

Abstract
Purpose of the Review
TGF-ß is required for tissue homeostasis but is also implicated in a disease processes including fibrosis, and thus represents a molecular target for therapy.
Recent Findings
Multiple strategies for inhibiting TGF-ß function exist. The three principal platforms are RNA-based technologies, monoclonal antibodies, and small molecules. Monoclonal antibodies targeting TGF-ß have been used in a clinical trial, with disappointing results to date. Antibodies to the αvß6 integrin prevent activation of latent TGF-ß and show promise in preclinical studies. Over a dozen small molecules inhibit the kinase activity of TGF-ß receptors. Several commonly used drugs have unanticipated anti-TGF-ß activity and may therefore have a role in anti-fibrotic therapy. Because TGF-ß has important physiologic functions, inhibiting its activity might lead to aberrant immune activation, epithelial hyperplasia and impaired wound healing; spontaneous autoimmunity in particular is a concern in an autoimmune disease such as SSc. Novel insights from DNA microarray analysis and genetic polymorphisms in TGF-ß signaling will aid in defining patient populations most likely to respond to anti-TGF-ß treatment.
Summary
Anti-TGF-ß therapies promise to have a major impact in SSc. Significant concerns regarding efficacy, safety, identification of optimal candidates for therapy, and of biomarkers of safety and efficacy, are critical challenges ahead.
Keywords: TGF-ß, fibrosis, scleroderma, avß6 integrin, ALK5, therapy
INTRODUCTION
Fibrosis, the hallmark of systemic sclerosis (SSc), continues to defy effective therapies, and accounts for much of the morbidity and mortality in this disease, along with those of diverse fibrosing conditions. The limited efficacy of immunosuppressive treatments reflects the complex pathogenesis of fibrosis and highlights the uncertain role of inflammation. Recent studies implicate transforming growth factor-ß (TGF-ß) as an essential mediator of fibrosis, and therefore a potential target for anti-fibrotic therapy. Most cell types both produce TGF-ß and express its surface receptors. This pleiotropic cytokine regulates cell proliferation, differentiation, migration, adhesion, survival. epithelial-mesenchymal transition (EMT) and collagen and extracellular matrix (ECM) synthesis, and is essential for angiogenesis, wound healing and immune regulation on the one hand, and cancer, metastasis, diabetes and fibrosis on the other. There is considerable variation among individuals in their basal level of endogenous TGF-ß signaling that is determined, in part, by genetic factors. While the complex biology of TGF-ß in cancer, where it has dual roles as both a potent tumor suppressor and as a stimulus for malignant conversion, invasion and metastasis, has been extensively investigated, its essential roles in autoimmunity and fibrosis are now coming into focus (1). Aberrant TGF-ß regulation and function are implicated in pulmonary fibrosis, glomerulonephritis and diabetic kidney disease, congestive heart failure, liver cirrhosis, Marfan syndrome hypertrophic scars and SSc, and the range of disorders linked to TGF-ß continues to expand (2). Understanding normal and perturbed regulation of TGF-ß synthesis, activation and signaling could lead to novel approaches for blocking pathological TGF-ß responses in the treatment of these diseases.
Currently, the three main strategies are: 1) blocking the TGF-ß ligand; 2) blocking TGF-ß receptor (TßR) activation and downstream signaling; and 3) selective inhibition of intracellular signal transduction by interfering with Smads or with coactivators (Table 1). The most promising advances to date have been achieved in cancer therapy. Relevant clinical trials can be found at http://clinicaltrials.gov. In this review we summarize the biology of TGF-ß in the context of fibrosis, and highlight recent progress toward TGF-ß targeting for fibrosis therapy. While the focus is on TGF-ß, this is not to imply that additional mediators (in particular connective issue growth factor, platelet-derived growth factor, endothelin-1, monocyte chemoattractant protein-1, interleukin-13 and adenosine) do not also have important roles in pathogenesis, and be potential targets for therapy.
Table 1.
Potential strategies for interfering with TGF-ß biology for fibrosis therapy
| Strategy | Example |
|---|---|
| Block ligand production or activity | Isotype-specific neutralizing antibodies |
| Soluble TßR1-3 receptors | |
| Antibodies to αvß6 integrin | |
| Natural TGF-ß binding proteins (eg. Decorin) | |
| Nucleic acid-based (antisense, ribozyme, siRNA) | |
| Block activation of TGF-ß receptors inhibitors | Orally active small molecule TßR kinase |
| Block Smad function | Physiologic endogenous inhibitor Smad7 |
| Block coactivator recruitment and function | Aptamers (Trx-SARA) |
TGF-ß signaling and regulation in the context of fibrosis and systemic sclerosis
Members of the large TGF-ß superfamily regulate cell proliferation and differentiation, apoptosis and migration, and are involved in organogenesis during embryogenesis, and in maintaining tissue homeostasis and immune regulation in the adult (3). Once secreted, TGF-ß interacts with latency-associated peptide (LAP) and latent TGF-ß binding proteins (Fig. 1). The inactive TGF-ß complex, called large latent complex, is sequestered in the ECM by binding to fibrillin-1. In response to injury, the latent TGF-ß complex undergoes activation catalyzed by thrombospondin or by the αvß6 integrins, and active TGF-ß binds to its ubiquitous serine/threonine kinase cell surface receptors. The activated type I TGF-ß receptor (TßR1) ALK5 phosphorylates the R-Smads Smad2 and Smad3. Recruitment of cytoplasmic R-Smads to the activated TßR1 requires SARA (Smad anchor for receptor activation). Upon phosphorylation, R-Smads are released from the SARA-TßR1 complex, heteromerize with Smad4 and translocate from the cytoplasm into the nucleus. Within the nucleus, Smads bind to Smad-binding DNA elements (SBE) containing a repeated AGAC sequence, and regulate transcription (4). Transcriptional profiling of human skin fibroblasts identifies over 300 genes that are modulated in response to TGF-ß (Sargent J, Whitfield M; unpublished). Since the sequence-specific DNA binding affinity of Smad3 is relatively weak, high affinity recruitment of Smad3/4 to specific DNA targets in the biological context depends on interaction with over two dozen DNA-binding factors and cofactors such as histone acetyltransferases p300/CBP and histone deacetylases (HDACs) (5).
Figure 1. TGF-ß signaling pathway.
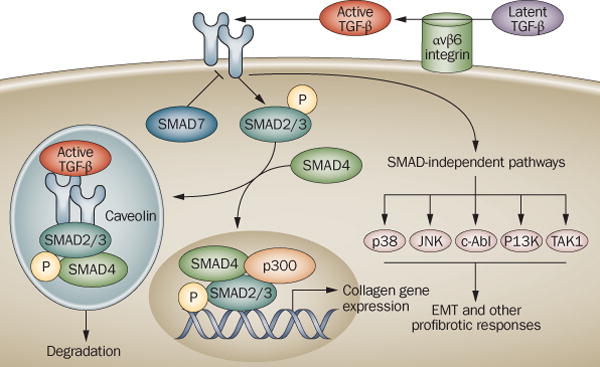
The major components of the TGF-ß signaling pathway in normal fibroblasts. TGF-ß secreted from a variety of cell types is sequestered within the ECM in a biologically inactive form. Activation of latent TGF-ß is catalyzed by αvß6 integrin. Activated TGF-ß binds to the TßR1-TßR2 complex of transmembrance serine/threonine kinases. Smad2/3 are recruited to TßR1 by SARA. Phosphorylated Smad2/3 forms a complex with Smad4 and translocates into the nucleus, where it binds to SBE sequences of TGF-ß-responsive target genes. Cofactors such as p300 are recruited to the SBE-Smad complex and transcription is activated. Smad7 is an endogenous physiologic inhibitor of Smad signaling. Excessive TGF-ß production or activation, or deregulated intracellular Smad signal transduction, result in collagen overproduction and fibrosis.
In addition to Smad2/3 and Smad4, a third class of Smads includes Smad7, an inhibitory Smad that negatively regulates the TGF-ß signaling pathway by inactivation of the TGF-ß receptor complex. Under normal conditions, the duration and intensity of Smad-mediated TGF-ß signaling is limited by multiple mechanisms. Physiologic negative regulators of TGF-ß signaling include Smad7, as well as PPM1A, a nuclear phosphatase that dephosphorylates activated R-Smads and promotes nuclear export and terminating their activity (6), and Man1 (also known as LEMD3), an inner nuclear membrane protein that inhibits TGF-ß signaling by sequestering R-Smads within the nucleus (7).
Substantial evidence implicates TGF-ß in the pathogenesis of SSc, with excessive TGF-ß production or activity, enhanced target cell sensitivity, or failure to terminate TGF-ß-mediated responses as potential mechanisms (reviewed in 8). A recent study with a transgenic mouse harboring a fibroblast-specific constitutively active TßR1 (TßR1ca) provides compelling evidence for the critical role of fibroblast TGF-ß signaling (9). Because clinical observations and animal models suggest that the contribution of TGF-ß is greatest in early-stage SSc, anti-TGF-ß therapies are most likely to be effective in early disease prior to the development of irreversible tissue fibrosis. Transcriptional profiling of lesional tissue from SSc patients demonstrated significant up-regulation of TGF-ß-regulated genes, including those for collagens, ECM and profibrotic cytokines (10). Interestingly, there was substantial patient-to-patient heterogeneity in the strength of the molecular “TGF-ß signature”, suggesting variable pathogenic influence of TGF-ß among individual SSc patients. DNA microarray analysis could ultimately be used as a biomarker for identifying those patients who would be most likely to respond to anti-TGF-ß therapy.
Targeting TGF-ß signaling by blocking the ligand
Targeting the expression and biological activity of a pathogenic cytokine using soluble receptors or neutralizing antibodies has proven to be highly successful in the case of TNF-alpha in rheumatoid arthritis. A comparable approach for the treatment of SSc could be the blockade of TGF-ß expression and function. Some of the strategies under investigation include neutralizing antibodies and soluble receptors, antisense oligonucleotides and RNA interference (Fig. 2).
Figure 2. Antagonism of TGF-ß signaling: targeting the extracellular pathway.
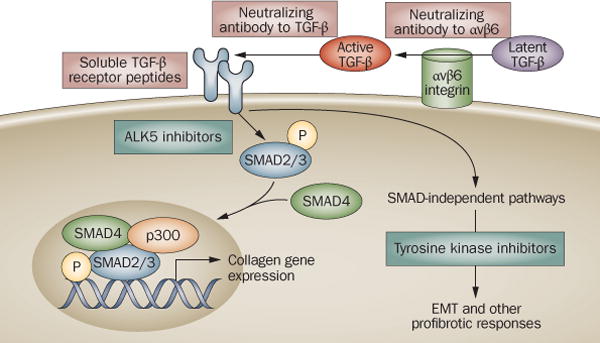
Neutralizing antibodies to αvß6 integrin block its catalytic activity, and inhibit latent TGF-ß activation only at sites where αvß6 integrin is expressed. Neutralizing antibodies to TGF-ß sequester the active ligand, and soluble TßRs prevent its binding to cell surface receptors.
Neutralizing antibodies are particularly effective as they bind directly to ligand and prevent receptor activation, and have been successful in several animal models of fibrosis. Long-term treatment of mice and rats with monoclonal antibody to TGF-ß appeared to be well tolerated, and was shown to prevent renal fibrosis (11–13). In transgenic mouse that harbor fibrillin-1 mutants identified in Marfan syndrome, there is increased TGF-ß signaling in multiple organs, including the lungs, heart and aorta. A neutralizing anti-TGF-ß antibody ameliorated the pulmonary and cardiac manifestations in this Marfan model (14).
The first clinical trial of anti-TGF-ß antibody used a human recombinant IgG antibody specific for the TGF-ß1 isoform (metelimumab) in SSc (15). Patients were randomized to intravenous infusions of antibody or placebo for an 18-week period. No significant improvement in the extent of skin involvement was noted in this dose-ranging pilot study (not powered to evaluate efficacy). Four patients in the antibody-treated group died, apparently from disease complications. A potential limitation of this trial was the isotype specificity of the antibody used. Additional clinical trials include a neutralizing antibody to TGF-ß2 (lerdelimumab) for preventing scarring after glaucoma surgery (16). A novel monoclonal antibody targeting all three TGF-ß isoforms (GC1008) is currently under evaluation in a Phase I trial in idiopathic pulmonary fibrosis.
Soluble receptors and receptor mimetics that abrogate TGF-ß signaling by binding to ligand and preventing it from activating TßRs are also being developed for therapy. Soluble TßRIII was effective in preventing diabetic renal fibrosis in mice (17). A topically administered small peptide fragment of TßRIII, an accessory TGF-ß receptor also called ß-glycan, was effective in ameliorating skin fibrosis in the mouse model of bleomycin-induced scleroderma (18). A recent study screened chemical libraries for small molecules that could specifically block TGF-ß binding to its receptors (19). Five molecules were found that reduced ligand binding to TßRII, and selectively abrogated cellular responses triggered by TGF-ß in vitro. An interesting take on reducing levels of circulating TGF-ß as a therapeutic strategy involved hemoperfusion using an adsorption column specifically reacting with the latent form of TGF-ß (20).
A novel anti-TGF-ß strategy is to selectively block latent TGF-ß activation just at sites where excess TGF-ß activation occurs. The αvß6 integrin, expressed on epithelial cells, is up-regulated by injury and catalyzes the in situ activation of ECM-bound latent TGF-ß in the local microenvironment. The fundamental importance of αvß6 in fibrosis is illustrated by the attenuation of bleomycin-induced lung fibrosis in mice with targeted deletion of this integrin (21). Administration of anti-αvß6 antibodies prevented the development of radiation- or bleomycin-induced fibrosis in mice (22,23). Because integrin-mediated TGF-ß activation is restricted to sites where αvß6 is expressed, a theoretical advantage of αvß6 blockade is that it would only block excessive TGF-ß, while preserving basal TGF-ß signaling required for homeostasis. This concept remains to be validated in the clinical setting.
Antisense oligonucleotides and RNA interference (RNAi) to reduce TGF-ß expression are becoming clinically applicable. Short hairpin RNA vectors that contain RNAi sequences that catalyze enzymatic cleavage of RNA can be stably transfected into cells and used to inhibit translation. Recent preclinical studies demonstrated the anti-fibrotic efficacy of siRNA targeting TGF-ß in mouse models of liver cirrhosis (24) and of peritoneal fibrosis (25). In these rodent studies, siRNA administration decreased collagen accumulation and myofibroblast transformation in the target organs.
Given TGF-ß’s multifunctionality and its critical role of physiological responses, global blockade of TGF-ß activity couldhave undesired consequences. In particular, since TGF-ß is the most potent natural immunosuppressor, complete abrogation of signaling could lead to spontaneous activation of T and B cells, inhibition of regulatory T cell (CD4+ CD25+) function, inflammation and autoimmunity. Indeed, local up-regulation of anti-tumor immunity by TGF-ß blockade may be desirable in cancers characterized by a failure to mount adequate immune responses (26). However, spontaneous autoimmunity has not been a problem to date in preclinical studies of TGF-ß inhibition with antibodies (27) or soluble receptors (28). Indeed, Singh et al. recently showed that even in lupus-probe NZB × NZW mice, anti-TGF-ß antibody surprisingly did not trigger or exacerbate autoimmunity (1). Absence of spontaneous immune activation could be because neutralizing antibodies, soluble receptors and natural antagonists achieve only partial TGF-ß deficiency, interfering with excess TGF-ß activity without altering basal homeostatic TGF-ß signaling. This theoretically could result in abrogation of pathological responses associated with excess TGF-ß such as fibrosis, while preserving physiological TGF-ß responses. While blockade of TGF-ß signaling by targeting the ligand may thus have acceptable toxicity, careful long-term observation in clinical trials will be required to validate this concept.
Inhibiting TGF-ß by blocking receptor kinase activity
Since TGF-ß responses are mediated through cell surface serine threonine kinases, inhibiting TGF-ß signaling at the receptor kinase level would result in abrogation of TGF-ß biological activity (Fig. 3). Several small molecules that recognize the ATP binding domain of the type I or type II TGF-ß receptor kinases and prevent substrate phosphorylation have been developed. Binding to the kinase domain renders the ALK5 receptor incapable of activating Smad2/3. SB431542 (Glaxo), SD-208 (Scios) and SM305 (Biogen-Idec) are each sufficient for blocking TGF-ß-mediated EMT, myofibroblast differentiation and collagen, laminin and fibronectin stimulation by TGF-ß (29–31). Using animal models of fibrosis, small molecule inhibitors of TßR have been shown to be effective in preventing renal (32), liver (33,34), and pulmonary fibrosis (35). A novel inhibitor, GW788388, was shown to block the activity of both TßR1 and TßR2, and prevent TGF-ß signaling and renal fibrosis in the db/db mouse model of spontaneous diabetic nephropathy (36).
Figure 3. Antagonism of TGF-ß signaling: targeting the intracellular pathway.
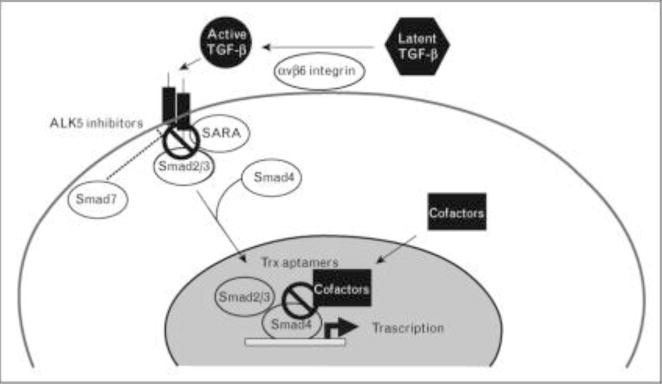
Small molecule inhibitors of TßR1 such as ALK4 blocks Smad2/3 activation by preventing their binding to the kinase domain of ALK5. Inability to phosphorylate Smad2/3 results in abrogation of Smad-dependent TGF-ß responses such as collagen synthesis and myofibroblast transformation. Peptide aptamers displaying the Smad-binding domains of particular Smad-interacting proteins such as SARA (Trx-SARA) prevent recruitment of essential cofactors to the activated Smad complex, and selectively prevent Smad-mediated transcriptional responses. Commonly used drugs such as the PPAR-gamma agonist rosiglitazone, and the Abl tyrosine kinase inhibitor imatinib mesylate block signal transduction downstream from TßR1.
Novel small molecule TßR inhibitors with increased binding affinity and specificity are under development. Whether such competitive ALK5 kinase antagonists will ultimately advance to clinical trials in fibrosis and in SSc remains uncertain. While the oral route of administration makes small molecules highly attractive, the cross-reactivity characteristically associated with kinase inhibitors raises the possibility that multiple pathways will be blocked, with off-target effects unrelated to TGF-ß signaling. Furthermore, blocking all TßR-mediated responses could interfere with the physiologically desirable effects of TGF-ß including immune regulation, cancer surveillance and wound healing.
Targeting downstream Smad signaling activity
Because profibrotic TGF-ß responses are mediated principally via the Smad pathway, antagonism of intracellular Smad signal transduction is of interest. Strategies for Smad targeting (Fig. 3) include overexpression of physiological inhibitors of R-Smads, sequestration or targeting of Smads for ubiquitination and degradation (37). In vivo overexpression of Smad7 by gene delivery has been shown to ameliorate pulmonary, renal and peritoneal fibrosis and vitreous retinopathy in animal models (38–41). Interestingly, some of the anti-fibrotic activities attributed to interferon-gamma could be due to its ability to induce endogenous Smad7 (42).
The selectivity of TGF-ß for activating distinct sets of target genes is determined in the cellular and tissue context by the repertoire of cofactors available for interacting with Smads. Disruption of selected protein-protein interactions offers the theoretical advantage of blocking specific biological responses. An exciting novel strategy for inhibiting selected TGF-ß responses involved the generation of so-called aptamers, which are peptide motifs expressed on a rigid protein scaffold. Peptide aptamers have been used to selectively block intracellular signal transduction by binding to target proteins and disrupt specific protein-protein interactions. Investigators have utilized Escherichia coli thioredoxin A (Trx) as a scaffold for displaying Smad-binding domains (SBD) from specific Smad-interacting cofactors such as p300/CBP (43,44). Introduction of individual Trx aptamers into mammalian cells blocked specific TGF-ß responses such as EMT without generally inhibiting Smad-dependent signaling. While substantial technical hurdles remain, aptamer-based cell therapy thus has the potential for blocking specific Smad interactions with cofactors, thus selectively preventing undesirable TGF-ß responses.
Drugs that antagonize TGF-ß signaling and may have anti-fibrotic activity
Of substantial interest is the anti-TGF-ß activity of commonly used drugs (Table 2). Four such drugs in current clinical use, tranilast, losartan, glitazones and imatinib mesylate, have been found to block the production, activation, or biological activity of TGF-ß. Pirfenidone and halofuginone are additional drugs with anti-TGF-ß activity that are under development.
Table 2.
Drugs that interfere with TGF-ß and may have potential anti-fibrotic activity
| Drug | Clinical indication | Putative anti-TGF-ß mechanism |
|---|---|---|
| Tranilast (approved in Asia) | allergy; prevents mast cell degranulation | inhibits TGF-ß secretion, activity |
| Losartan | Hypertension AT1 receptor blocker | blocks angiotensin II induction of TGF-ß; reduced levels of TGF-ß |
| HMG CoA reductase inhibitors (statins) | hypercholesterolemia | blocks TGF-ß stimulation of collagen synthesis |
| Imatinib mesylate | chronic myelogeneous Leukemia (CML) | blocks TGF-ß signaling; also blocks PDGF receptor activation, Bcr-Abl activity |
| PPAR-γ agonists (rosiglitazone) | Type II diabetes Insulin sensitizing | disrupts intracellular TGF-ß-Smad signal transduction |
| Pirfenidone | none | inhibits TGF-ß production, latent TGF-ß activation |
| Halofuginone | none | unknown |
Tranilast is an anthranilic acid derivative that prevents mast cell degranulation, and under the name Rizaben has been extensively used in Japan for the treatment of asthma, allergic rhinitis and atopic dermatitis. Tranilast has potent anti-fibrotic effects in normal and SSc fibroblasts (45), and in vivo in animal models of fibrosis (46,47). In cultured cells, tranilast prevented the secretion of TGF-ß, down-regulated the expression of TßR, and blocked TGF-ß-induced activation of Smad2 as well as ERK1 (48). Furthermore, tranilast prevented the stimulation of CTGF activity (46). Importantly, tranilast showed anti-inflammatory activity in a mouse model (49). A pilot study in Crohn’s disease demonstrated some clinical efficacy in preventing the progression of strictures (50). The established safety profile of tranilast, together with its combined anti-fibrotic and anti-inflammatory activities, make it an appealing drug for clinical evaluation in SSc.
Pirfenidone is an orally active pyridone small molecule that inhibits collagen synthesis and exerts anti-fibrotic activites in vitro and in vivo through poorly-understood mechanisms. Pirfenidone has been shown to reduce the activation of latent TGF-ß by inhibiting furin, a TGF-ß-activating convertase (51). In a randomized multicenter clinical trial in idiopathic pulmonary fibrosis, pirfenidone treatment was associated with a modest improvement in pulmonary function (52).
Halofuginone is a small molecule plant alkaloid that inhibits collagen synthesis, and blocks TGF-ß signaling via inhibition of Smad3 phosphorylation. Halofuginone was effective in reducing fibrosis in murine sclerodermatous graft versus host disease and in a mouse model of pulmonary fibrosis (53,54).
The antihypertensive angiotensin II type 1 receptor blocker losartan has been found to antagonize TGF-ß signaling through inhibition of the renin-angiotensin axis. In a mouse model of Marfan syndrome induced by fibrillin-1 mutation, losartan reduced aortic wall thickness by suppression of tissue TGF-ß signaling (55), and restored normal muscle architecture (56). Because losartan is commonly used in SSc for the treatment of Raynaud phenomenon, its anti-TGF-ß activity may indicate a potential additional therapeutic benefit.
Imatinib mesylate (Gleevec) is a small molecule inhibitor of Bcr-Abl kinase. Bcr-Abl is an oncogenic fusion protein that incorporates the nonreceptor tyrosine kinase c-Abl and Bcr, a multidomain protein of unknown function. Bcr-Abl results from the t(9;22) reciprocal translocation in hematopoietic stem cells, creating the Philadelphia chromosome found in >90% of cases of chronic myelogeneous leukemia. Recent studies demonstrate that c-Abl is activated by TGF-ß and mediates profibrotic responses in fibroblasts (57, 58). Imatinib potently blocks TGF-ß-induced c-Abl activity and stimulation of fibrotic gene responses, and reduced basal collagen levels in SSc fibroblasts (57–59). The anti-TGF-ß effects are Smad-independent and may be due to blockade of the early growth response gene Egr-1 (Bhattacharyya S and Varga J, unpublished). Imatinib has been shown to prevent the development of fibrosis in mouse models of lung fibrosis (60), renal fibrosis (61) and bleomycin-induced scleroderma (62), but failed to arrest progression of established lung fibrosis when treatment was initiated following bleomycin (63). Interestingly, inhibition of TGF-ß signaling by imatinib was not associated with inflammation or spontaneous autoimmunity, making imatinib an attractive potential therapeutic agent. Clinical trials of imatinib as an anti-fibrotic therapy are currently underway for SSc and pulmonary fibrosis.
Conclusions and Perspectives
The pleiotropic biological activities of TGF-ß contribute to physiologic and pathological processes and can be beneficial or detrimental. Fibrotic disorders including SSc are associated with perturbed TGF-ß expression and functions, and thus TGF-ß is the molecular tagert of choice for therapy. It is remarkable that although TGF-ß was discovered just over 20 years ago, multiple platforms for its inhibition have been already developed and moved along the clinical development pipeline. Strategies for inhibition of TGF-ß include suppressing ligand production and activation, blocking receptor binding, and disrupting intracellular signal transduction and biological responses. Selectivity may be achieved spatially, for instance using antibodies to block TGF-ß activation only at sites where integrin αvß6 is expressed; or by selective inhibition of deleterious biological responses by blocking target gene-specific coactivator-Smad interactions using aptamers. Both large and small molecules that effectively block TGF-ß are in preclinical evaluation or in early clinical trials. Furthermore, a number of drugs in current clinical use for a variety of indications show anti-TGF-ß activity and might therefore have anti-fibrotic potential.
While the functional pleiotropy of TGF-ß suggests that blocking its activity could be associated with significant potential risks, including spontaneous autoimmunity and epithelial hyperplasia, studies to date have not found this to be the case; possibly due to incomplete abrogation of TGF-ß biology with current anti-TGF-ß strategies. The modest toxicities associated to date with anti-TGF-ß strategies provide encouragement for further clinical development and evaluation of such therapies for SSc. The selection of appropriate target populations of patients who may be responders, such as those who show a strong “TGF-ß signature” in affected tissues, elevated levels of circulating TGF-ß, or genetically-determined elevations in basal levels of endogenous TGF-ß signaling, and the optimal timing of anti-TGF-ß intervention, most likely to be effective in early-stage disease when fibrosis is TGF-ß-dependent and reversible, will require carefully-designed clinical trials incorporating biomarkers of biological response and of clinical efficacy, and depend on extensive collaborations among SSc investigators.
References
- 1.Saxena V, Lienesch DW, Zhou M, Bommireddy R, Azhar M, Doetschman T, Singh RR. Dual roles of immunoregulatory cytokine TGF-beta in the pathogenesis of autoimmunity-mediated organ damage. J Immunol. 2008 Feb 1;180(3):1903–12. doi: 10.4049/jimmunol.180.3.1903. This study in a lupus-prone mouse strain highlights the complex biological duality of TGF-ß in the context of chronic autoimmunity. Whereas in lymphoid organs there was reduced TGF-ß signaling, contributing to spontaneous autoimmunity and inflammation, in target organs such as the kidney, glomerulosclerosis and renal fibrosis were associated with increased TGF-ß expression and activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008 Apr;1782(4):197–228. doi: 10.1016/j.bbadis.2008.01.006. Epub 2008 Feb 11. [DOI] [PubMed] [Google Scholar]
- 3.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003 Jun 13;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 4.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998 Mar;1(4):611–7. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol. 2007 Dec;213(3):663–71. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- 6.Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC, Shi Y, Chen YG, Meng A, Feng XH. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006 Jun 2;125(5):915–28. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin F, Morrison JM, Wu W, Worman HJ. MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet. 2005 Feb 1;14(3):437–45. doi: 10.1093/hmg/ddi040. Epub 2004 Dec 15. [DOI] [PubMed] [Google Scholar]
- 8.Trojanowska M, Varga J. Molecular pathways as novel therapeutic targets in systemic sclerosis. Curr Opin Rheumatol. 2007 Nov;19(6):568–73. doi: 10.1097/BOR.0b013e3282e6f495. [DOI] [PubMed] [Google Scholar]
- 9.Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, Vanpelt CS, Geng YJ, Deng JM, Behringer RR, de Crombrugghe B. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007 Jan;56(1):334–44. doi: 10.1002/art.22328. This paper describes a novel transgenic mouse strain that develops spontaneous skin fibrosis driven by inducible expression of fibroblast-specific constitutively active TßR2. The results clearly establish the pivotal role of TGF-ß signaling in fibroblasts as a mechanism underlying fibrosis in scleroderma. [DOI] [PubMed] [Google Scholar]
- 10.Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, McCalmont TH, Brown PO, Botstein D, Connolly MK. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003 Oct 14;100(21):12319–24. doi: 10.1073/pnas.1635114100. Epub 2003 Oct 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000 Jul 5;97(14):8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCormick LL, Zhang Y, Tootell E, Gilliam AC. Anti-TGF-beta treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. J Immunol. 1999 Nov 15;163(10):5693–9. [PubMed] [Google Scholar]
- 13.Fukasawa H, Yamamoto T, Suzuki H, Togawa A, Ohashi N, Fujigaki Y, Uchida C, Aoki M, Hosono M, Kitagawa M, Hishida A. Treatment with anti-TGF-beta antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-beta signaling. Kidney Int. 2004 Jan;65(1):63–74. doi: 10.1111/j.1523-1755.2004.00393.x. [DOI] [PubMed] [Google Scholar]
- 14.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003 Mar;33(3):407–11. doi: 10.1038/ng1116. Epub 2003 Feb 24. [DOI] [PubMed] [Google Scholar]
- 15.Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Akesson A, Coppock J, Hoogen F, Herrick A, Mayes MD, Veale D, Haas J, Ledbetter S, Korn JH, Black CM, Seibold JR, Cat-192 Study Group; Scleroderma Clinical Trials Consortium Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007 Jan;56(1):323–33. doi: 10.1002/art.22289. [DOI] [PubMed] [Google Scholar]
- 16.Mead AL, Wong TT, Cordeiro MF, Anderson IK, Khaw PT. Evaluation of anti-TGF-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest Ophthalmol Vis Sci. 2003 Aug;44(8):3394–401. doi: 10.1167/iovs.02-0978. [DOI] [PubMed] [Google Scholar]
- 17.Juárez P, Vilchis-Landeros MM, Ponce-Coria J, Mendoza V, Hernández-Pando R, Bobadilla NA, López-Casillas F. Soluble betaglycan reduces renal damage progression in db/db mice. Am J Physiol Renal Physiol. 2007 Jan;292(1):F321–9. doi: 10.1152/ajprenal.00264.2006. Epub 2006 Sep 5. [DOI] [PubMed] [Google Scholar]
- 18.Santiago B, Gutierrez-Cañas I, Dotor J, Palao G, Lasarte JJ, Ruiz J, Prieto J, Borrás-Cuesta F, Pablos JL. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J Invest Dermatol. 2005 Sep;125(3):450–5. doi: 10.1111/j.0022-202X.2005.23859.x. [DOI] [PubMed] [Google Scholar]
- 19.Burmester JK, Salzman SA, Zhang KQ, Dart RA. Small molecule antagonists of the TGF-beta1/TGF-beta receptor binding interaction. Med Oncol. 2006;23(4):553–62. doi: 10.1385/MO:23:4:553. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto Y, Ueda Y, Itoh T, Iwamoto A, Yamagishi H, Shimagaki M, Teramoto K. A novel immunotherapeutic modality with direct hemoperfusion targeting transforming growth factor-beta prolongs the survival of tumor-bearing rats. Oncol Rep. 2006 Dec;16(6):1277–84. [PubMed] [Google Scholar]
- 21.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999 Feb 5;96(3):319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 22.Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, Devitt ML, Horan GS, Weinreb PH, Lukashev ME, Violette SM, Grant KS, Colarossi C, Formenti SC, Munger JS. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008 Jan 1;177(1):82–90. doi: 10.1164/rccm.200706-806OC. Epub 2007 Oct 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ, Goyal J, Feghali-Bostwick CA, Matteson EL, O’Hara C, Lafyatis R, Davis GS, Huang X, Sheppard D, Violette SM. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008 Jan 1;177(1):56–65. doi: 10.1164/rccm.200706-805OC. Epub 2007 Oct 4. This paper demonstrates that integrin alpha(v)beta6 has a key role in the pathogenesis of fibrosis via activation of ECM-bound latent TGF-ß. Inhibition of integrin alpha(v)beta6 using monoclonal antibody leads to partial TGF-ß blockade and substantial amelioration of experimental fibrosis in the mouse. [DOI] [PubMed] [Google Scholar]
- 24.Hwang M, Kim HJ, Noh HJ, Chang YC, Chae YM, Kim KH, Jeon JP, Lee TS, Oh HK, Lee YS, Park KK. TGF-beta1 siRNA suppresses the tubulointerstitial fibrosis in the kidney of ureteral obstruction. Exp Mol Pathol. 2006 Aug;81(1):48–54. doi: 10.1016/j.yexmp.2005.11.005. Epub 2006 Jan 27. [DOI] [PubMed] [Google Scholar]
- 25.Liu FY, Xiao L, Peng YM, Duan SB, Liu H, Liu YH, Ling GH, Yuan F, Chen JX, Fu X, Zhu JL. Inhibition effect of small interfering RNA of connective tissue growth factor on the expression of vascular endothelial growth factor and connective tissue growth factor in cultured human peritoneal mesothelial cells. Chin Med J (Engl) 2007 Feb 5;120(3):231–6. [PubMed] [Google Scholar]
- 26.Pennison M, Pasche B. Targeting transforming growth factor-beta signaling. Curr Opin Oncol. 2007 Nov;19(6):579–85. doi: 10.1097/CCO.0b013e3282f0ad0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruzek MC, Hawes M, Pratt B, McPherson J, Ledbetter S, Richards SM, Garman RD. Minimal effects on immune parameters following chronic anti-TGF-beta monoclonal antibody administration to normal mice. Immunopharmacol Immunotoxicol. 2003 May;25(2):235–57. doi: 10.1081/iph-120020473. [DOI] [PubMed] [Google Scholar]
- 28.Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, Patel SC, Khozin S, Liu ZY, Green J, Anver MR, Merlino G, Wakefield LM. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002 Jun;109(12):1607–15. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tojo M, Hamashima Y, Hanyu A, Kajimoto T, Saitoh M, Miyazono K, Node M, Imamura T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci. 2005 Nov;96(11):791–800. doi: 10.1111/j.1349-7006.2005.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori Y, Ishida W, Bhattacharyya S, Li Y, Platanias LC, Varga J. Selective inhibition of activin receptor-like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum. 2004 Dec;50(12):4008–21. doi: 10.1002/art.20658. [DOI] [PubMed] [Google Scholar]
- 31.Ishida W, Mori Y, Lakos G, Sun L, Shan F, Bowes S, Josiah S, Lee WC, Singh J, Ling LE, Varga J. Intracellular TGF-beta receptor blockade abrogates Smad-dependent fibroblast activation in vitro and in vivo. J Invest Dermatol. 2006 Aug;126(8):1733–44. doi: 10.1038/sj.jid.5700303. Epub 2006 Jun 1. This study demonstrates that a small molecule inhibitor of ALK5 kinase activity effectively blocks TGF-ß-dependent transcriptional responses in normal fibroblasts, and reduces the stimulation of collagen and other profibrotic proteins. Furthermore, the inhibitor induces partial normalization of gene expression in SSc skin fibroblasts. [DOI] [PubMed] [Google Scholar]
- 32.Moon JA, Kim HT, Cho IS, Sheen YY, Kim DK. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006 Oct;70(7):1234–43. doi: 10.1038/sj.ki.5001775. Epub 2006 Aug 23. [DOI] [PubMed] [Google Scholar]
- 33.de Gouville AC, Boullay V, Krysa G, Pilot J, Brusq JM, Loriolle F, Gauthier JM, Papworth SA, Laroze A, Gellibert F, Huet S. Inhibition of TGF-beta signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br J Pharmacol. 2005 May;145(2):166–77. doi: 10.1038/sj.bjp.0706172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu K, Corbley MJ, Sun L, Friedman JE, Shan F, Papadatos JL, Costa D, Lutterodt F, Sweigard H, Bowes S, Choi M, Boriack-Sjodin PA, Arduini RM, Sun D, Newman MN, Zhang X, Mead JN, Chuaqui CE, Cheung HK, Zhang X, Cornebise M, Carter MB, Josiah S, Singh J, Lee WC, Gill A, Ling LE. SM16, an orally active TGF-beta type I receptor inhibitor prevents myofibroblast induction and vascular fibrosis in the rat carotid injury model. Arterioscler Thromb Vasc Biol. 2008 Apr;28(4):665–71. doi: 10.1161/ATVBAHA.107.158030. Epub 2008 Jan 17. [DOI] [PubMed] [Google Scholar]
- 35.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, Protter AA, Gauldie J. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med. 2005 Apr 15;171(8):889–98. doi: 10.1164/rccm.200405-612OC. Epub 2004 Nov 24. [DOI] [PubMed] [Google Scholar]
- 36.Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F, de Gouville AC, Huet S, ten Dijke P, Laping NJ. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008 Mar;73(6):705–15. doi: 10.1038/sj.ki.5002717. Epub 2007 Dec 12. [DOI] [PubMed] [Google Scholar]
- 37.Itoh S, ten Dijke P. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol. 2007 Apr;19(2):176–84. doi: 10.1016/j.ceb.2007.02.015. Epub 2007 Feb 2. [DOI] [PubMed] [Google Scholar]
- 38.Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest. 1999 Jul;104(1):5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nie J, Dou X, Hao W, Wang X, Peng W, Jia Z, Chen W, Li X, Luo N, Lan HY, Yu XQ. Smad7 gene transfer inhibits peritoneal fibrosis. Kidney Int. 2007 Dec;72(11):1336–44. doi: 10.1038/sj.ki.5002533. Epub 2007 Sep 12. [DOI] [PubMed] [Google Scholar]
- 40.Saika S, Yamanaka O, Nishikawa-Ishida I, Kitano A, Flanders KC, Okada Y, Ohnishi Y, Nakajima Y, Ikeda K. Effect of Smad7 gene overexpression on transforming growth factor beta-induced retinal pigment fibrosis in a proliferative vitreoretinopathy mouse model. Arch Ophthalmol. 2007 May;125(5):647–54. doi: 10.1001/archopht.125.5.647. [DOI] [PubMed] [Google Scholar]
- 41.Lan HY. Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci. 2008 May 1;13:4984–92. doi: 10.2741/3057. [DOI] [PubMed] [Google Scholar]
- 42.Weng H, Mertens PR, Gressner AM, Dooley S. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J Hepatol. 2007 Feb;46(2):295–303. doi: 10.1016/j.jhep.2006.09.014. Epub 2006 Nov 3. [DOI] [PubMed] [Google Scholar]
- 43.Cui Q, Lim SK, Zhao B, Hoffmann FM. Selective inhibition of TGF-beta responsive genes by Smad-interacting peptide aptamers from FoxH1, Lef1 and CBP. Oncogene. 2005 Jun 2;24(24):3864–74. doi: 10.1038/sj.onc.1208556. [DOI] [PubMed] [Google Scholar]
- 44.Zhao BM, Hoffmann FM. Inhibition of transforming growth factor-beta1-induced signaling and epithelial-to-mesenchymal transition by the Smad-binding peptide aptamer Trx-SARA. Mol Biol Cell. 2006 Sep;17(9):3819–31. doi: 10.1091/mbc.E05-10-0990. Epub 2006 Jun 14. This study describes an experimental approach for selectively disrupting the interaction of activated cytoplasmic Smads with individual signaling partners using aptamers. The strategy has the potential for selectively blocking deleterious TGF-ß responses without inhibiting the physiologically beneficial homeostatic activitiers of TGF-ß. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada H, Tajima S, Nishikawa T. Tranilast inhibits collagen synthesis in normal, scleroderma and keloid fibroblasts at a late passage culture but not at an early passage culture. J Dermatol Sci. 1995 Jan;9(1):45–7. doi: 10.1016/0923-1811(94)00355-i. [DOI] [PubMed] [Google Scholar]
- 46.Qi W, Chen X, Twigg S, Polhill TS, Gilbert RE, Pollock CA. Tranilast attenuates connective tissue growth factor-induced extracellular matrix accumulation in renal cells. Kidney Int. 2006 Mar;69(6):989–95. doi: 10.1038/sj.ki.5000189. [DOI] [PubMed] [Google Scholar]
- 47.Xu Q, Norman JT, Shrivastav S, Lucio-Cazana J, Kopp JB. In vitro models of TGF-beta-induced fibrosis suitable for high-throughput screening of antifibrotic agents. Am J Physiol Renal Physiol. 2007 Aug;293(2):F631–40. doi: 10.1152/ajprenal.00379.2006. Epub 2007 May 9. This study describes an animal model suitable for evaluating anti-fibrotic therapies in vivo, and highlights the efficacy of the small molecule tranilast in inhibiting TGF-ß-dependent fibrotic process. [DOI] [PubMed] [Google Scholar]
- 48.Platten M, Wild-Bode C, Wick W, Leitlein J, Dichgans J, Weller M. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) inhibits transforming growth factor-beta relesase and reduces migration and invasiveness of human malignant glioma cells. Int J Cancer. 2001 Jul 1;93(1):53–61. doi: 10.1002/ijc.1289. [DOI] [PubMed] [Google Scholar]
- 49.Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, Gupta R, Lee LY, Kidd BA, Robinson WH, Sobel RA, Selley ML, Steinman L. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. 2005 Nov 4;310(5749):850–5. doi: 10.1126/science.1117634. Erratum in: Science. 2006 Feb 17;311(5763):954. [DOI] [PubMed] [Google Scholar]
- 50.Oshitani N, Yamagami H, Watanabe K, Higuchi K, Arakawa T. Long-term prospective pilot study with tranilast for the prevention of stricture progression in patients with Crohn’s disease. Gut. 2007 Apr;56(4):599–600. doi: 10.1136/gut.2006.115469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burghardt I, Tritschler F, Opitz CA, Frank B, Weller M, Wick W. Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem Biophys Res Commun. 2007 Mar 9;354(2):542–7. doi: 10.1016/j.bbrc.2007.01.012. Epub 2007 Jan 10. [DOI] [PubMed] [Google Scholar]
- 52.Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, Sato A, Kudoh S. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005 May 1;171(9):1040–7. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 53.Halevy O, Nagler A, Levi-Schaffer F, Genina O, Pines M. Inhibition of collagen type I synthesis by skin fibroblasts of graft versus host disease and scleroderma patients: effect of halofuginone. Biochem Pharmacol. 1996 Oct 11;52(7):1057–63. doi: 10.1016/0006-2952(96)00427-3. [DOI] [PubMed] [Google Scholar]
- 54.Nagler A, Firman N, Feferman R, Cotev S, Pines M, Shoshan S. Reduction in pulmonary fibrosis in vivo by halofuginone. Am J Respir Crit Care Med. 1996 Oct;154(4 Pt 1):1082–6. doi: 10.1164/ajrccm.154.4.8887611. [DOI] [PubMed] [Google Scholar]
- 55.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006 Apr 7;312(5770):117–21. doi: 10.1126/science.1124287. This study used the mouse model of Marfan syndrome induced by transgenic expression of a mutant form of fibrillin-1 to establish the ability of losartan to block TGF-ß signaling and prevent the development of connective tissue abnormalities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007 Feb;13(2):204–10. doi: 10.1038/nm1536. Epub 2007 Jan 21. Erratum in: Nat Med. 2007 Apr;13(4):511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004 Nov;114(9):1308–16. doi: 10.1172/JCI19603. This study provides the first indication that the nonreceptor tyrosine kinase c_Abl is downstream of the activated TßR1, and plays a pivotal role in fibrotic responses elicited by TGF-ß in normal lung fibroblasts. Furthermore, by blocking Smad-independent TGF-ß signaling as well as inhibiting PDGF receptor activation, imatinib prevented the development of bleomycin-induced lung fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishida W, Bhattacharyya S, Hinchcliff M, Mori Y, Takehara K, Varga J. Novel role of c-Abl tyrosine kinase in profibrotic TGF-Beta responses: Selective modulation by the anticancer drug imatinib methylate (gleevec) Arthitis Rheum. 2006;54:S776. [Google Scholar]
- 59.Soria A, Cario-André M, Lepreux S, Rezvani HR, Pasquet JM, Pain C, Schaeverbeke T, Mahon FX, Taïeb A. The effect of imatinib (Glivec) on scleroderma and normal dermal fibroblasts: a preclinical study. Dermatology. 2008;216(2):109–17. doi: 10.1159/000111507. Epub 2008 Jan 23. [DOI] [PubMed] [Google Scholar]
- 60.Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, Izumi K, Sone S. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2005 Jun 1;171(11):1279–85. doi: 10.1164/rccm.200404-531OC. Epub 2005 Feb 25. [DOI] [PubMed] [Google Scholar]
- 61.Wang S, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005 Jan;19(1):1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- 62.Distler JH, Jüngel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, Michel BA, Hauser T, Schett G, Gay S, Distler O. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007 Jan;56(1):311–22. doi: 10.1002/art.22314. [DOI] [PubMed] [Google Scholar]
- 63.Vittal R, Zhang H, Han MK, Moore BB, Horowitz JC, Thannickal VJ. Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: implications for treatment of fibrotic diseases. J Pharmacol Exp Ther. 2007 Apr;321(1):35–44. doi: 10.1124/jpet.106.113407. Epub 2007 Jan 11. [DOI] [PubMed] [Google Scholar]