

Abstract
Lack of molecular targets in estrogen receptor-negative (ER-negative) breast cancer is a major therapeutic hurdle. We studied NF-κB activation in human breast tumors and in carcinoma cell lines. Activated NF-κB was detected predominantly in ER-negative vs. ER-positive breast tumors and mostly in ER-negative and ErbB2-positive tumors (86%). These in vivo results demonstrate association of activated NF-κB with a subgroup of human breast tumors and are consistent with previously reported in vitro observations using similar classes of human breast cancer cell lines. Finding such an association suggested functional and biological significance. Immunofluorescence demonstrated increased nuclear p65, a component of the active NF-κB complex, in cytokeratin 19 (CK19)-positive epithelial cells of ER-negative/ErbB2-positive tumor samples. In contrast, nuclear NF-κB was detected mostly in stroma of ER-negative and ErbB2-negative tumors, suggesting a role of activated NF-κB in intercellular signaling between epithelial and stromal cells in this type of breast cancers. To elucidate roles of activated NF-κB, we used an ER-negative and ErbB2-positive human breast tumor cell line (SKBr3). The polypeptide heregulin β1 stimulated, and herceptin, the anti-ErbB2 antibody, inhibited, NF-κB activation in SKBr3 cells. The NF-κB essential modulator (NEMO)-binding domain (NBD) peptide, an established selective inhibitor of IκB-kinase (IKK), blocked heregulin-mediated activation of NF-κB and cell proliferation, and simultaneously induced apoptosis only in proliferating and not resting cells. These results substantiate the hypothesis that certain breast cancer cells rely on NF-κB for aberrant cell proliferation and simultaneously avoid apoptosis, thus implicating activated NF-κB as a therapeutic target for distinctive subclasses of ER-negative breast cancers.
Human breast cancers are phenotypically heterogeneous and frequently pursue unpredictable clinical courses. Multiple and distinct molecular alterations presumably determine the diversity of histological patterns, pathological grades, and behaviors observed in these cancers (1, 2). Clinicians rely on the expression of two important growth factor receptors, the nuclear estrogen receptor (ER) and the membrane receptor tyrosine kinase, ErbB2 (HER2/neu) to classify human breast cancers into therapeutic and prognostic groups (2–5). Between 20% and 30% of human breast cancers express high levels of the ErbB2 receptor protein, and at least 30% of ER-negative breast cancers contain overexpressed ErbB2. ErbB2 belongs to the family of tyrosine kinase-coupled dimeric receptors and signals by forming heterodimers with ErbB1, ErbB3, and ErbB4 in response to ligands, including the heregulins, which are members of the neuregulin or neu-differentiating factor (NDF) family of natural ligands (5–7).
Although ER-positive breast cancers respond to hormonal therapy, the prognosis for ER-negative breast cancers is poorer because of the lack of target-directed therapies and a more clinically aggressive biology (4, 8–11). Treatment of human breast cancers, containing an amplified ErbB2 gene, with a humanized monoclonal antibody to the extracellular domain of this receptor (trastuzumab, marketed as herceptin) produces remissions in 11–15% of patients with metastatic breast cancer failing standard chemotherapy (12–14). Improvements in treatment depend on discovering molecular pathways and gene products that play central roles in cancer growth. Tailoring treatment to specific subclasses of cancer, defined by vital molecular targets, should improve the survival of breast cancer patients.
NF-κB stimulates proliferation and blocks programmed cell death (apoptosis) in different cell types, including human breast cancers (15–17). We have reported that activated NF-κB is detected in ER-negative human breast cancer cells harboring overexpressed ErbB1 [epidermal growth factor receptor (EGFR)] (17–19). Treatment with the protein kinase C (α/β) inhibitor Go6976 was strongly therapeutic against murine tumors generated from cells with activated NF-κB (17). Furthermore, selective inhibition of NF-κB activation by stable expression of the dominant negative IκB kinase (dnIKKβ) incapacitates tumor formation in a mouse mammary adenocarcinoma model (17–19). Activated NF-κB was frequently detected in cultured cells from ER-negative breast cancers (20). We proposed that NF-κB in ER-negative breast cancer cells functions by participating in proliferative pathways and regulating cell death signals (17–19).
In the current study, we found activated NF-κB predominantly in the ER-negative/ErbB2-positive subclass in comparison with ER-positive human breast cancers. Selective inhibition of NF-κB activation in the SKBr3 cell line blocked heregulin-induced proliferation and resulted in apoptosis. We propose that active NF-κB signaling disables cell death pathways and permits cell proliferation in ER-negative tumors harboring amplified ErbB2. These results qualify NF-κB as a potential therapeutic target for this subgroup of breast cancer patients.
Methods
Human Tissues. A research tissue bank belonging to the Dana–Farber/Harvard Specialized Programs of Research Excellence (SPORE) in Breast Cancer is approved to collect and distribute discarded tissue for research purposes. Content of ER and ErbB2 is determined by a widely used, clinically established scale, which annotates the anonymous specimens in the tissue bank. ER-positive refers to tumors with >10% of nuclei staining by immunohistochemistry for this nuclear receptor. ErbB2-positive tumors are those graded 3+ by immunohistochemistry (on a scale from 0–3+) or positive by fluorescence in situ hybridization (FISH) and reported clinically. Individual tumors are referred to in the text by their tissue bank number (BOT number).
Detection of Active NF-κB in Tissue and Nuclear Extracts. The level of active NF-κB was determined by electrophoretic mobility-shift assay (EMSA) of protein extracts of breast cancer specimens frozen shortly after surgical removal, prepared from whole tissues as described (17–22). The 32P–DNA–protein complex was detected as a retarded radioactive band and was characterized in a supershift assay by interaction with specific antibodies against specific Rel family proteins.
Double Immunofluorescence Assay on Tissue Sections. Thawed sections were fixed, permeabilized, and incubated with a mixture of either anti-p50 (SC-7178) rabbit polyclonal and anti-p65 (SC-8008) mouse monoclonal antibodies, or with a mixture of anti-cytokeratin 19 (SC-6278) mouse monoclonal and anti-p65 (SC-8008) rabbit polyclonal antibodies (antibodies from Santa Cruz Biotechnology) (23). Secondary antibodies were FITC-conjugated goat anti-mouse IgG and rhodamine-conjugated goat anti-rabbit IgG, generating green fluorescence and red fluorescence for monoclonal and polyclonal antibodies, respectively. Nuclei were stained with the fluorescent dye 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence microscopy was performed on a Zeiss Axioskop 2 (MOT), and images were captured on a Zeiss AxioCam HRC camera by using openlab software as described (19).
Cell Proliferation and Apoptosis. Cell proliferation was monitored by the MTT assay (18). The level of ErbB1 [epidermal growth factor receptor (EGFR)], ErbB2 (HER2/neu), and ER was measured in SKBr3 cells by Western blot analysis (17). Apoptosis was assayed by FITC-conjugated antibodies to annexin V following a modified protocol from the supplier (Oncogene Research Products, Cambridge, MA), and apoptotic cell numbers were determined by flow cytometry as described (17, 19). The NF-κB essential modulator (NEMO)-binding domain (NBD) peptides (24) (WT drqikiwfqnrrmkwkkTALDWSWLQTE; and mutant drqikiwfqnrrmkwkkTALDASALQTE) were synthesized and purified by reverse-phase HPLC and quantified (25) at the Core Facilities of Dana–Farber Cancer Institute. Lowercase designates the antennapedia peptide sequence of NBD that facilitates its cell entry. Underlined letters designate amino acid replacements, W (trp) in the wild-type with A (ala) in the mutant peptides.
Results
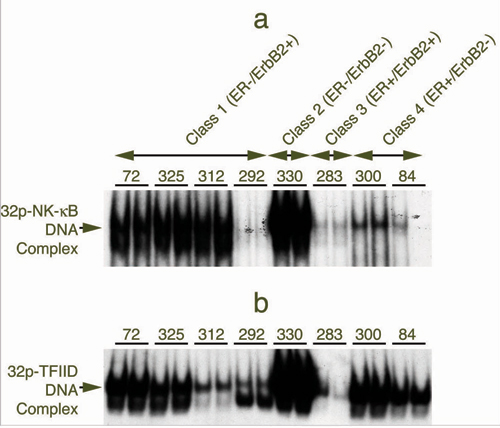
NF-κB Activation in Human Breast Cancer Specimens. We measured the level of activated NF-κB in extracts of 31 human breast tumor specimens by EMSA (Fig. 1) (17–21) and by double immunofluorescence assay (Fig. 2) (23). The tissue specimens are classified by their level of expression of ER and ErbB2: class 1, ER-negative/ErbB2-positive (n = 7); class 2, ER-negative/ErbB2-negative (n = 9); class 3, ER-positive/ErbB2-positive (n = 8); and class 4, ER-positive/ErbB2-negative (n = 7). Active NF-κB was detected predominantly in ER-negative human breast tumor specimens (Fig. 1 a and b), whereas this NF-κB activation is detected comparatively in a smaller proportion of ER-positive tumors (Fig. 1 c and d). Activated NF-κB was detected in 6 of 7 tumor extracts of class 1 (86%), 3 of 9 tumors of class 2 (33%), and in 2 of 15 ER-positive tumors (13%) of classes 3 and 4 (Table 1, which is published as supporting information on the PNAS web site). Supershift assays with antibodies against three Rel family proteins identified p50/p65 subunits but not the cRel in all of the NF-κB-positive tumors from class 1 and class 2. Results of one representative from class 1 (BOT 72) and one from class 2 (BOT 288) tumors are shown in Fig. 1 e and f. The cRel protein is not a part of the NF-κB complex in any one of these tissues.
Fig. 1.

Detection of active NF-κB in extracts of human breast cancer tissues by EMSA. Tumor specimens are designated with numbers indicated in each panel (tissue bank numbers) and classified by their ER and ErbB2 receptor levels. Arrows to the left of each representative gel show the 32P–NF-κB–DNA complex. (a) EMSA-binding activity for six of the seven class 1 (ER-negative/ErbB2-positive) tumors. (b) Three tumors with positive binding results were chosen from nine tumors in class 2 (ER-negative/ErbB2-negative). (c and d) Five representative tumors from eight in class 3 (ER-positive/ErbB2-positive) are displayed (c) and seven of seven from class 4 (ER-positive/ErbB2-negative) are shown (d). Each panel displays duplicates for each tumor determination. (e and f) Supershifted 32P–NF-κB–DNA complexes. (e) Tumor BOT 72 from class 1. (f) Tumor BOT 288 from class 2, each shifted with three anti-rel protein antibodies (against p50, p65, and c-rel). The upper arrow shows the supershifted band.
Fig. 2.

Localization of active NF-κB in frozen sections from human breast cancers. Localization of NF-κB complexes was performed in frozen sections from each EMSA-positive tumor (Fig. 1). (a–k) Sections from the ER-negative/ErbB2-positive and NF-κB-EMSA-positive sample BOT 312 were stained by double antibody immunofluorescence (23). (a) DAPI-stained nuclei in a representative section from BOT 312 at low power (×10). (b) The epithelial cell marker CK19 is demonstrated by green fluorescence and shown in the same section at the same magnification as in a. Higher power images of regions containing epithelial cells are shown at ×40 (c–f). A field of cells is shown stained with DAPI (c), with antibodies to CK19 (d), and with antibodies detecting p65 (e). Superimposed images by using photoshop software (Adobe Systems, San Jose, CA) demonstrate colocalization of nuclear p65 (red fluorescence) encircled by green fluorescence, generated by cytoplasmic CK19 in the same cell (f). These signals are further elaborated by digital enlargements of the superimposed CK19 and nuclear p65 containing epithelial cells from the boxed area of panels c–f (g–k). DAPI-stained nuclei (g) are superimposed with p65 staining to demonstrate the nuclear localization of this NF-κB component (h), and colocalization of cytoplasmic CK19 (i) and nuclear p65 (j) is demonstrated in merged images (k). Levels of CK19 and p65 in a similarly analyzed frozen section of the class 2 ER-negative/ErbB2-negative/NF-κB-positive tumor specimen, BOT 288, is shown (l–o). (l) The distribution of DAPI-stained nuclei shows the overall cellularity of the section at ×40. (m) CK19-stained epithelial cells show cords off cells infiltrating tumor. (n) p65-positive cells appear distinct from CK19-positive epithelial cells, and (o) the merged image shows that CK19- and p65-positive cells are in separate islands. (p) An enlarged presentation of panel o shows CK19 cells with vacant, unstained nuclei. (q–t) BOT 288 was also examined for the presence of the p50/p65 complex, studied by using mixtures of antibodies to p50 and p65 with double immunofluorescence. A low-power hematoxylin and eosin-stained section (q) and the corresponding DAPI-stained nuclei (r) show the border of the tumor pushing against the surrounding stroma. The p50 and p65 associated merged red and green fluorescence generated the brownish yellow signals in the nuclei of the cells. Few nuclei in the tumor stained for both components of active NF-κB(s); however, more frequent colocalization was seen in merged images of the stromal cells (t).
We also determined the level of an unrelated DNA-binding protein TFIID in NF-κB-positive and NF-κB-negative samples and observed its binding activity in almost every sample, independent of NF-κB activation status, confirming the uniform quality of the extracts (Fig. 6, which is published as supporting information on the PNAS web site). Variable intensities of the TFIID DNA binding may be a reflection of the transcriptional state and proliferation in these tissues. Lymphocytic infiltration of tumor tissue may contribute to the DNA-binding activity of NF-κB. Microscopic examination of frozen sections from the same tissue did not show a significant correlation of NF-κB activation with the extent of infiltration (Table 1).
Localization of Activated NF-κB in Frozen Tumor Sections. To confirm the biochemical assessment of NF-κB activation and to identify the cell types contributing to active NF-κB, a double immunofluorescence assay was performed with antibodies to p65 and the epithelial cytokeratin 19 (CK19) in all of the class 1 and class 2 EMSA-positive and several EMSA-negative samples from class 3 and 4 tumors. Nuclear localization of the p65 subunit was used for the recognition of the active state of NF-κB (26–29). We observed nuclear localization of p65 in a majority of the DAPI-stained nuclei of the EMSA-positive samples (>50%, Fig. 2). All EMSA-negative tumors are uniformly negative by nuclear p65 immunofluorescence. Results of one of these tumors (BOT 312 of class 1) are shown in Fig. 2 a–k). FITC-conjugated antibody against CK19 identified islands of epithelial cells, and rhodamine red-conjugated anti-p65 antibody localized nuclear p65 in the same CK19-positive epithelial cells. DAPI-stained nuclei (Fig. 2a) and green fluorescence emitting CK19 (Fig. 2b) under low magnification (×10) demonstrated the organization of the epithelial cells with a core necrotic region and the surrounding stroma. A region of CK19-positive epithelial cells (indicated by the arrow) examined under higher magnification (×40) identified p65 as a cluster of red fluorescence-emitting nuclei (Fig. 2e) seen within the green fluorescence-emitting CK19-positive cells in merged images (Fig. 2f). Digitally enlarged boxed areas of Fig. 2 c–f demonstrate nuclear localization of p65 in merged images (Fig. 2 g and h). The epithelial nature of nuclear p65 is shown in Fig. 2 i–k. Nuclear p65 indicates activated NF-κB in the nucleus of these breast carcinoma cells, presumably as a p50–p65 complex bound to the specific DNA sequences of responsive genes (26, 28). The NF-κB–DNA complex detected in extracts by EMSA is, at least in part, contributed by the epithelial population of the mixture of cells as demonstrated by the colocalization of p65 in CK19-positive epithelial cells of all of the six class 1 tumors with elevated NF-κB–DNA-binding activity (Fig. 1). One specimen in class 1 that did not show DNA-binding activity (Table 1, BOT 292) also did not show nuclear p65 by immunofluorescence assay.
A notable difference was revealed by similar immunofluorescence analysis of frozen sections from three class 2 samples with elevated levels of activated NF-κB determined by EMSA (Fig. 1b). In contrast to class 1 tumors, strong signals with anti-p65 were detected mostly in CK19-negative regions of these three class 2 tumors. A tumor section of BOT 288 showing DAPI-stained nuclei (Fig. 2l) was stained for CK19 to highlight epithelial cells (Fig. 2m) and for p65 for NF-κB activation (Fig. 2n). Merged images (Fig. 2 o and p) reveal distinct and discrete nonoverlapping staining for p65 and CK19 (Fig. 2k). These results were verified by independent detection of nuclear p50/p65 complexes with rhodamine red-conjugated anti-p50 and FITC-conjugated anti-p65 antibodies in a section of the same BOT 288 tissue (Fig. 2 q–t). The p50/p65 complexes (identified by brown merged signals) were detected in very few epithelial cells (Fig. 2s, indicated by the arrow), although such p50/p65 complexes were detected in numerous nuclei within a stromal region of the same tissue section (Fig. 2t).
Heregulin β1 (Heregulin)-Induced Activation of NF-κB. SKBr3 is an ER-negative human breast cancer cell line with ErbB2 gene amplification and highly expressed receptor levels and is also ErbB3- and ErbB4-positive (28, 30, 31). Heregulin treatment of SKBr3 cells in serum-free medium stimulated NF-κB–DNA-binding activity could be detected within 2 h and persisted as long as 18 h (Fig. 3a, lanes 2–4). Quantification of the autoradiographic signals revealed a 4-fold increase in the NF-κB–DNA-binding activity by 18 h. Treatment of cells with the anti-ErbB2 antibody herceptin blocked heregulin-induced NF-κB activation in a concentration-dependent manner (Fig. 3b).
Fig. 3.

Modulation of heregulin-induced activation of NF-κB by herceptin and NBD peptide in SKBr3 cells. Nuclear extracts from SKBr3 cells were prepared and NF-κB–32P–DNA-binding activity was determined by EMSA. (a) Binding of 32P-labeled oligonucleotides to NF-κB at the indicated times after treatment of the cells with 1 nM heregulin is shown in duplicate (lanes 1–4). The identities of the NF-κB components were determined by supershift assays with antibodies to rel-family proteins p50 and p65 (data not shown). (b) NF-κB–32P–DNA-binding activity was inhibited by simultaneous treatment of SKBr3 cells with the indicated concentrations of heregulin and herceptin, measured at 18 h after application. (c) Heregulin-stimulated NF-κB–32P–DNA-binding activity in SKBr3 cells and simultaneous treatment with WT NBD (lanes 2–4) and mutant NBD (lanes 5–7). The DMSO concentration at the highest NBD concentration was 1%, and the control reaction (lane 1) contained the same amount of DMSO.
To verify the role of IκB-kinase (IKK) in heregulin-induced activation of NF-κB, and eventually to determine the proportion of heregulin-stimulated growth contributed by active NF-κB, we treated SKBr3 cells simultaneously with the NBD peptide and with heregulin. The WT NBD peptide blocked heregulin-induced activation of NF-κB (Fig. 3c, lanes 2–4), suggesting that NF-κB activation by heregulin in SKBr3 cells is mediated by means of the action of IKK. The specificity of this action was further verified by treating cells with a mutant NBD and observing no effect on heregulin-induced activation of NF-κB at the same concentrations (Fig. 3c, lanes 5–7). Therefore, NF-κB is activated by ErbB2 signaling and can be blocked at the level of the receptor or at the level of activation by IKK. Selective inhibition of NF-κB activation by NBD reduced binding activity to the basal level (Fig. 3a) suggesting minimal contribution by other kinases, such as CK2 (32–34), or alternative pathways (35) in these cells.
Influence of Selective Inhibition of NF-κB Activation on Cell-Cycle Progression and Apoptosis. In rich medium with 10% FBS, SKBr3 cells progress through the cell cycle (Fig. 4a), and, in serum-free medium, cells were arrested in G0/G1 (Fig. 4b). The addition of heregulin (1 nM) to cells in serum-free medium reestablished S-phase progression, recycling the growing cells in the appropriate proportions in different phases of the cell cycle (Fig. 4c). Selective inhibition of NF-κB activation with NBD (100 μM) blocked heregulin-induced cell-cycle progression, with accumulation of cells in G0/G1 and also in a sub-G0/G1 phase (Fig. 4d). No significant increase in this sub-G0/G1 population was detected in cells treated with mutant NBD (Fig. 4e).
Fig. 4.

Cell-cycle progression and apoptosis in heregulin and NBD peptide-treated SKBr3 cells. Growth of cells and drug treatment conditions were the same as in Fig. 3. The fraction of cells in different phases of the cell cycle was measured by propidium iodide (PI) staining followed by FACS (Becton Dickinson) analysis. Cell-cycle distribution of cells grown in rich medium (a), in serum-free medium (b), in serum-free medium in the presence of heregulin (1 nM) (c), in serum-free medium in the presence of heregulin (1 nM) and WT NBD (100 μM) (d), and in serum-free medium in the presence of heregulin (1 nM) and mutant NBD (100 μM) (e). Numbers in each panel show the percent distribution of cycling cells (excluding the dead sub G0 population) in different phases under these treatment conditions. The apoptotic fraction of cells detected by annexin V staining after different treatments is shown, with numerals in the lower right-hand panel of each figure showing the annexin V-positive fraction (f–j). (f) Cells grown in serum-free medium in the presence of heregulin (1 nM). (g) Cells grown in serum-free medium in the presence of WT NBD (100 μM). (h and i) Cells grown in serum-free medium in the presence of heregulin (1 nM) and WT NBD at 50 μM (h) and 100 μM (i). (j) Treatment with the mutant NBD peptide at 100 M showed minimal or no effect. All treatments were for 18 h.
Measurement of Apoptosis by Annexin V Binding. Cultures of SKBr3 cells growing in rich medium, in serum-free medium, or in serum-free medium with heregulin contain a low number (3.7%) of apoptotic cells (Fig. 4f and data not shown). In the presence of WT NBD (100 μM) alone (in the absence of heregulin), a slightly elevated number of apoptotic cells were detected (Fig. 4g, 9.4%). The apoptotic cell number increased 9- to 10-fold after simultaneous treatment with heregulin (1 nM) and 50 μM (Fig. 4h, 32.4%) or 100 μM WT NBD (Fig. 4i, 41%). In cells treated with heregulin and mutant NBD (100 μM), the apoptotic fraction was not much different (Fig. 4j, 7%) from cells treated only with heregulin (Fig. 4f). During heregulin stimulation, the apoptotic fraction of cells appreciably increased when NF-κB signaling was specifically interrupted by NBD treatment.
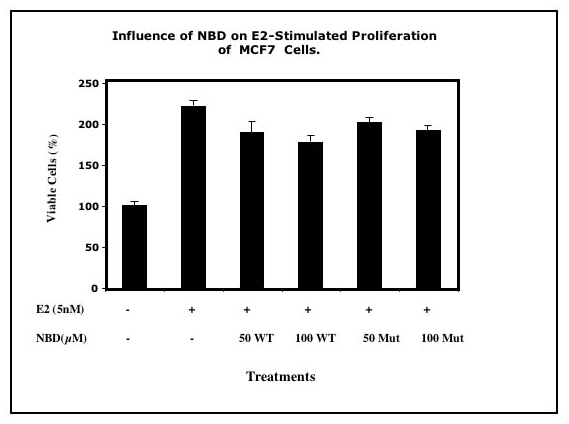
Influence of Selective Inhibition of Heregulin-Induced NF-κB Activation and Cell Proliferation. Cell proliferation was stimulated 2-fold after treatment of SKBr3 cells with 1–2 nM heregulin in serum-free medium for 72 h (Fig. 5a). Simultaneous treatment of cultures with WT NBD at the indicated concentrations blocked heregulin-induced cell proliferation (Fig. 5b, WT). In contrast, cells treated with mutant NBD at the same concentrations responded normally to heregulin (Fig. 5b, MUT). Neither WT nor mutant NBD affected cell proliferation in the absence of heregulin (Fig. 5c). Selective inhibition of IKK with WT NBD blocked NF-κB activation and heregulin-induced cell proliferation without any effect on resting cells. NF-κB activation is necessary and required for heregulin-stimulated growth, and at least some of the decrease in cell proliferation was due to the increased fraction of cells undergoing programmed death. In contrast, E2-induced cell proliferation in the ER-positive breast cancer cell line MCF7 was not blocked by NBD (Fig. 7, which is published as supporting information on the PNAS web site). Our previously reported results (17, 18) demonstrated that E2 treatment of MCF7 cells did not induce NF-κB activation. This observation is in accord with the concept that E2-induced cell proliferation that does not involve NF-κB activation also is not blocked by the specific inhibition of the latter.
Fig. 5.

Influence of selective inhibition of NF-κB activation by NBD peptide on heregulin-induced cell proliferation. Duplicate cultures of cells were seeded in 96-well plates in rich medium. Twenty-four hours later, the rich medium was replaced with serum-free medium and treated with indicated agents, and cell numbers were determined by MTT assay after 72 h of treatment. Assays were normalized to cell counts without any treatments, taken as 100%. (a) SKBr3 cell proliferation in serum-free medium in the presence of the indicated concentrations of heregulin. (b) The same number of cells grown in the presence of heregulin (1 nM) and the indicated concentrations of WT or mutant (MUT) NBD peptide. (c) Cell proliferation in the presence of WT or mutant (MUT) NBD peptide in the absence of heregulin.
Discussion
Homeostasis in higher organisms is maintained by fine-tuning positive and negative signal-induced cellular regulatory mechanisms responsible for cell proliferation and death, which are apparently disturbed in tumor cells (36). Inappropriate gene expression and function, leading to uncontrolled cell proliferation and loss of regulated cell death, contributes significantly to the tumor cell phenotype (1, 36–38). The proliferative and antiapoptotic properties of activated NF-κB qualify this transcription factor as a key cellular regulatory molecule, potentially contributing to both normal and neoplastic phenotypes (36, 37). Although elevated levels of this transcription factor in breast tumors have been reported (20, 31, 38), the results herein are, to our knowledge, the first to show a strict association of activated NF-κB with distinctive subclasses of human breast cancers. The detection of positive DNA-binding activity of the transcriptional coactivator TFIID in all four classes of tumor samples validates the observed NF-κB DNA-binding activity in the extracts of the same tumor samples. A striking finding was the association of NF-κB activity with ER-negative breast cancers, and presumably with the inactivity of this pathway in ER-positive cancers. This reciprocal relationship suggests the importance of NF-κB signaling in certain cancers, and not in others. The class 2 ER-negative cancers without ErbB2 overexpression did contain activated NF-κB in some members of this class (three of nine, 33%). However, in contrast to class 1 tumors, activated NF-κB in each of these three samples of class 2 tumors is located in stromal and not epithelial cells. We speculate that NF-κB activation plays a role in the crosstalk between two cell systems such as stromal and epithelial cells. An analogous role of NF-κB activation influencing two separate systems by means of overexpression of cytokines has been reported (39).
Breast cancer is not one disease, and screening for drug activity that does not account for the molecular diversity of this malignancy are liable for failure. Finding such a faithful association of NF-κB activation in one subclass of breast cancer implies a functional role of this transcription factor in the growth and survival of this particular class. In addition, the finding will have practical importance when directing treatment at steps in NF-κB activation; it is necessary to tailor such treatment to relevant subclasses of breast cancers. In fact, reducing the heterogeneity of breast cancer, by determining salient growth and antiapoptotic pathways and by treating molecularly defined subgroups, is an important emerging concept in breast cancer treatment.
Unregulated cell proliferation and the loss of regulated cell death are two characteristic phenotypes of cancer cells. Both processes are intertwined and important for cancer cell growth (36–38). NF-κB activation and inhibition of apoptosis are consistent with the prevalent concept that cancer cells require sustained proliferative signals to grow continually, but also must acquire antiapoptotic assets to survive (36). Proliferative signals induced by growth factors such as heregulin are proposed to increase or decrease apoptosis, depending on stimulation of one or another signaling pathway linked to the cell cycle (36). Our results are consistent with the suggestion that cancer cells subjected to unregulated proliferative stimuli may be especially sensitive to agents that block their antiapoptotic signals (40).
Responsiveness of ErbB2-overexpressing human breast cancer tumors to herceptin suggests that receptor-mediated signaling pathway is activated in these tumors. This observation is further supported by the observed distinct gene expression profile of ErbB2-positive and ErbB2-negative human breast tumor samples (41). However, it seems that treatment with herceptin in combination with other chemotherapeutic agents is prognostically better than herceptin alone, suggesting that blockade of other downstream events of the ErbB2-mediated cell signaling will add to the efficacy of herceptin therapy.
A connection between ErbB2 and NF-κB signaling was explored in a cell culture system by using ER-negative and ErbB2-positive SKBr3 cells (i) by treatment with the polypeptide growth factor heregulin, elevating NF-κB activation and stimulating cell proliferation, (ii) by reversing this stimulation by herceptin, and (iii) by the inhibition of NF-κB activation with NBD, a specific inhibitor of IKK. Supporting a proximate cause for NF-κB in proliferation or apoptosis after ErbB2 stimulation, selective inhibition by NBD blocked not only heregulin-stimulated NF-κB activation, but also cell proliferation (Fig. 5). Under conditions of heregulin stimulation, this selective blockade also induced substantial apoptotic cell death (Fig. 4). NBD did not cause apoptosis in resting cells, suggesting that inhibition of the apoptotic signal is associated with heregulin-induced cell proliferation. Absence of NF-κB activation in ER-positive, ErbB2-positive tumors is consistent with the observation that stimulation of the ER-positive pathway in breast cancer cells blocks NF-κB activation and presumably the signaling pathway mediated by its activation in this class of breast cancer cells (42).
Previous work from our laboratory and others has highlighted the participation of NF-κB signaling in mammary epithelial cells, particularly in highly aggressive ER-negative cells that overexpress the epidermal growth factor receptor (EGFR) family of receptors (17–20). These tumors do not respond to hormone receptor-based treatment, effective for ER-positive breast cancers. Treatments that target cell-surface receptors, such as herceptin and newer small molecule tyrosine kinase inhibitors, are encouraging, but their full potential will be achieved when combined with other agents, targeting sequential or parallel pathways. We propose that activated NF-κB is a therapeutic target worthy of testing in those specific classes of breast cancers shown to harbor activated NF-κB, or in subgroups of ER-negative tumors with elevated levels of ErbB1 as shown earlier (17), and ErbB2 as shown in this study.
Supplementary Material
Acknowledgments
We thank Shridar Ganeson, Christine Merrese, Mike Boxen, and Eric Smith of the Dana–Farber Cancer Institute for assistance and advice with immunofluorescence microscopy and Lyndsay Harris (Dana–Farber Cancer Institute) for providing clinical samples of herceptin. We thank Dr. Andrea Richardson and Gabriela Lodeiro of Brigham and Women's Hospital for providing the human breast cancer tissue samples. This work is supported by funds from Department of Defense Grant DAMD17-02-1-0692, by an award from Friends of Dana–Farber Cancer Institute, and by the National Cancer Institute Specialized Programs of Research Excellence in Breast Cancer at Brigham and Women's Hospital and the Dana–Farber Cancer Institute. We also acknowledge support from the Women's Cancer Program at the Dana–Farber Cancer Institute.
Abbreviations: ER, estrogen receptor; EMSA, electrophoretic mobility-shift assay; DAPI, 4′,6-diamidino-2-phenylindole; NBD, NF-κB essential modulator (NEMO)-binding domain; CK19, cytokeratin 19; IKK, IκB-kinase.
References
- 1.Perou, C. M., Sorlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., Pollack, J. R., Ross, D. T., Johnsen, H., Akslen, L. A., et al. (2000) Nature 406, 747-752. [DOI] [PubMed] [Google Scholar]
- 2.Schnitt, S. J. & Guidi, A. J. (2000) in Diseases of the Breast, eds. Harris, J. R., Lippman, M. E., Morrow, M. & Osborne, C. K. (Lippincott Williams & Wilkins, Philadelphia), 2nd Ed., pp. 425-470.
- 3.Menard, S., Tagliabue, E., Campiglio, M. & Pupa, S. M. (2000) J. Cell Physiol. 182, 150-162. [DOI] [PubMed] [Google Scholar]
- 4.Elledge, R. M. & Fuqua S. A. W. (2000) in Diseases of the Breast, eds. Harris, J. R., Lippman, M. E., Morrow, M. & Osborne, C. K. (Lippincott Williams & Wilkins, Philadelphia), 2nd Ed., pp. 471-488.
- 5.Yarden, Y. & Sliwkowski, M. X. (2001) Nat. Rev. Mol. Cell Biol. 2, 127-137. [DOI] [PubMed] [Google Scholar]
- 6.Chen, X., Levkowitz, G., Tzahar, E., Karunagaran, D., Lavi, S., Ben-Baruch, N., Leitner, O., Ratzkin, B. J., Bacus, S. S. & Yarden, Y. (1996) J. Biol. Chem. 271, 7620-7629. [PubMed] [Google Scholar]
- 7.Graus-Porta, D., Beerli, R. R., Daly, J. M. & Hynes, N. E. (1997) EMBO J. 16, 1647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bardou, V.-J., Arpino, G., Elledge R. M., Osborne, C. K. & Clark, G. M. (2003) J. Clin. Oncol. 21, 1973-1979. [DOI] [PubMed] [Google Scholar]
- 9.Ali, S. & Coombes, R. C. (2002) Nat. Rev. Cancer 2, 101-115. [DOI] [PubMed] [Google Scholar]
- 10.Howat, J. M. T., Harris, M., Swindell, R. & Barnes, D. M. (1985) Br. J. Cancer 51, 263-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shek, L. L., Godolphin, W. & Spinelli, J. J. (1987) Br. J. Cancer 56, 825-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisenhauer, E. A. (2001) N. Engl. J. Med. 344, 841-842. [DOI] [PubMed] [Google Scholar]
- 13.Harries, M. & Smith, I. (2002) Endocr. Relat. Cancer 9, 75-85. [DOI] [PubMed] [Google Scholar]
- 14.Ligibel, J. A. & Winer, E. P. (2002) Semin. Oncol. 29, 38-43. [DOI] [PubMed] [Google Scholar]
- 15.Barkett, M. & Gilmore, T. D. (1999) Oncogene 18, 6910-6924. [DOI] [PubMed] [Google Scholar]
- 16.Karin, M. & Lin, A. (2002) Nat. Immunol. 3, 221-227. [DOI] [PubMed] [Google Scholar]
- 17.Biswas, D. K., Martin, K. J., McAlister, C., Cruz, A. P., Graner, E., Dai, S. C. & Pardee, A. B. (2003) Cancer Res. 63, 290-295. [PubMed] [Google Scholar]
- 18.Biswas, D. K. & Cruz, A. P. (2000) Proc. Natl. Acad. Sci. USA 97, 8542-8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biswas, D. K., Dai, S. C., Cruz, A., Weiser, B., Graner, E. & Pardee, A. B. (2001) Proc. Natl. Acad. Sci. USA 98, 10386-10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakshatri, H. & Goulet, R. J. (2002) in Current Problems in Cancer, ed. Johnstone, A. S. pp. 282-300. [DOI] [PubMed]
- 21.Biswas, D. K., Cruz, A., Pettit, N., Mutter, G. L. & Pardee, A. B. (2001) Mol. Med. 7, 59-67. [PMC free article] [PubMed] [Google Scholar]
- 22.Dignam, J. D., Lebovitz, R. M. & Roeder, R. D. (1983) Nucleic Acids Res. 11, 1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scully, R., Ganesan, S., Brown, M., DeCaprio, J. A., Canistra, S. A., Feunteum, J., Schnitt, S. & Livingston, D. M. (1996) Science 272, 123-126. [DOI] [PubMed] [Google Scholar]
- 24.May, M. J., D'Acquisto, F., Madge, L. A., Glockner, J., Pober, J. S. & Ghosh, S. (2000) Science 289, 1550-1554. [DOI] [PubMed] [Google Scholar]
- 25.Bidlingmeyer, B. A., Cohen, S. A. & Tarvin, T. L. (1984) J. Chromatogr. 336, 93-104. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin, A. S., Jr. (1996) Annu. Rev. Immunol. 14, 649-683. [DOI] [PubMed] [Google Scholar]
- 27.Schmid, J. A., Birbach, A., Hofer-Warbinek, R., Pengg, M., Burner, U., Furtmuller, P. G., Binder, B. R. & de Martin, R. (2000) J. Biol. Chem. 275, 17035-17942. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh, S. & Karin, M. (2002) Cell 109, S81-S96. [DOI] [PubMed] [Google Scholar]
- 29.Sen, R. & Baltimore, D. (1986) Cell 46, 705-716. [DOI] [PubMed] [Google Scholar]
- 30.Kraus, M. H., Popescu, N. C., Amsbaugh, S. C. & King, C. R. (1987) EMBO J. 6, 605-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esparis-Ogando, A., Diaz-Rodriguez, E., Montero, J. C., Yuste, L., Crespo, P. & Pandiella, A. (2002) Mol. Cell. Biol. 22, 270-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romieu-Mourez, R., Landesman-Bollag, E., Seldin, D. C. & Sonenshein, G. E. (2002) Cancer Res. 62, 6770-6778. [PubMed] [Google Scholar]
- 33.Romieu-Mourez, R., Landesman-Bollag, E., Seldin, D. C., Abdulmaged, T. M., Mercurio, F. & Sonenshein, G. E. (2001) Cancer Res. 61, 3810-3818. [PubMed] [Google Scholar]
- 34.Kato, T., Delhase, M., Hoffmann, N. & Karin, M. (2003) Mol. Cell 12, 829-839. [DOI] [PubMed] [Google Scholar]
- 35.Tergaonkar, V., Bottero, V., Ikawa, M., Li, Q. & Verma, I. M. (2003) Mol. Cell. Biol. 23, 8070-8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vermeulin, K., Berneman, Z. N. & Van Bockstale, D. R. (2003) Cell Prolif. 36, 165-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinstein, I. B. (2000) Carcinogenesis 2, 857-864. [DOI] [PubMed] [Google Scholar]
- 38.Cogswell, P. C., Guttridge, D. C., Funkhouser, W. K. & Baldwin, A. S. (2000) Oncogene 19, 1123-1131. [DOI] [PubMed] [Google Scholar]
- 39.Biswas, D. K., Salas, T. R., Wang, F., Ahlers, C. M., Dezube, B. J. & Pardee, A. B. (1995) J. Virol. 69, 7437-7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keyomarssii, K. & Pardee, A. B. (2003) Prog. Cell Cycle Res. 5, 527-532. [PubMed] [Google Scholar]
- 41.Wang, Z. C., Lin, M., Wei, L. J., Li, C., Miron, A., Lodeiro, G., Harris, L., Ramaswamy, S., Tanenbaum, D. M., Meyerson, M., et al. (2004) Cancer Res. 64, 64-71. [DOI] [PubMed] [Google Scholar]
- 42.Liu, H., Lee, E. S., Gajdos, C., Pearce, S. T., Chen, B., Osipo, C., Loweth, J., McKian, K., De Los Reyes, A., Wing, L. & Jordan, V. C. (2003) J. Natl. Cancer Inst. 95, 1586-1597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}