± 0.25- and 0.54 ± 0.22-fold vs CTL, respectively, and U0126 pretreatment reduced it to 0.46 ± 0.1- and 0.56 ± 0.23-fold vs CTL. These results clearly show that SREBP activation is downstream of cAMP/PKA-ERK1/2-miR-122 pathways in the events leading to LH/hCG-induced LHR down-regulation.
Figure 4.

Inhibition of miR-122, PKA, and ERK1/2 abrogates hCG-induced increases in SREBP activation in the superovulated rat ovaries. Superovulated rats were injected either with (A) miR-122 antagomir on day 4 or with H-89 (100 mg/kg b.w.) or U0126 (10 mg/kg b.w.) (B) 1 hour before hCG injection on day 5, and ovaries were collected 4 hours later. S10 fractions were prepared using RIPA buffer. Equal amounts of protein from the CTL or hCG-treated S10 fractions were subjected to Western blot analysis using SREBP-1a antibody. The blots were stripped and reprobed with SREBP-2, LRBP, and finally with tubulin antibody. The blots shown are representative of 3 independent experiments. The graphs represent SREBP levels normalized to tubulin and are shown as percent change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG.
miR-122 antagomir blocks hCG-mediated increases in the recruitment of SREBP to the putative SRE element in the proximal promoter region of LRBP
The binding of SREBP-1a to the promoter region of LRBP was examined using ChIP assay followed by real-time PCR. The binding of SREBP-1a to the LRBP promoter was increased 3.46-fold after hCG treatment, when compared with the CTL (P < .05 vs CTL) (Figure 5, lanes 1 and 2, lower panel). This increase was almost completely abrogated by pretreatment with miR-122 antagomir, before hCG treatment (1.4 ± 0.09 vs CTL, P < .05 vs hCG). Negative CTL (DNA isolated from mouse IgG immunoprecipitates) did not show any detectable Ct (Threshold cycle) values in real-time PCR assay. This showed that SREBP binds to the putative SRE element in the promoter region of LRBP gene, which then increases the LRBP gene transcription. Inhibition of miR-122 by the antagomir treatment resulted in the inhibition of LRBP gene expression as shown in Figure 1, by decreasing the association of active SREBP to the LRBP promoter seen in Figure 5, lane 3. As shown in the upper panel of Figure 5, positive CTL (DNA from chromatin immunoprecipitates using Pol II antibody) showed a strong signal for GAPDH promoter in agarose gel but negative CTL (ChIP using mouse IgG) did not give any signal. The input DNA from all the samples showed equal levels of GAPDH promoter.
Figure 5.

HCG-induced increases in the binding of SREBP to the LRBP promoter was abrogated on pretreatment with miR-122 antagomir. LNA-conjugated miR-122 antagomir was injected into the bursa of the ovaries of superovulated rats on day 4, followed by hCG on day 5. Ovaries were collected 4 hours later and were processed for ChIP assay as described in detail in Materials and Methods. Upper panel, Tissues were cross-linked, and ChIP assay was performed with mouse IgG (negative CTL) or Pol II antibody (positive CTL), as described in Materials and Methods. The purified DNA samples from ChIP assay (positive and negative CTLs) as well as input DNA were amplified by PCR using rat primer sequences for the GAPDH gene promoter, and the PCR products were analyzed using 1.2% agarose gel electrophoresis. Lower panel, The immunoprecipitated DNA, along with input DNA, was subjected to real-time PCR quantitation using specific primers and probes for MVK promoter and GAPDH. The graph represents MVK promoter levels in the immunoprecipitated samples normalized to input DNA and shown as fold change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG; n = 3.
Inhibition of SREBP activation using fatostatin abrogated hCG-induced up-regulation of LRBP mRNA and protein
An alternate approach was employed to examine the role of SREBPs in hCG-induced LRBP up-regulation, by inhibiting SREBP activation using a chemical agent, fatostatin. Fatostatin has been shown to bind the protein SREBP cleavage-activating protein (SCAP) and inhibits the translocation of SREBPs from the endoplasmic reticulum to the Golgi, thereby preventing its proteolytic cleavage and activation (30). To test the effect of SREBP processing on LRBP, rats were treated with fatostatin 4 hours before hCG treatment and inhibition of SREBP activation in the cytosolic fractions were confirmed using Western blotting. As shown in Figure 6A, pretreatment with fatostatin completely abrogated hCG-mediated activation of both SREBP-1a and SREBP-2. A 3.2-fold increase in the levels of active SREBP-1a was seen after hCG treatment, which was reduced significantly (0.4-fold vs CTL) with fatostatin pretreated animals. Similarly the increase in the levels of active form of SREBP-2 was also abrogated by fatostatin (hCG, 3.9-fold vs CTL and Fato+hCG, 0.72-fold vs CTL; P < .05). Next, we analyzed the effects of inhibition of SREBP activation on hCG-mediated LRBP up-regulation. Fatostatin pretreatment significantly inhibited hCG-mediated increase in both LRBP mRNA and protein levels. HCG treatment produced a 100% increase in LRBP mRNA when compared with CTL, but in fatostatin-treated groups, this increase was reduced to 10% (P < .05 vs hCG) (Figure 6B). The same trend was observed in the protein levels also (Figure 6C), because a 1.9-fold increase in response to hCG was inhibited significantly by fatostatin treatment (1.1-fold vs CTL). These results further support that hCG-mediated LRBP up-regulation is downstream of SREBP activation.
Figure 6.

Inhibition of hCG-induced increases in SREBP activation and LRBP expression using fatostatin (Fato) pretreatment. Superovulated rats were injected with fatostatin (30 mg/kg b.w.) 4 hours before hCG injection on day 5, and ovaries were collected 4 hours later. S10 fractions were prepared using RIPA buffer. Equal amounts of protein from the S10 fractions were subjected to Western blot analysis using (A) SREBP-1a antibody followed by stripping and reprobing with SREBP-2 and tubulin antibodies or with (C) LRBP antibody followed by stripping and reprobing with tubulin. The blots shown are representative of 3 independent experiments. The graphs represent active SREBP (68 kDa) levels normalized to tubulin and are shown as percent change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG. B. Separate set of ovaries were processed for total RNA isolation, which were then reverse transcribed, and the resulting cDNAs subjected to real-time PCR quantitation using specific primers and probes for LRBP. The graph represents changes in mRNA levels normalized to 18S rRNA and shown as fold change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL and **, P < .05 vs hCG; n = 3.
Inhibition of SREBP activation reversed hCG-induced increase in mRNP complex formation and decreased LHR mRNA down-regulation
Because SREBP inhibition was found to reverse hCG-induced LRBP up-regulation, it is expected to reflect in the extent of the association of LRBP with LHR mRNA. We examined the changes in the formation of LRBP-LHR mRNP complex using ovarian S100 fractions by REMSA. The results clearly showed that although hCG treatment increased the intensity of the 45-kDa band representing LHR mRNA-LRBP complex (Figure 7A, lane 2), pretreatment with fatostatin reduced this complex formation between LRBP and LHR mRNA (lane 4). Fatostatin alone did not produce any significant change (Figure 7A, lane 3). Similarly, because SREBP activation is upstream of LH/hCG-induced LRBP expression and its association with LHR mRNA, its inhibition would be expected to reduce LRBP-mediated LHR down-regulation. To examine this, we studied the effect of inhibition of SREBP activation on LHR mRNA down-regulation. Analysis of LHR mRNA levels showed that inhibition of SREBP activation significantly suppressed LHR mRNA down-regulation. Consistent with earlier results, hCG treatment caused a 90% decrease in LHR mRNA levels, which was restored partially (50% change) but significantly, by fatostatin pretreatment (Figure 7B).
Figure 7.

Fatostatin (Fato) pretreatment results in the reversal of hCG-induced increases in LHR mRNA binding activity of LRBP and LHR down-regulation. Superovulated rats were injected with fatostatin (30 mg/kg b.w.) 4 hours before hCG injection on day 5, and ovaries were collected 6 hours later. Ovaries were processed for total RNA isolation or REMSA. A, Gel mobility shift analysis was performed using S100 fractions isolated from ovaries from different treatment groups, containing equal amounts of total protein, after incubating with [32P]-labeled LBS (1.5 × 105 c.p.m). The autoradiogram shown is representative of 3 independent experiments. B, Total RNAs were reverse transcribed, and the resulting cDNAs were subjected to real-time PCR quantitation using specific primers and probes for LHR. The graph represents changes in mRNA levels normalized to 18S rRNA and shown as fold change vs CTL. Error bars represent mean ± SE. *, P < .05 vs CTL; **, P < .05 vs hCG; n = 4.
Discussion
Down-regulation of LHR occurs in response to preovulatory LH surge or when exposed to supraphysiological concentration of LH or hCG to mimic the preovulatory LH surge (3–8, 13, 14). This phenomenon occurs in the rodent as well as in the human (12, 20, 23). The present report is an extension of our previous studies to determine the mechanism of the rapid loss of LHR mRNA during this process, using a model system. More recently, we have demonstrated that a microRNA, miR-122, participates in this process (26). The present study unravels the molecular mechanism involved in miR-122-mediated regulation of LRBP and subsequent LHR mRNA degradation after the administration of LH/hCG to superovulated rats, specifically focusing on the role played by SREBPs.
microRNAs in general are thought to elicit their effect by silencing the expression of target genes (24). But recent studies show that they can also increase gene expression of key proteins in several systems indirectly by suppressing other RNAs in the pathway. miR-122 belongs to the latter group that has been linked to the regulation of cholesterol and lipid metabolism (31–37) possibly by targeting repressors of these target proteins. miR-122 was previously thought to be a liver specific noncoding RNA. We have shown that miR-122 is expressed in both rat and human ovarian tissues (26). Other studies have also shown that miR-122 is expressed in other nonhepatic tissues (38–40). In our previous studies, we have shown that miR-122 is expressed in both rat and human ovaries during conditions of LHR down-regulation and mediates hCG-mediated LRBP protein expression (26). In the present study, we present data showing that inhibition of miR-122 inhibits LRBP gene induction as well as LHR mRNA degradation. Treatment with miR-122 antagomir has been shown to inhibit mevalonate kinase and other oxysterol responsive gene products, such as HMG-CoA reductase in the liver (37). Due to these reasons, it has been suggested to be a potentially important clinical target for regulating cholesterol synthesis (37). We now extend these studies showing that miR-122-mediated LRBP induction is an integral part of the ligand-mediated LHR down-regulation.
Our results show that regulation of LRBP (mevalonate kinase) by miR-122 is mediated through the activation of SREBPs. Although there are other studies that show that miR-122 regulates SREBP genes, this is the first study that shows a link between miR-122 and SREBPs in the regulation of LHR expression. SREBP processing and its regulation of cholesterol metabolism have been delineated in detail (see Ref. 27 for review). After processing the inactive precursor in the endoplasmic reticulum, the active SREBP translocate to the nucleus and activates transcription by binding to nonpalindromic SREs in the promoter/enhancer regions of multiple target genes. Our previous studies showed that SREBP-1a and SREBP-2 are activated by hCG (26). In the present study, we used a specific inhibitor of SREBP activation, fatostatin, to determine the effect of inhibiting SREBP activation on LBRP and LHR mRNA regulation. This diarylthiazole derivative impairs the activation of SREBPs, by binding to SCAP and inhibiting the endoplasmic reticulum-Golgi translocation of SREBPs (30). These studies showed that inhibition of SREBP activation inhibits LH/hCG-mediated LRBP up-regulation, which then leads to abrogation of LHR mRNA down-regulation. It is noteworthy that miR-122 antagomir pretreatment inhibited the activation of SREBPs, thereby reducing the availability as well as binding of active SREBP-1a to the putative SRE element in the promoter region of LRBP after hCG treatment. This suggested that miR-122 probably directly targets an inhibitory gene in the SREBP activation pathway, thereby increasing the activation of SREBPs, which then binds to LRBP promoter leading to increased LRBP gene expression. The role of SREBPs in LRBP up-regulation is supported by other studies which show that genes involved in cholesterol biosynthesis containing classic SREs in their promoters are strongly and efficiently activated by both SREBP-1a and SREBP-2 (41). Our results are consistent with these reports and further highlight the role of SREBPs in the regulation of LH receptor expression in the ovaries. This is supported by the recent studies showing that SREBPs are involved in the regulation of a large number of genes that are at the crossroads of different functional pathways, some of them unrelated to cholesterol or lipid metabolism (42).
Our demonstration that inhibition of cAMP/PKA-ERK1/2 as well as miR-122 inhibits SREBP activation provides further insights into the mechanism of LH/hCG-mediated LRBP induction. Furthermore, both miR-122 and SREBP have been shown to play crucial role in the regulation of cholesterol biosynthetic pathway in liver (27, 35). Our results further show that they also regulate an important physiological process by controlling the expression of LHR mRNA in the ovary which is crucial in mammalian reproduction. The implications of our findings is that during preovulatory LH surge, miR-122, expressed in response to LH/hCG stimulation activates SREBPs to induce up-regulation of LRBP, which then binds to LHR mRNA to induce its degradation leading to LHR down-regulation (Figure 8). With the termination of the down-regulation, the suppressive effect of hCG on LHR down-regulation is reversed by a decline in expression of miR-122 and SREBP followed by LRBP. This results in resumption of normal ovarian function. In summary, the present studies provide a mechanism by which miR-122 regulates LHR expression in the ovary.
Figure 8.
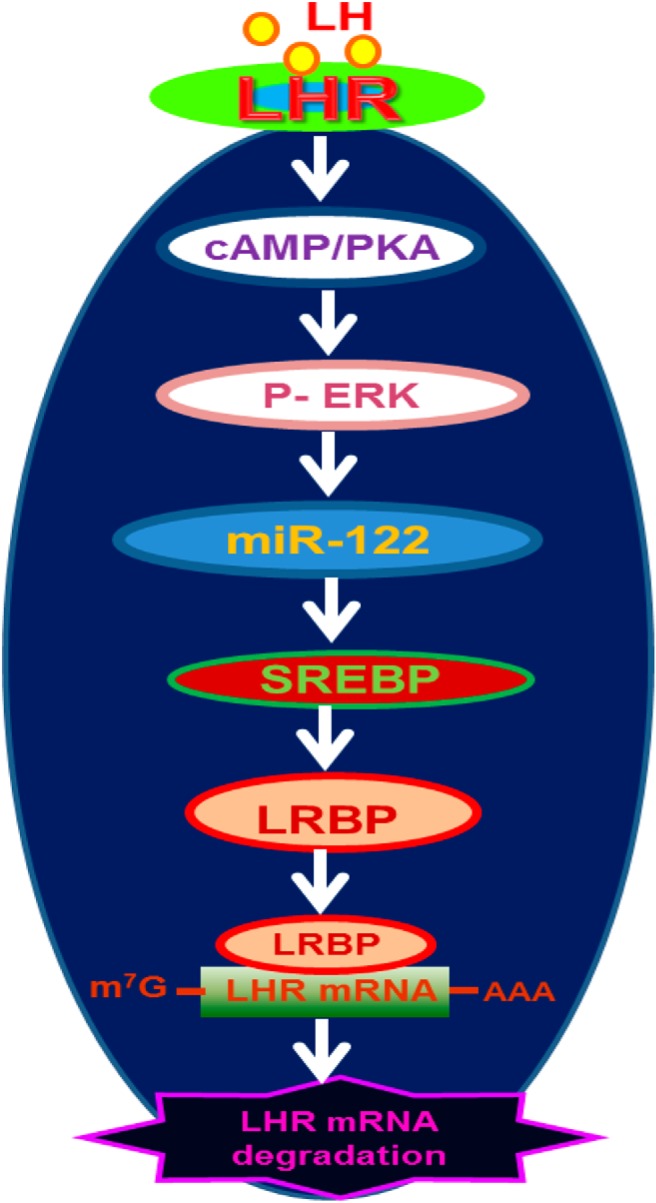
Schematic model depicting the proposed signaling pathway in LH/hCG-induced miR-122-mediated LHR mRNA down-regulation. Binding of ligand to LH receptor induces activation of cAMP/PKA signaling followed by ERK1/2. This leads to an increase in the expression of miR-122, which causes the activation of SREBPs. Activated SREBPs increase the gene expression of LRBP, and the increase in translated protein is expressed as an increase in its LHR mRNA binding activity, ultimately leading to the degradation of LHR mRNA.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 HD06656.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- b.w.
- body weight
- ChIP
- chromatin immunoprecipitation
- CTL
- control
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- hCG
- chorionic gonadotropin
- LBS
- [α-32P]UTP-labeled LRBP binding sequence
- LHR
- LH/human gonadotropin hormone receptor
- LNA
- locked nucleic acid
- LRBP
- LHR mRNA binding protein
- mRNP
- mRNA-protein complex
- MVK
- mevalonate kinase
- PKA
- protein kinase A
- Pol II
- polymerase II
- REMSA
- RNA EMSA
- SRE
- sterol response element
- SREBP
- sterol regulatory element binding protein.
References
- 1. McFarland KC, Sprengel R, Phillips HS, et al. Lutropin-choriogonadotropin receptor: an unusual member of the G protein-coupled receptor family. Science. 1989;245(4917):494–499. [DOI] [PubMed] [Google Scholar]
- 2. Loosfelt H, Misrahi M, Atger M, et al. Cloning and sequencing of porcine LH-hCG receptor cDNA: variants lacking transmembrane domain. Science. 1989;245(4917):525–528. [DOI] [PubMed] [Google Scholar]
- 3. LaPolt PS, Oikawa M, Jia XC, Dargan C, Hsueh AJ. Gonadotropin-induced up- and down-regulation of rat ovarian LH receptor message levels during follicular growth, ovulation and luteinization. Endocrinology. 1990;126(6):3277–3279. [DOI] [PubMed] [Google Scholar]
- 4. Segaloff DL, Wang HY, Richards JS. Hormonal regulation of luteinizing hormone/chorionic gonadotropin receptor mRNA in rat ovarian cells during follicular development and luteinization. Mol Endocrinol. 1990;4(12):1856–1865. [DOI] [PubMed] [Google Scholar]
- 5. Hoffman YM, Peegel H, Sprock MJ, Zhang QY, Menon KMJ. Evidence that human chorionic gonadotropin/luteinizing hormone receptor down-regulation involves decreased levels of receptor messenger ribonucleic acid. Endocrinology. 1991;128(1):388–393. [DOI] [PubMed] [Google Scholar]
- 6. Lu DL, Peegel H, Mosier SM, Menon KMJ. Loss of lutropin/human choriogonadotropin receptor messenger ribonucleic acid during ligand-induced down-regulation occurs post transcriptionally. Endocrinology. 1993;132(1):235–240. [DOI] [PubMed] [Google Scholar]
- 7. Peegel H, Randolph J, Jr, Midgley AR, Menon KMJ. In situ hybridization of luteinizing hormone/human chorionic gonadotropin receptor messenger ribonucleic acid during hormone-induced down-regulation and the subsequent recovery in rat corpus luteum. Endocrinology. 1994;135(3):1044–1051. [DOI] [PubMed] [Google Scholar]
- 8. Nair AK, Kash JC, Peegel H, Menon KMJ. Post-transcriptional regulation of luteinizing hormone receptor mRNA in the ovary by a novel mRNA-binding protein. J Biol Chem. 2002;277(24):21468–21473. [DOI] [PubMed] [Google Scholar]
- 9. Minegishi T, Tano M, Abe Y, Nakamura K, Ibuki Y, Miyamoto K. Expression of luteinizing hormone/human chorionic gonadotrophin (LH/HCG) receptor mRNA in the human ovary. Mol Hum Reprod. 1997;3(2):101–107. [DOI] [PubMed] [Google Scholar]
- 10. Zeleznik AJ. The physiology of follicle selection. Reprod Biol Endocrinol. 2004;2:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ikeda S, Nakamura K, Kogure K, et al. Effect of estrogen on the expression of luteinizing hormone-human chorionic gonadotropin receptor messenger ribonucleic acid in cultured rat granulosa cells. Endocrinology. 2008;149(4):1524–1533. [DOI] [PubMed] [Google Scholar]
- 12. Nair AK, Peegel H, Menon KMJ. The role of luteinizing hormone/human chorionic gonadotropin receptor-specific mRNA binding protein in regulating receptor expression in human ovarian granulosa cells. J Clin Endocrinol Metab. 2006;91(6):2239–2243. [DOI] [PubMed] [Google Scholar]
- 13. Kash JC, Menon KMJ. Identification of a hormonally regulated luteinizing hormone/human chorionic gonadotropin receptor mRNA binding protein. Increased mrna binding during receptor down-regulation. J Biol Chem. 1998;273(17):10658–10664. [DOI] [PubMed] [Google Scholar]
- 14. Kash JC, Menon KMJ. Sequence-specific binding of a hormonally regulated mRNA binding protein to cytidine-rich sequences in the lutropin receptor open reading frame. Biochemistry. 1999;38(51):16889–16897. [DOI] [PubMed] [Google Scholar]
- 15. Nair AK, Menon KMJ. Isolation and characterization of a novel trans-factor for luteinizing hormone receptor mRNA from ovary. J Biol Chem. 2004;279(15):14937–14944. [DOI] [PubMed] [Google Scholar]
- 16. Nair AK, Menon KMJ. Regulation of luteinizing hormone receptor expression: evidence of translational suppression in vitro by a hormonally regulated mRNA-binding protein and its endogenous association with luteinizing hormone receptor mRNA in the ovary. J Biol Chem. 2005;280(52):42809–42816. [DOI] [PubMed] [Google Scholar]
- 17. Nair AK, Young MA, Menon KMJ. Regulation of luteinizing hormone receptor mRNA expression by mevalonate kinase–role of the catalytic center in mRNA recognition. FEBS J. 2008;275(13):3397–3407. [DOI] [PubMed] [Google Scholar]
- 18. Wang L, Gulappa T, Menon KMJ. Identification and characterization of proteins that selectively interact with the LHR mRNA binding protein (LRBP) in rat ovaries. Biochim Biophys Acta. 2010;1803(5):591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang L, Menon KMJ. Regulation of luteinizing hormone/chorionic gonadotropin receptor messenger ribonucleic acid expression in the rat ovary: relationship to cholesterol metabolism. Endocrinology. 2005;146(1):423–431. [DOI] [PubMed] [Google Scholar]
- 20. Wang L, Nair AK, Menon KMJ. Ribonucleic acid binding protein-mediated regulation of luteinizing hormone receptor expression in granulosa cells: relationship to sterol metabolism. Mol Endocrinol. 2007;21(9):2233–2241. [DOI] [PubMed] [Google Scholar]
- 21. Menon B, Peegel H, Menon KMJ. Evidence for the association of luteinizing hormone receptor mRNA-binding protein with the translating ribosomes during receptor downregulation. Biochim Biophys Acta. 2009;1793(11):1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menon B, Sinden J, Menon KMJ. Association of luteinizing hormone receptor (LHR) mRNA with its binding protein leads to decapping and degradation of the mRNA in the p bodies. Biochim Biophys Acta. 2013;1833(5):1173–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menon B, Franzo-Romain M, Damanpour S, Menon KMJ. Luteinizing hormone receptor mRNA down-regulation is mediated through ERK-dependent induction of RNA binding protein. Mol Endocrinol. 2011;25(2):282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–531. [DOI] [PubMed] [Google Scholar]
- 25. Valinezhad Orang A, Safaralizadeh R, Kazemzadeh-Bavili M. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int J Genomics. 2014;2014:970607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Menon B, Sinden J, Franzo-Romain M, Botta RB, Menon KMJ. Regulation of LH receptor mRNA binding protein by miR-122 in rat ovaries. Endocrinology. 2013;154(12):4826–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang T, Espenshade PJ, Wright ME, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489–500. [DOI] [PubMed] [Google Scholar]
- 29. Barnett KR, Tomic D, Gupta RK, et al. The aryl hydrocarbon receptor is required for normal gonadotropin responsiveness in the mouse ovary. Toxicol Appl Pharmacol. 2007;223(1):66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kamisuki S, Mao Q, Abu-Elheiga L, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol. 2009;16(8):882–892. [DOI] [PubMed] [Google Scholar]
- 31. Nie YQ, Cao J, Zhou YJ, et al. The effect of miRNA-122 in regulating fat deposition in a cell line model. J Cell Biochem. 2014;115(5):839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L, Tang W, Yan S, et al. Efficient delivery of miR-122 to regulate cholesterol metabolism using a non-covalent peptide-based strategy. Mol Med Rep. 2013;8(5):1472–1478. [DOI] [PubMed] [Google Scholar]
- 33. Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122(8):2884–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hsu SH, Wang B, Kota J, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122(8):2871–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moore KJ, Rayner KJ, Suárez Y, Fernández-Hernando C. microRNAs and cholesterol metabolism. Trends Endocrinol Metab. 2010;21(12):699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98. [DOI] [PubMed] [Google Scholar]
- 37. Krützfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. [DOI] [PubMed] [Google Scholar]
- 38. Sun JG, Liao RX, Qiu J, et al. Microarray-based analysis of microRNA expression in breast cancer stem cells. J Exp Clin Cancer Res. 2010;29:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang B, Wang H, Yang Z. MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS One. 2012;7(10):e47053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abu-Halima M, Hammadeh M, Schmitt J, et al. Altered microRNA expression profiles of human spermatozoa in patients with different spermatogenic impairments. Fertil Steril. 2013;99(5):1249–1255.e16. [DOI] [PubMed] [Google Scholar]
- 41. Amemiya-Kudo M, Shimano H, Hasty AH, et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res. 2002;43(8):1220–1235. [PubMed] [Google Scholar]
- 42. Rome S, Lecomte V, Meugnier E, et al. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol Genomics. 2008;34(3):327–337. [DOI] [PubMed] [Google Scholar]