

Abstract
Founder mutations are an important cause of Lynch syndrome and facilitate genetic testing in specific ethnic populations. Two putative founder mutations in MSH6 were analyzed in 2685 colorectal cancer (CRC) cases, 337 endometrial cancer (EnCa) cases and 3310 healthy controls of Ashkenazi Jewish (AJ) descent from population-based and hospital-based case-control studies in Israel, Canada and the USA. The carriers were haplotyped and the age of the mutations was estimated. MSH6*c.3984_3987dupGTCA was found in 8/2685 CRC cases, 2/337 EnCa cases, and 1/3310 controls, consistent with a high risk of CRC (odds ratio (OR) = 9.9, 95% confidence interval (CI) = 1.2–78.9, p=0.0079) and a very high risk of EnCa (OR = 19.6, 95%CI = 1.8–217.2, p = 0.0006). MSH6*c.3959_3962delCAAG was identified in 3/2685 CRC cases, 2/337 EnCa cases and no controls. Each mutation was associated with separate conserved haplotypes. MSH6*c.3984_3987dupGTCA and MSH6*c.3959_3962delCAAG likely arose around 585 CE and 685 CE respectively. No carriers were identified in Sephardi Jews (450 cases and 490 controls). Truncating mutations MSH6*c.3984_3987dupGTCA and MSH6*c.3959_3962delCAAG cause Lynch syndrome and are founder mutations in Ashkenazi Jews. Together with other AJ founder mutations, they contribute substantially to the incidence of CRC and EnCa and are important tools for the early diagnosis and appropriate management of AJ Lynch syndrome patients.
Keywords: MSH6, founder mutation, Lynch syndrome, Ashkenazi Jews
INTRODUCTION
Lynch syndrome is the most common autosomal dominant condition predisposing to colorectal cancer. It is characterized by early onset cancers of the colorectum, endometrium, small bowel, ureter and renal pelvis [1], as well as other malignancies [2]. Lynch syndrome is caused by germline mutations in mismatch-repair (MMR) genes, the common of which occur in MLH1 [3] and MSH2 [4], and are less frequently seen in MSH6 [5, 6] and PMS2 [7].
The MSH6 gene is localized at chromosome 2p15–16 and consists of 10 exons encoding a 1360 amino acid protein. The MSH6 protein interacts with the MSH2 protein to form the MutSα heterodimer [5]. The primary function of MutSα is to initiate the repair process by binding to DNA mismatches detected by MSH6. The characteristic role of this heterodimer is to correct single mispaired bases or small insertion or deletion loops [8]. Most reports indicate an attenuated phenotype of MSH6-related Lynch syndrome in comparison with mutations of MLH1 or MSH2 genes. Although fewer families with MSH6 mutations fulfill the Amsterdam criteria than families with either MLH1 or MSH2 mutations, the characteristic features of MSH6 mutation families comprise a higher risk for CRC, endometrial, ovarian, upper urinary tract, and stomach cancers [9]. Moreover, families with MSH6 mutations have a higher incidence of extracolonic cancers in comparison with other Lynch families [10].
Several studies have investigated the frequency of MSH6 mutations in CRC and endometrial cancers. About 10% of Lynch syndrome colorectal cancers and 0.3% of all colorectal cancers are explained by the mutations in MSH6 [11, 12]. The prevalence of MSH6 mutations in endometrial cancer patients who were not selected for family history is about 1.1% [13]. A recent study of 113 MSH6 families estimated cumulative risks to age 80 years for colorectal cancer to be 44% for men and 20% for women; for endometrial cancer, the risk was 44%. MSH6 mutation carriers had an eightfold increased incidence of colorectal cancer in comparison with incidence in general population and women with MSH6 mutations had a 26-fold increased incidence of endometrial cancer [14]. In families with MSH6 mutations, the onset of CRC and endometrial cancer is significantly delayed compared to families with MLH1 or MSH2 mutations (MSH6 vs. MSH2/MLH1 = 55 years vs. 44/41 years for CRC and 55 years vs. 49/48 years for endometrial cancer) [15].
Over 500 unique variants have been identified in MSH6 gene [16]. Two recently described mutations in MSH6 exon 9 are truncating mutations identified in several families around the world: the mutation c.3959_3962delCAAG (rs63751151) (p.Ala1320GlufsX6) was first reported in an endometrial cancer patient of unknown ethnicity from the United States, where her MSI-positive tumor with an unmethylated MLH1 promoter was diagnosed at the age 71 [17] and the mutation c.3984_3987dupGTCA (p.Leu1330ValfsX12), was first reported to occur in an Ashkenazi Jewish (AJ) family with a history of CRC and adenomatous polyps [18] and using slightly different nomenclature was later reported in two families; one from the Netherlands [19] and another family from the United States [20] with recently published Israeli study finding c.3984_3987dupGTCA in 19 members from four AJ families [21].
The aim of the present study was to characterize the MSH6 mutations c.3984_3987dupGTCA (referred below as MSH6*dup) and c.3959_3962delCAAG (referred below as MSH6*del) and estimate the magnitude of the contribution of these mutations to the Lynch syndrome in the AJ population. The frequency of these mutations was evaluated in a large, population-based case-control study in northern Israel and in series of AJ individuals with colorectal, endometrial, or ovarian cancer, ascertained in North America. Furthermore, we sought to establish whether these two mutations are founder mutations and to calculate the age of these mutations in the AJ population.
MATERIALS AND METHODS
Study samples
The present study included individuals of AJ descent from centers in Israel, Canada, and the United States. We studied the frequency of these two mutations in several different types of datasets - a) population-based series of cases and controls (Israel); b) individuals with a personal and/or family history of colorectal cancer and other Lynch syndrome-associated tumors referred to high-risk clinics (Montreal); c) familial gastrointestinal cancer registry-based cases (Toronto); d) hospital-based series of cases and controls (New York); e) unaffected control individuals (Montreal and Toronto) and f) carriers identified through a CLIA-certified commercial laboratory (Mayo clinic). These datasets are described briefly below –
a) Population-based – the MECC case-control study from Israel
DNA samples from 2685 cases and 1591 healthy controls from the large Israeli population-based study of colorectal cancer were genotyped in the University of Michigan. The Molecular Epidemiology of Colorectal Cancer (MECC) study is a population-based case-control study of colorectal cancer in northern Israel. The cases include histologically confirmed incident colorectal cancer patients between 1998 and 2009. Population-based controls were enrolled from the Clalit Health Services database (the largest Israeli health care provider) and matched by year of birth, gender, clinic, and Jewish/Non-Jewish ethnicity.
b) High-risk clinics
Montreal: Individual J2205-2 (Table 1) was found to have the MSH6*dup mutation after full sequencing of MSH6. She was diagnosed with colon and endometrial cancer. The family history met Bethesda guidelines, but did not fulfill Amsterdam criteria. Family members of the index case were analyzed for the mutation and haplotyped at the Jewish General Hospital (JGH). Additionally, 22 CRC cases and 22 unaffected individuals from other high-risk colorectal cancer families referred to the cancer genetics clinic at the JGH were analyzed for the two MSH6 mutations. All cases were negative for other known AJ founder mutations in CRC genes (MLH1*c. 394C>G (common name D132H), MSH2*c.1906G>C (common name A636P), APC*c.3920T>A (common name I1307K), BLM*c.2207_2212delATCTGAinsTAGATTC (common name BLMAsh).
Table 1.
Clinicopathological characteristics of affected Ashkenazi Jewish carriers of the MSH6* c.3984_3987dupGTCA and MSH6* c.3959_3962delCAAG mutations identified in the participating centers
| Center/Study | Family | Patient | Sex | Age at ascertainment | MSH6 ex9 indel | Clinical Features | Amsterdam Criteria | MSI | IHC |
|---|---|---|---|---|---|---|---|---|---|
| McGill | 1 | 1 | female | 68 | c.3984_3987dupGTCA | Endometrial cancer (66) | No | N/A | MSH6(−) |
| McGill | 1 | 2 | female | 44 | c.3984_3987dupGTCA | Colorectal cancer (40), Endometrial cancer (43) | No | MSS | N/A |
| McGill | 2 | 1 | female | 87 | c.3984_3987dupGTCA | Metachronous Colorectal cancer (64, 65), SCC (86), BCC (87) | No | N/A | N/A |
| McGill | 2 | 2 | female | 54 | c.3984_3987dupGTCA | Colorectal cancer (50), Endometrial cancer (50) | No | N/A | MSH6(−) |
| MECC_case | 3 | 1 | male | 70 | c.3984_3987dupGTCA | Colorectal cancer (70) | No | MSS | N/A |
| MECC_case | 4 | 1 | female | 86 | c.3984_3987dupGTCA | Endometrial cancer (47), Ovarian cancer (66), Colorectal cancer (86) | No | MSI-High (mono) | N/A |
| MECC_case | 5 | 1 | female | 79 | c.3984_3987dupGTCA | Colorectal cancer (79) | No | MSI-High (mono) | N/A |
| MECC_case | 6 | 1 | male | 55 | c.3984_3987dupGTCA | Colorectal cancer (55) | No | MSI-High | N/A |
| MECC_case | 7 | 1 | female | 73 | c.3984_3987dupGTCA | Endometrial cancer (58), Cecal cancer (73) | Amsterdam II | N/A | MSH6(−) |
| MECC_case | 8 | 1 | female | 52 | c.3984_3987dupGTCA | Colorectal cancer (52) | N/A | N/A | N/A |
| MECC_case | 9 | 1 | male | 56 | c.3984_3987dupGTCA | Colorectal cancer (56) | No | MSI-High (mono) | N/A |
| MECC_control | 10 | 1 | female | 71 | c.3984_3987dupGTCA | Endometrial cancer (61), Cecal cancer (76) | No | N/A | N/A |
| Toronto | 11 | 1 | female | 85 | c.3984_3987dupGTCA | Breast cancer (75), Colorectal cancer (81) | No | N/A | N/A |
| Toronto | 11 | 2 | female | 54 | c.3984_3987dupGTCA | Colorectal cancer (50), Endometrial cancer (51) | No | N/A | N/A |
| Mayo | 12 | 1 | male | 75 | c.3984_3987dupGTCA | SA (71), SCC (71), BCC (71) | No | N/A | MSH6(−) |
| Mayo | 12 | 2 | male | 75 | c.3984_3987dupGTCA | SCC (71), SA (71), BCC (75), SA (75) | No | N/A | N/A |
| MSKCC | 19 | 1 | female | 57 | c.3984_3987dupGTCA | Endometrial cancer (57) | No | N/A | MSH6(+) |
| MSKCC | 20 | 1 | female | 68 | c.3984_3987dupGTCA | Endometrial cancer (68) | No | N/A | MSH6(−) |
|
| |||||||||
| MECC_case | 13 | 1 | male | 69 | c.3959_3962delCAAG | Colorectal cancer (69) | No | MSS | N/A |
| MECC_case | 14 | 1 | male | 82 | c.3959_3962delCAAG | Colorectal cancer (60), Colorectal cancer (82) | No | N/A | N/A |
| MECC_case | 15 | 1 | female | 84 | c.3959_3962delCAAG | Colorectal cancer (84) | N/A | N/A | N/A |
| Toronto | 16 | 1 | male | 73 | c.3959_3962delCAAG | Colorectal cancer (70) | No | MSI-High | MSH6(−) |
| Toronto | 17 | 1 | male | 68 | c.3959_3962delCAAG | Colorectal cancer (62) | No | MSI-High | N/A |
| Toronto | 18 | 1 | male | 72 | c.3959_3962delCAAG | Rhabdomyosarcoma (39), Colorectal cancer (64), Cancer of pancreas (71) | Amsterdam II | MSI-High | N/A |
| MSKCC | 21 | 1 | female | 43 | c.3959_3962delCAAG | Endometrial cancer (43) | No | N/A | N/A |
| MSKCC | 22 | 1 | female | 49 | c.3959_3962delCAAG | Endometrial cancer (49) | No | N/A | N/A |
CRC – colorectal cancer; SCC – squamous cell carcinoma; BCC – basal cell carcinoma; SA – sebaceous carcinoma; N/A- not available; Mono – MSI in mononucleotide microsatellite markers only.
c) Familial Gastrointestinal Cancer Registry
Total of 490 AJ were collected through the Familial Gastro-Intestinal Cancer Registry (FGICR) in Toronto, Ontario, Canada and their DNA extracted from blood was tested for MSH6 exon 9 mutations. This registry constitutes a provincial resource that registers individuals with suspected or verified hereditary cancers and, for the Jewish population, also those who underwent GI cancer screening irrespective of their family history of cancer. Family histories are obtained through a standardized questionnaire.
d) Hospital series
A total of 337 hospital-based AJ women with endometrial cancer and 285 AJ female controls with a mean age of 58 years from Memorial Sloan-Kettering Cancer Center (MSKCC) were tested for both MSH6 mutations.
e) Volunteering controls
Women’s College Hospital, Toronto: 1011 AJ female controls were genotyped for both MSH6 exon 9 mutations. The mean age of the controls was 48.5 years, ranging from 25 years to 78 years.
Jewish General Hospital, Montreal: 423 unaffected AJ controls (215 females, 201 males, 1 unknown) were tested for both MSH6 mutations. The mean age of 402 controls was 54.3 years with a range from 18 years to 93.9 years. The age was unknown for 21 controls.
f) Clinical Laboratory series
Two carriers of MSH6*dup mutation were identified in the Mayo Medical Laboratory mutation database. These carriers were invited to participate in this research through the University of Michigan Cancer Genetics Registry. The patients signed written, informed consent and donated additional blood samples for haplotype analysis.
Genotyping of MSH6 exon 9 and haplotype analysis
Methods for screening of MSH6 exon 9 for deletions and duplications varied between different centers, but all carriers were re-analyzed and haplotyped at the University of Michigan. DNA fragments containing exon 9 and microsatellite markers were amplified using PCR. PCR product size was determined by fragment length analysis using ABI3730 instrument (Applied Biosystems, Foster City, CA) and compared to the positive control carrying MSH6*dup and negative control (tested by sequencing) using GeneMarker (State College, PA) software.
Samples with the fragment size different from expected 164 base pairs (bp) were sequenced using the same set of primers from both sides. Single nucleotide polymorphisms (SNPs) were analyzed by direct sequencing. Primers and reaction conditions are described in Supplementary Table 1.
Microsatellite Instability analysis and Immunohistochemistry
Microsatellite instability assays were performed as described elsewhere [22] using microdissected DNA from paraffin-embedded tissue blocks. Five markers (BAT25, BAT26, D2S123, D5S346, and D17S250, often called the Bethesda panel) were PCR amplified using radioactively labeled primers. The patterns of the microsatellite markers in normal tissue and tumor were compared to find changes in marker length. Samples were categorized according to established criteria [7]: MSI-High (two or more markers are unstable), MSI-Low (one marker is unstable), microsatellite stable (MSS) ( no markers being unstable).
Tumor samples from MSH6 exon 9 mutation carriers were retrieved and tested for the expression of MLH1, MSH2, MSH6 and PMS2 proteins. Immunohistochemistry (IHC) was performed on 4-μm sections of formalin-fixed and paraffin-embedded tissues. Sections were dried, de-paraffinized and re-hydrated. Slides were stained with monoclonal antibodies. The protein expression in normal lymphocytes and colonocytes adjacent to the tumor served as an internal positive control.
Statistical Analysis
Association between MSH6 mutations and risk of colorectal and endometrial cancers was estimated using Fisher exact test at SAS 9.1 (The SAS Institute, Cary, NC). Haplotype reconstruction was performed using PHASE v.2.1.1 [23, 24]. Uncertain genotype phases in haplotype estimates were excluded from the analysis. The age of the mutation was estimated using a Bayesian MCMC linkage disequilibrium mapping approach (DMLE+ software) [25]. Genetic distance was estimated at 1 Mb corresponding to 1 cM. We assumed population growth rate 1.125-fold per generation, considering the population of Jewish people in a north-eastern European region of 11,000 in the year 600 and 5 million in the year 1900. Considering a lifetime risk of colorectal cancer of 6.85%, a worldwide population of Ashkenazi Jews of 13 million [26], and the mutation prevalence among CRC cases of 0.3% (duplication) and 0.1% (deletion) estimated from the MECC study, we calculated the number of disease chromosomes in AJ population: 2,672 for duplication and 891 for deletion. Accordingly, the proportion of mutation carrying chromosomes sampled was 0.00299 (8/2672) and 0.00337 (3/891) for duplication and deletion respectively.
RESULTS
Identification and screening for MSH6 exon 9 indel mutations
An AJ family found to carry MSH6*dup was evaluated at McGill University for Lynch Syndrome. Based on this case and prior reports of this mutation in the literature, we hypothesized that this variant might represent a founder mutation identifiable among Ashkenazi Jews. This led to a comprehensive search among investigators with large series of AJ colorectal cancer cases, endometrial cancer cases, and controls. One further family from Montreal, one family from Toronto, eight families from northern Israel, and two families from MSKCC were found to carry the same mutation. One family ascertained through Mayo Medical Labs with MSH6*dup mutation was also ascertained. Since the mutation screening relied on fragment size analysis of PCR products, a second mutation within the same region was also recognized corresponding to a truncating mutation, MSH6*del in three families from northern Israel, three families from Toronto, and two families from MSKCC (Table 1).
Frequency of MSH6 exon 9 dup-del mutations in Ashkenazi Jews and risk of colorectal cancer
We evaluated the frequency of MSH6*dup and MSH6*del mutations in the MECC study with the addition of control AJ individuals from multiple centers in Israel, Canada, and the United States. MSH6*dup was found in 8 of 2685 (0.3%) cases and 1 of 3310 (0.03%) controls. The only control carrying MSH6*dup had an endometrial cancer at age 61 and developed cecal cancer at age 76, several years after being ascertained as a control without CRC. Considering this fact and epidemiologic principles [27], we included this carrier to both case and control groups to calculate the odds ratio (OR). To evaluate the risk of colorectal cancer associated with these two MSH6 mutations, we used MECC cases (n = 2685) and controls (n = 1591) as well as large series of healthy Ashkenazi Jews from a study carried out at Women’s College Research Institute, Toronto (n =1011), Memorial Sloan-Kettering Cancer Center, New York (n = 285) and Jewish General Hospital, Montreal (n = 423). The carriers of MSH6 had a very high risk of CRC (OR = 9.9, 95% confidence interval (CI) = 1.2–78.6, p = 0.0079) (Table 2). The MSH6*del was less common, identified in 3 of 2685 (0.11%) cases and no controls. No carriers of either mutation were identified in 450 Sephardi Jewish cases or 490 Sephardi Jewish controls from Israel.
Table 2.
Risk of colorectal and endometrial cancers among MSH6 exon 9 indel mutation carriers in an Ashkenazi Jewish population
| Mutation | Cases | Controls | OR | 95%CI | p-value |
|---|---|---|---|---|---|
| Colorectal cancer | |||||
| c.3984_3987dupGTCA | 8/2685 (0.3%) | 1/3310 (0.03%) | 9.9 | 1.2–78.9 | 0.0079 |
| c.3962_3965delCAAG | 3/2685 (0.1%) | 0/3310 | ∞ | ∞ | 0.0546 |
| MSH6 exon 9 dup or del | 11/2685 (0.4%) | 1/3310 (0.03%) | 13.6 | 1.8–105.1 | 0.0011 |
| Endometrial cancer* | |||||
| c.3984_3987dupGTCA | 2/337 (0.6%) | 1/3310 (0.03%) | 19.6 | 1.8–217.2 | 0.0006 |
| c.3962_3965delCAAG | 2/337 (0.6%) | 0/3310 | ∞ | ∞ | <0.0001 |
| MSH6 exon 9 dup or del | 4/337 (1.2%) | 1/3310 (0.03%) | 39.3 | 4.4–352.5 | <0.0001 |
Endometrial cancer risk was estimated from MSKCC hospital-based series.
MSH6*dup and MSH6*del mutations in patients and controls from hospital-based series
At the Jewish General Hospital, Montreal, one of the 22 (4.5%) CRC affected probands from high-risk colorectal cancer families carried MSH6*dup, and there were no MSH6*del mutations found.
One carrier of MSH6*dup and three carriers of MSH6*del were identified in 490 AJ patients from Toronto registry. After testing family members of the identified carriers, two carriers of MSH6*dup (family 11) and four MSH6*del carriers (families 16 and 17) were detected (Figs. 1 and 2). In 337 hospital-based cases of AJ patients with endometrial cancer from MSKCC, two carriers (0.6%) of MSH6*dup and two carriers (0.6%) of MSH6*del mutations were identified. Roughly estimated risk of endometrial cancer in AJ MSH6*dup mutation carriers is high (OR = 19.6, 95%CI = 1.8–217.2, p = 0.0006) (Table 2).
Figure 1.

Representative families carrying c.3984_3987dupGTCA. Mutation carriers are indicated by “+/−“. None of the families meet Amsterdam criteria, but history of family cancers is suggestive of mismatch-repair mutations.
Figure 2.

Representative families carrying c.3959_3962delCAAG. RMS – Rhabdomyosarcoma. Mutation carriers are indicated by “+/−“. None of the families meet Amsterdam criteria, but history of family cancers is suggestive of mismatch-repair mutations.
Both affected twins from Mayo clinic were diagnosed with sebaceous adenoma, squamous cell carcinoma, and basal cell carcinoma, but did not have any Lynch syndrome associated cancers. In one of the brothers, IHC analysis of sebaceous adenoma showed impaired expression of MSH6 protein. Testing for mutations in exon 9 in MSH6 showed MSH6*dup in both brothers.
Clinicopathological features of MSH6 founder mutation carriers
Among all carriers the mean age at diagnosis of colorectal cancer was 66 years (median 67 years), and the mean age at diagnosis of endometrial cancer was 54 years (median 52 years). Microsatellite instability and immunohistochemistry staining against mismatch repair proteins were performed in carriers with available tumor tissue (Table 1). MSI status of the tumors was determined for 37% of carriers (10/27) and IHC was available for three colorectal cancers, three endometrial tumors, and 1 sebaceous adenoma. In our data, 33% (3/10) of carriers had MSS tumors and 67% (7/10) had MSI-High tumors. It is noteworthy that 3 out of 7 MSI-High tumors were unstable in mononucleotide markers only. In six out of seven tumors with IHC, the MSH6 protein was not expressed, while other mismatch repair proteins were intact (MSH2, MLH1, PMS2).
Characteristics of MSH6 mutation-positive families
A total of 19 MSH6*dup carriers and 12 MSH6*del carriers from 22 unrelated families were identified (Table 3, Figs. 1 and 2). Among all carriers, 18 MSH6*dup carriers and 8 MSH6*del carriers were affected with colorectal or endometrial cancers (Table 1). None of the families met Amsterdam criteria I and only two families met Amsterdam criteria II (families 7 and 18). Although families did not fulfill Amsterdam criteria, the family history of cancer is suggestive of mutations in the mismatch-repair genes. In family 11, both mother (I.2) and daughter (II.4) had colorectal cancer, while the daughter was diagnosed at the age of 50 followed by the diagnosis of endometrial cancer in a year. In families 16 and 17, the patients (I.1 and I.1) had two synchronous colorectal tumors; the proband in the family 7 and the patient I.2 from family 2 were diagnosed with metachronous colorectal cancer. It is noteworthy that three MSH6*dup carriers had squamous cell carcinoma (SCC) and basal cell carcinoma (BCC).
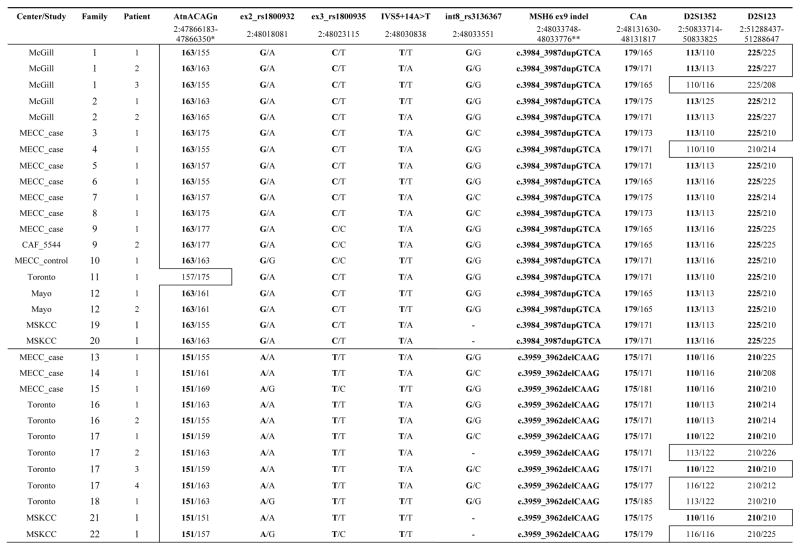
Table 3.
Haplotype analysis of the carriers of MSH6* c.3984_3987dupGTCA and MSH6* c.3959_3962delCAAG mutations
|
Chromosomal location is taken from ENST00000234420 Ensembl transcript of MSH6 (Genome assembly GRCh37). Both affected and unaffected carriers were used for haplotype analysis. The common haplotypes associated with MSH6*dup and MSH6*del mutations are highlighted by an outlined box.
Chromosomal location includes both MSH6*del and MSH6*dup.
Haplotype analysis
To characterize the haplotypes associated with MSH6 exon 9 deletion and duplication we analyzed four microsatellite markers (AtnACAGn, CAn, D2S1352, D2S123) and four SNPs (rs1800932, rs1800935, c.1146+14A>T, rs3136367) in all identified carriers (Table 3). The markers cover about 3.4 Mb including MSH6 sequence. The marker AtnACAGn is approximately 144 kb upstream of MSH6, followed by SNPs in the coding region of MSH6, two polymorphic microsatellite loci (CAn, D2S1352), with the last marker D2S123 located 32 Mb away from the 3’-UTR of the MSH6. The physical order of the markers is tel - AtnACAGn - rs1800932 - rs1800935 - c.1146+14A>T - rs3136367 - CAn - D2S1352 - D2S123 – cen. Haplotype analysis demonstrated separate conserved haplotypes for each mutation. The haplotype tel-163-G-C-T-G-MSH6*dup-179-113-225-cen was observed with MSH6 exon 9 dup in 16/19 carriers. Three MSH6*dup carriers had recombination events involving the markers at the ends of the haplotype (AtnACAGn, D2S1352, D2S123). The MSH6*del was associated with the haplotype tel-151-A-T-T-G-MSH6*del-175-110-210-cen found in 8/12 carriers. Four MSH6*del carriers had recombination events involving the marker D2S1352.
Age of MSH6 exon 9 indel mutations
We estimated the age of MSH6*dup and MSH6*del using an established method of combined markers that utilizes rate of recombination and population frequency of flanking marker alleles [25]. Using the growth rate of Ashkenazi population of 0.125 per generation, the estimated age of MSH6*dup is about 57 (45–83) generations and the age of MSH6*del is about 53 (40–76) generations. Considering that one generation approximately equals to 25 years, the age of the mutations are 1425 (1125–2075) and 1325 (1000–1900) years for MSH6*dup and MSH6*del respectively. In other words, the duplication mutation likely arose around 585 CE and the deletion mutation probably originated in 685 CE.
DISCUSSION
In the present study we characterized two MSH6 truncating founder mutations c.3984_3987dupGTCA and c.3959_3962delCAAG (rs63751151) that cause Lynch syndrome in Ashkenazi Jews. Both MSH6*dup and MSH6*del create premature stop codons and likely result in nonfunctional proteins. We found 19 MSH6*dup carriers from 14 unrelated AJ families who all share the same haplotype associated with c.3984_3987dupGTCA, while all 12 MSH6*del carriers from 8 unrelated families share a different haplotype associated with c.3959_3962delCAAG. These results confirm that both MSH6*dup and MSH6*del are founder mutations in the AJ population.
The case-control settings of the MECC study allowed us to estimate frequency and CRC related risk of MSH6*dup and MSH6*del in Ashkenazi and Sephardi Jews. MSH6*dup was found in 0.3% and MSH6*del in 0.1% of Ashkenazi Jews with CRC, and was not observed in 940 Sephardi Jewish cases and controls. We identified one MSH6*dup carrier among the MECC controls, who developed colorectal cancer after being ascertained as a control. This carrier was included in the analysis both as a control and a case. Our data indicate that carriers of MSH6*dup have an almost 10 times higher risk to develop colorectal cancer than do non-carriers (OR = 9.9, 95% CI = 1.2–78.9, p = 0.0079). The frequency of Lynch syndrome among unselected CRC cases is ~ 3% – 5%, which corresponds to 81 to 134 cases of Lynch syndrome in the 2685 MECC CRC cases. Therefore, although the combined frequency of MSH6*dup and MSH6*del mutations is only 0.4% in CRC patients of AJ origin, the frequency of these mutations in AJ patients with Lynch syndrome is likely to be 8–14% (11 of 134 to 81 cases). Endometrial cancer risk, roughly estimated from the hospital-based series, was about 20 times higher in the carriers of MSH6*dup than in non-carriers (OR = 19.6, 95% CI = 1.8–217.2, p = 0.0006). We used the only identified AJ control carrying MSH6*dup from the MECC study for approximate calculation of the risk of endometrial cancer, although this patent had an endometrial cancer before being ascertained as a control. Despite the wide confidence intervals, the point estimates of the risk of colorectal and endometrial cancer in our study are, however, very comparable to the risk demonstrated in the recent analysis of 113 families with MSH6 mutations [14]. This study found an eight-fold increase in risk of colorectal cancer and 26-fold increase in risk of endometrial cancer. In addition, the fact that we only observed one carrier in 3310 controls, and this person later developed a Lynch-related cancer is supportive of our conjecture that these are highly penetrant alleles.
In our data the mean age at diagnosis of colorectal cancer among carriers of MSH6 exon 9 mutations was 66 years, 20 years older than in carriers of MSH2 or MLH1 mutations (44 years) [28]. The mean age at diagnosis of endometrial cancer was 54 years, comparable with previous studies [29]. This later age of onset may explain the fact that only two families meet Amsterdam criteria in our data.
About half of the MSI-High tumors (3/7) were instable in mononucleotide markers only (BAT25, BAT26, beta-catenin) in line with previous observations that mononucleotide microsatellite markers are more frequently affected in tumors harboring MSH6 germline mutations [30].
Recombination events allowed estimation of the age of the mutations. MSH6*dup arose ~1425 years ago in the 6th century CE, and MSH6*del arose ~1325 years ago in the 7th century CE, both at the time of formation of the AJ ethnicity in Western Europe. These mutations are much older than other previously described Ashkenazi founder mutations in MMR genes, such as MSH2 c.1906G>C, that probably arose in 15th–18th century CE, but more recent than BRCA2 c.6174delT (3rd century BCE) and APC p.I1307K (7th century BCE) [31]. The estimated age of the founder mutations can in part explain their frequency in Ashkenazi Jews, more recent mutations (MSH6*dup, MSH6*del) have lower than 0.03% frequency in general population, while the older mutations (BRCA2 c.6174delT and APC p.I1307K) have much higher frequency of 1.5% and 6% respectively [32, 33].
Only a few AJ founder mutations leading to colorectal and endometrial cancers have been described until now. In addition to the MSH6 exon 9 deletion and duplication characterized here, previous studies report another mismatch-repair gene mutation, MSH2*c.1906G>C (common name A636P), associated with increased risk of both colorectal [34] and endometrial cancers [34, 35], as well as mutations in APC*c.3920T>A (common name I1307K) [33] and BLM*c.2207_2212delATCTGAinsTAGATTC (common name BLMAsh) [36] associated with increased risk of colorectal cancer only. The reported frequency of APC*I1307K, BLMAsh and MSH2*A636P among AJ colorectal cancer patients is 12% [37], 1%–3% [36], and 1% [34] respectively. The frequency of MSH2*A636P in AJ endometrial cancer patients is about 1% [34]. Altogether AJ founder mutations (APC*I1307K, BLMAsh, MSH2*A636P, MSH6*dup, and MSH6*del) can be involved in approximately 14%–16% of all colorectal cancer cases and 2.2% of all endometrial cancer cases in Ashkenazi Jews. The frequency of these mutations in AJ patients with family history of colorectal and/or endometrial cancers can be significantly higher. A panel designed to detect known AJ founder mutations consisted of APC*I1307K, BLMAsh, MSH2*A636P, MSH6*dup, and MSH6*del could have value as a first-line screen in all AJ colorectal and/or endometrial cancer cases, irrespective of family history, IHC or MSI status.
Supplementary Material
Acknowledgments
We would like to thank Philip H. Gordon for this helpful assistance with this study.
Grant support: This work was supported in part by the Jewish General Hospital Weekend to End Women’s Cancers. WDF holds a Fonds de la Recherche en Santé du Québec (FRSQ) national scientist award and MT holds a FRSQ clinician-scientist award. SBG is supported by NCI 1R01CA81488 and University of Michigan Comprehensive Cancer Center Core Support grant (NIH 5P30CA46592).
Footnotes
Disclosure/Conflict of interest: None
References
- 1.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–6. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 2.Watson P, Riley B. The tumor spectrum in the Lynch syndrome. Familial Cancer. 2005;4(3):245–8. doi: 10.1007/s10689-004-7994-z. [DOI] [PubMed] [Google Scholar]
- 3.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368(6468):258–61. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 4.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75(5):1027–38. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 5.Drummond JT, Li GM, Longley MJ, et al. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268(5219):1909–12. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 6.Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17(3):271–2. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 7.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7(3):153–62. doi: 10.1038/nrclinonc.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marsischky GT, Filosi N, Kane MF, et al. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10(4):407–20. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 9.Hendriks YMC, Wagner A, Morreau H, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127(1):17–25. doi: 10.1053/j.gastro.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 10.Plaschke J, Engel C, Krüger S, et al. Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol. 2004;22(22):4486–94. doi: 10.1200/JCO.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 11.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–8. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–60. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 13.Hampel H, Panescu J, Lockman J, et al. Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res. 2007;67(19):9603. doi: 10.1158/0008-5472.CAN-07-2308. [DOI] [PubMed] [Google Scholar]
- 14.Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2009;102(3):193–201. doi: 10.1093/jnci/djp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner A, Hendriks Y, Meijers-Heijboer EJ, et al. Atypical HNPCC owing to MSH6 germline mutations: analysis of a large Dutch pedigree. J Med Genet. 2001;38(5):318–22. doi: 10.1136/jmg.38.5.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woods MO, Williams P, Careen A, et al. A new variant database for mismatch repair genes associated with Lynch syndrome. Hum Mutat. 2007;28(7):669–73. doi: 10.1002/humu.20502. [DOI] [PubMed] [Google Scholar]
- 17.Goodfellow PJ, Buttin BM, Herzog TJ, et al. Prevalence of defective DNA mismatch repair and MSH6 mutation in an unselected series of endometrial cancers. Proc Natl Acad Sci U S A. 2003;100(10):5908–13. doi: 10.1073/pnas.1030231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterlongo P, Nafa K, Lerman GS, et al. MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer. 2003;107(4):571–9. doi: 10.1002/ijc.11415. [DOI] [PubMed] [Google Scholar]
- 19.Hendriks YM, Wagner A, Morreau H, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127(1):17–25. doi: 10.1053/j.gastro.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 20.Hegde M, Blazo M, Chong B, et al. Assay validation for identification of hereditary nonpolyposis colon cancer-causing mutations in mismatch repair genes MLH1, MSH2, and MSH6. J Mol Diagn. 2005;7(4):525–34. doi: 10.1016/S1525-1578(10)60584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberg Y, Porat RM, Kedar I, et al. An Ashkenazi founder mutation in the MSH6 gene leading to HNPCC. Fam Cancer. 2009;9(2):141–50. doi: 10.1007/s10689-009-9298-9. [DOI] [PubMed] [Google Scholar]
- 22.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57. [PubMed] [Google Scholar]
- 23.Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76(3):449–62. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reeve JP, Rannala B. DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinformatics. 2002;18(6):894–5. doi: 10.1093/bioinformatics/18.6.894. [DOI] [PubMed] [Google Scholar]
- 26.Sun S, Greenwood CM, Thiffault I, et al. The HNPCC associated MSH2*1906G-->C founder mutation probably originated between 1440 CE and 1715 CE in the Ashkenazi Jewish population. J Med Genet. 2005;42(10):766–8. doi: 10.1136/jmg.2005.030999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothman KJ, Greenland S. Modern epidemiology. 2. Philadelphia, PA: Lippincott-Raven; 1998. [Google Scholar]
- 28.Plaschke J, Engel C, Kruger S, et al. Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol. 2004;22(22):4486–94. doi: 10.1200/JCO.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 29.Wagner A, Hendriks Y, Meijers-Heijboer EJ, et al. Atypical HNPCC owing to MSH6 germline mutations: analysis of a large Dutch pedigree. J Med Genet. 2001;38(5):318–22. doi: 10.1136/jmg.38.5.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wijnen J, de Leeuw W, Vasen H, et al. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet. 1999;23(2):142–4. doi: 10.1038/13773. [DOI] [PubMed] [Google Scholar]
- 31.Greenwood CM, Sun S, Veenstra J, et al. How old is this mutation?- a study of three Ashkenazi Jewish founder mutations. BMC Genet. 2010;11(1):39. doi: 10.1186/1471-2156-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roa BB, Boyd AA, Volcik K, et al. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996;14(2):185–7. doi: 10.1038/ng1096-185. [DOI] [PubMed] [Google Scholar]
- 33.Niell BL, Long JC, Rennert G, et al. Genetic anthropology of the colorectal cancer-susceptibility allele APC I1307K: evidence of genetic drift within the Ashkenazim. Am J Hum Genet. 2003;73(6):1250–60. doi: 10.1086/379926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foulkes WD, Thiffault I, Gruber SB, et al. The founder mutation MSH2*1906G-->C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population. Am J Hum Genet. 2002;71(6):1395–412. doi: 10.1086/345075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavie O, Gruber SB, Lejbkowicz F, et al. Gynecologic malignancies in Ashkenazi families with the MSH2 A636P founder mutation. Am J Obstet Gynecol. 2008;199(2):148 e1–3. doi: 10.1016/j.ajog.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Gruber SB, Ellis NA, Scott KK, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297(5589):2013. doi: 10.1126/science.1074399. [DOI] [PubMed] [Google Scholar]
- 37.Locker GY, Kaul K, Weinberg DS, et al. The I1307K APC polymorphism in Ashkenazi Jews with colorectal cancer: clinical and pathologic features. Cancer Genet Cytogenet. 2006;169(1):33–8. doi: 10.1016/j.cancergencyto.2006.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.