

Abstract
ONC201/TIC10 is a small molecule inducer of the TRAIL gene under current investigation as a novel anticancer agent. In this study, we identify critical molecular determinants of ONC201 sensitivity offering potential utility as pharmacodynamic or predictive response markers. By screening a library of kinase siRNAs in combination with a subcytotoxic dose of ONC201, we identified several kinases that ablated tumor cell sensitivity, including the MAPK pathway inducer KSR1. Unexpectedly, KSR1 silencing did not affect MAPK signaling in the presence or absence of ONC201, but instead reduced expression of the anti-apoptotic proteins FLIP, Mcl-1, Bcl-2, cIAP1, cIAP2, and survivin. In parallel to this work, we also conducted a synergy screen in which ONC201 was combined with approved small molecule anticancer drugs. In multiple cancer cell populations, ONC201 synergized with diverse drug classes including the multi-kinase inhibitor sorafenib. Notably, combining ONC201 and sorafenib led to synergistic induction of TRAIL and its receptor DR5 along with a potent induction of cell death. In a mouse xenograft model of hepatocellular carcinoma, we demonstrated that ONC201 and sorafenib cooperatively and safely triggered tumor regressions. Overall, our results established a set of determinants for ONC201 sensitivity that may predict therapeutic response, particularly in settings of sorafenib co-treatment to enhance anticancer responses.
Keywords: ONC201, TIC10, FLIP, Mcl-1, sorafenib, KSR1
Introduction
ONC201/TIC10 is a small molecule that was discovered due to its ability to engage the endogenous TNF-related apoptosis-inducing ligand (TRAIL) cell death pathway that triggers cell death in tumor cells with a favorably wide therapeutic window. We previously reported the discovery and selective advantages of this small molecule compared to other TRAIL-based agents along with preclinical efficacy, safety, and mechanism of action studies. These studies revealed substantial monoagent efficacy in several tumors types that appeared to be driven by a late stage dual inactivation of Akt and MAPK signaling to boost downstream production of TRAIL through activation of Foxo3a (1). ONC201 is undergoing clinical introduction in advanced cancer patients to evaluate monoagent safety and preliminary antitumor activity in select solid and liquid tumors.
We previously reported genetic determinants of sensitivity to TRAIL (2) and therapeutic strategies to increase cancer cell sensitivity to TRAIL such as combination therapy with sorafenib (3). Our previous studies found that ONC201-mediated apoptosis is Bax-dependent in HCT116 cells, as is the case with recombinant TRAIL (1, 2). In this study, unbiased genetic and small molecule approaches to identify determinants of ONC201 sensitivity were pursued in parallel and to augment the antitumor activity of ONC201 through combination therapy. In addition to basic findings that include a novel relationship that we identify between KSR1 and regulators of apoptosis, these studies also identify several predictive markers of ONC201 response and drug combinations that warrant investigation in future monoagent and combinatorial clinical studies.
Materials and Methods
Cell culture and reagents
Cell lines were obtained from American Tissue Type Culture Collection and cultured under recommended conditions in log-phase growth. Sorafenib was kindly provided by Charles D. Smith. Sorafenib was formulated in DMSO (Sigma-Aldrich) for in vitro studies and in 50% ethanol (Sigma-Aldrich) and 50% Cremophor EL (Sigma-Aldrich) for in vivo studies. ONC201 was synthesized as a salt by Provid Pharmaceuticals and structural integrity was validated by NMR and X-Ray crystallography. ONC201 was formulated in DMSO for in vitro studies and phosphate-buffered saline for in vivo studies. The FDA Approved Oncology Set IV was obtained from the NCI.
siRNA kinase library screen
HCT116 p53−/− cells were transfected at 20,000 cells/well in 96-well plates. Cells were transfected with the Stealth RNAi human kinase library (Life Technologies) using RNAiMax (Life Technologies) in Optimem (Life Technologies). Scramble and AllStars Hs Cell Death Control siRNA (Qiagen) were used as negative and positive controls, respectively. Transfection was carried out overnight and the following day complete media containing ONC201 or DMSO was added to the plates following removal of the transfection media. CellTiterGlo (Promega) analysis was performed according to the manufacturer’s instructions at indicated time points.
Small molecule synergy screen
Procedures were performed with a Biomek 2000 robot using pin tools for drug treatment. Cells were seeded at 5×104 cells/mL in 96-well black-wall plates and treatment was performed 12 hours later. Combinatorial activity was initially assessed by calculating the difference between the observed activity with the combination and the sum of the monoagent activities.
Cell-based assays
Cell TiterGlo, Western blot analysis, and cell cycle flow cytometry analysis were performed as previously described (1). Cell surface TRAIL and DR5 were assessed following fixation with 4% paraformaldehyde in PBS for 20 minutes, rinsed in PBS, incubated overnight at 1:200 with anti-TRAIL (Abcam) or anti-DR5 antibodies, rinsed in PBS, incubated with fluorophore-conjugated secondary antibodies (Thermo Scientific Pierce) at 1:250 for 30 minutes, and analyzed by flow cytometry. pERK was assessed with an antibody for p-T202/Y204 (Cell Signaling) and pAkt was assessed with antibody for pT308 (Cell Signaling).
In vivo studies
6-week-old athymic nude mice were obtained from Charles River Laboratories. 107 HepG2 cells in 200 µL (1:1, PBS: Matrigel) were injected into each rear flank. Measurable tumors were assessed 1 week later. Treatment was then initiated as indicated. Sorafenib and ONC201 were administered as 100 µL solutions by oral gavage. ONC201 was administered 12 hours following sorafenib treatment. Tumor volume was assessed with digital calipers and calculated as a spheroid. Tumor dimensions and body weights were assessed twice a week. IHC (Vector Labs) and TUNEL (Millipore) analyses were performed as previously described (1).
Statistical analysis
Pairwise comparisons were assessed by Student’s two-tailed t test. Combination indices were calculated using Chou-Talalay method and Calcusyn software.
Results
Identification of kinase regulators of ONC201 sensitivity
We conducted a siRNA screen to identify kinases that affect ONC201 response in cancer cells to potentially gain mechanistic insight regarding ONC201 and identify molecular targets for combination therapy to improve the activity of ONC201 and/or predict response. A siRNA library targeting 636 kinases were employed for the screen along with 1 µM dose of ONC201 in HCT116 p53−/− cells. The screen identified a number of candidate positive and negative regulators of ONC201 sensitivity at 12 and 36 hours-post treatment (Figure 1A). As expected there was a general trend toward decreased cell viability with knockdown of most kinases themselves, though the decreased cell viability was typically modest in magnitude (<30%). The top 3–4 positive and negative modulators of ONC201 sensitivity at each evaluated time point were selected for validation studies (Table S1), which identified 4 kinases that enhance response to ONC201 following knockdown at 36 hours-post treatment in HCT116 p53−/− cells: DGKD, SGKL, STK123, and KSR1 (Table S2; Figure S1). Enhanced ONC201 efficacy with knockdown of these 4 kinases is also apparent at 48 but not 24 hours post-treatment (Figure 1B), suggesting that the molecular mechanism of sensitivity may occur involve the late apoptotic effects rather than the early signaling effects.
Figure 1. siRNA screen identifies kinase regulators of ONC201 sensitivity.
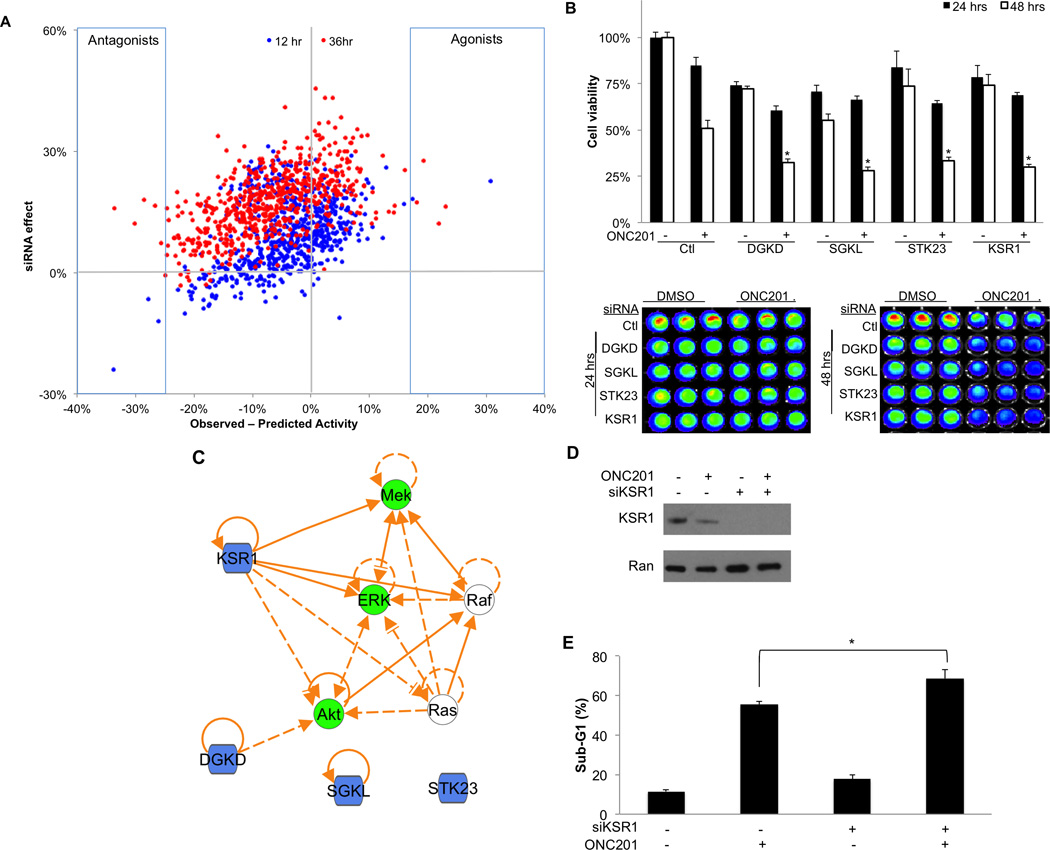
A) Decrease in cell viability in HCT116 cells associated with the siRNA alone (y-axis) or the difference in observed and predicted activity in cell viability associated with the combination of ONC201 treatment (1 µM) and knockdown by siRNA (x-axis). (B) Cell viability in HCT116 cells following ONC201 treatment (1 µM) and/or siRNA knockdown at 24 or 48 hours post-treatment (n=3). Quantification (top panel) and raw data (bottom panels) are shown. *P < 0.05 compared to 48 hours post-ONC201 treatment and control siRNA by Student’s two-tailed t test. (C) Network analysis of ONC201 kinase regulators (blue) and putative mechanism of action (green). (D) Western blot analysis of HCT116 cells treated with DMSO or ONC201 (5 µM) with or without siRNA-mediated knockdown of KSR1 (60 hours). (E) Sub-G1 DNA content analysis following treatment with ONC201 (5 µM) or sorafenib at indicated concentrations (72 hours, n=3). * P< 0.05 by student’s two-tailed t test.
Network analysis of these 4 kinases that regulate ONC201 sensitivity and the previously described mechanism of ONC201 revealed rational overlap of signaling pathways with some kinases. Among the 4 identified kinases, KSR1 possessed the most direct connections to the putative mechanism of ONC201 that involves the dual inhibition of Akt and the MAPK pathway (Figure 1C). KSR1 is described as a MAPK scaffold protein that positively regulates the MAPK signaling pathway (4). Western blot analysis confirmed knockdown of KSR1 at the protein level by siRNA and that ONC201 has a modest negative effect on KSR1 expression as a monoagent (Figure 1D). Cell death assays with ONC201 and siRNA targeting KSR1 revealed that KSR1 knockdown significantly enhanced cell death induced by ONC201 (Figure 1E). Also in support a MAPK-independent effect, knockdown of KSR1 did not enhance the effects of the dual EGFR/HER2 small molecule inhibitor (Figure S2).
We next investigated if KSR1 knockdown affected the amount or kinetics of TRAIL or DR5 production that is stimulated downstream by ONC201. No effect was observed on these parameters with knockdown of KSR1 (Figure S3). Consistent with this notion, the concentration threshold and magnitude of MAPK and Akt signaling inhibition was not affected by KSR1 knockdown (Figure S4). These observations suggest that the ability of KSR1 to sensitize cancer cells to ONC201 involves factor(s) that lie downstream of TRAIL production.
Identification of small molecule anti-cancer drugs that synergize with ONC201
In parallel to the siRNA screen, we screened for FDA approved drugs that synergize with ONC201 in a panel of human cancer cell lines that represent a diverse array of solid tumors (brain, breast, colon, liver, lung, ovarian) to potentially improve the antitumor activity of ONC201. For this screen we utilized the NCI DTP Approved Set IV that includes 101 small molecules approved by the FDA for an indication in oncology. To enable sufficient evaluation of synergism, we chose to screen using a subcytotoxic dose of ONC201 (1 µM) and tested multiple doses of the approved oncology drugs (10 nM, 100 nM, 1 µM), given their wide range of GI50 values amongst each other and across cell lines. Employing cell viability assays following 72 hours of treatment, we identified 33 approved oncology drugs that combined with ONC201 to give a >20% reduction in cell viability over the expected reduction in cell viability, which was calculated as the sum of the observed monoagent activity (Figure 2A). Twelve of these agents synergized under more than a single condition tested in the screen (Figure S5A).
Figure 2. Identification of synergistic combinations of FDA-approved small molecule anti-cancer drugs with ONC201.
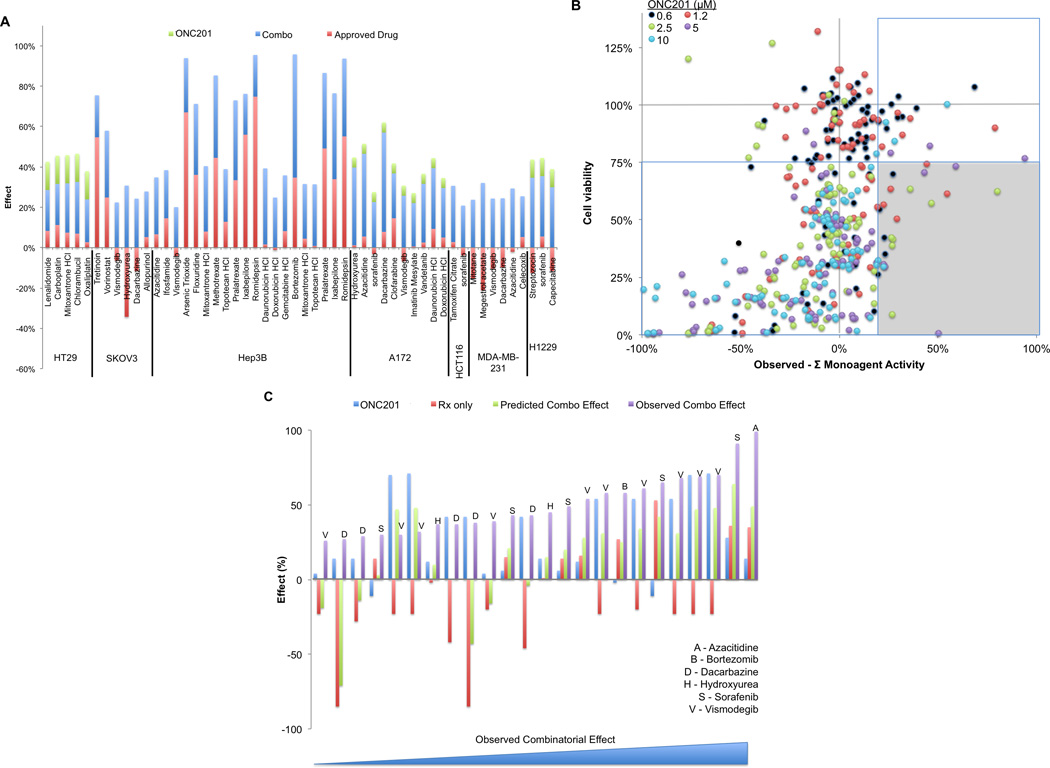
A) Reduction in cell viability in response to ONC201 (1 µM), approved agents, or the combination (72 hours, n=3). The difference between the observed activity and the sum of the monoagent activities are shown in blue. Only combinations yielding >20% observed activity over the predicted activity are shown. (B) Prioritization of combinatorial data by efficacy and synergistic activity (n=2). The prioritization criterion was defined as the lower right shaded quadrant. Data for the prioritized monoagent and combinations are shown in (C). Each set of 4 bars represents a distinct synergistic data point under a given combination of drug concentrations, along with its associated monoagent and vehicle controls. The data sets are sorted with increasing combinatorial efficacy from left to right.
Analyzing the classes of drugs tested in the screen revealed that ONC201 synergizes with a wide array of drug classes, though synergy was not ubiquitous within any particular class (Figure S5B). Notable exceptions were the absence of synergy observed with any specific mTOR or EGFR/MAPK signaling inhibitors. This observation is in accordance with previous studies of ONC201 that demonstrated the ablation of Akt and MAPK signaling following ONC201 monoagent treatment in HCT116 cells (1). Lack of synergy with specific anti-angiogenic agents were also noted, which is expected as angiogenesis is not represented in standard cell culture models. Alkylating agents and nucleotide synthesis inhibitors possessed the largest number of agents that synergized with ONC201, though this can be attributed to the increased prevalence of these agents among the approved oncology drugs (Figure S5C).
We selected eight approved oncology drugs for validation based on the amount of synergy and the number of instances of synergy observed in the screen: azacitidine, bortezomib, dacarbazine, hydroxyurea, pralatrexate, sorafenib, topotecan, and vismodegib. We evaluated the combinatorial activity of these agents with ONC201 in multi-dose cell viability assays in the cell lines where synergy was previously demonstrated (Figure S6A). The combination of ONC201 with these agents generally resulted in a complete elimination of tumor cells at the higher tested doses.
We prioritized the validation as a function of synergistic activity as well as overall efficacy (Figure 2B). Six of the nine validated agents met the prioritization criteria of imparting >20% increased reduction in cell viability over the expected observations and a >25% reduction in absolute cell viability with the combination: azacitidine, bortezomib, dacarbazine, hydroxyurea, sorafenib, and vismodegib (Figure 2C). Synergy between TRAIL and bortezomib, dacarbazine, hydroxyurea, or sorafenib has been previously reported (3, 5, 6). Examining monoagent and combinatorial activity, we found that adding vismodegib to ONC201 did not result in improved activity compared to monoagent ONC201, but rather reversed surprising agonistic growth effects of monoagent vismodegib that we observed in several cell lines. Sorafenib possessed the highest number of data points in the validation data set that met our prioritization criteria, demonstrating synergy in HCT116 and A172 cell lines at multiple doses (Figure S6B).
ONC201 synergizes with sorafenib in vitro and in vivo
We next explored the activity of ONC201 alone and in combination with sorafenib in its approved indications, hepatocellular carcinoma and renal cell carcinoma. The combination of sorafenib and ONC201 resulted in synergistic reduction of cell viability in HepG2 cells, but not other cell lines in the panel (Figure 3A; Figure S7; Table S3). Synergy was objectively confirmed by combination indices using the Chou Talalay method (Table S4).
Figure 3. ONC201 synergizes with sorafenib in HepG2 human HCC cells.
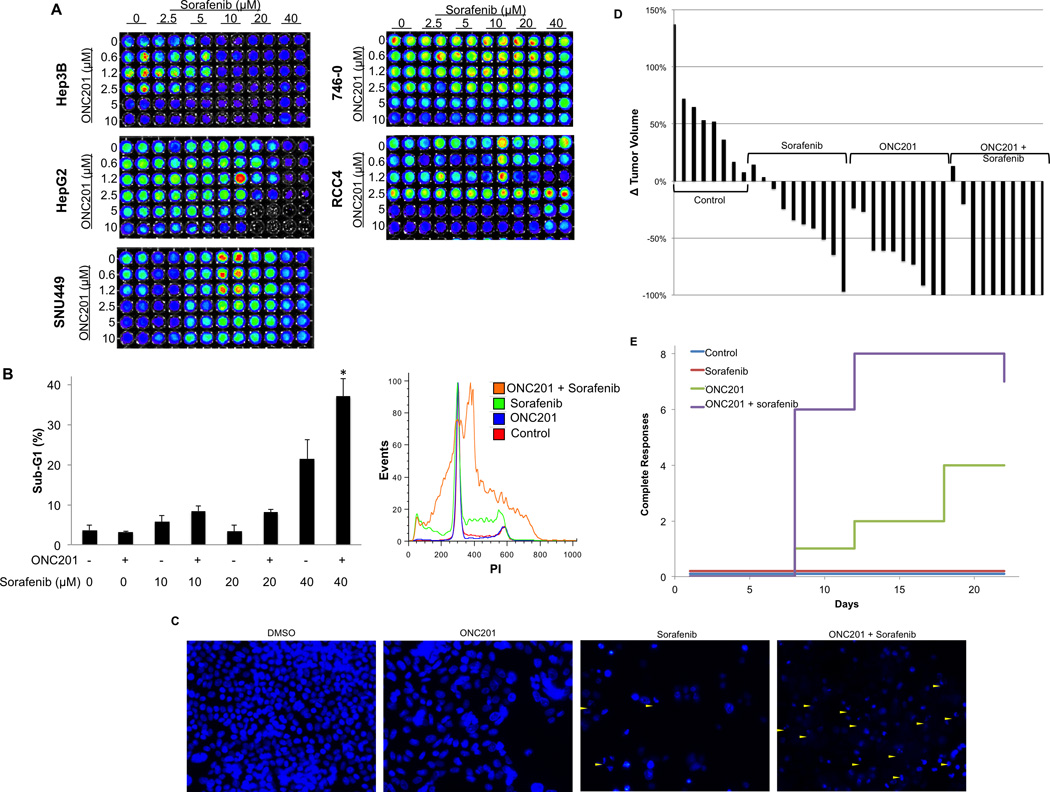
A) Cell viability of hepatocellular and renal cell carcinoma cell lines (72 hours, n=3). (B) Sub-G1 DNA content analysis in HepG2 cells following treatment with ONC201 (5 µM) or sorafenib at indicated concentrations (72 hours, n=3). * P< 0.05 by student’s two-tailed t test. (C) DAPI staining of HepG2 cells treated with ONC201 (5 µM), sorafenib (40 µM), or the combination (72 hours). Apoptotic nuclei are indicated by yellow triangles. (D) Waterfall plot showing tumor volume in HepG2 xenografts at 14 days following treatment initiation relative to tumor size prior to treatment initiation. Treatments were ONC201 (25 mg/kg PO on day 1 and 7) and sorafenib (40 mg/kg PO on days 1–5, 8–12) (n≥8). (E) Number of mice with complete tumor regressions (n=10).
We next investigated the possibility that ONC201 and sorafenib synergistically induce apoptosis in HepG2 cells. We found that ONC201 in combination with sorafenib cooperatively induce cell death that was significantly greater than either of the monoagents (Figure 3B). Further supporting evidence of apoptosis was obtained by Western blot analysis of caspase-8 activation and cleavage of PARP and DNA fragmentation (Figure 3C; Figure S8). Together this evidence indicates that ONC201 and sorafenib synergistically induce apoptosis in HepG2 human hepatocellular carcinoma cells.
We then evaluated the antitumor activity of ONC201 and sorafenib alone and in combination in HepG2 subcutaneous xenografts in athymic nude mice. Sorafenib was administered once a day for days 1–5 and days 8–12 at 40 mg/kg orally and ONC201 was administered once a week at 25 mg/kg. ONC201 and sorafenib monoagent therapies induced partial regressions of these xenograft tumors, which do not grow aggressively in vivo, as previously reported (Figure 3D; Figure S9A). In contrast to in vitro results, ONC201 exhibited a more potent antitumor effect compared to sorafenib under these conditions. The combination produced a significantly increased rate of complete tumor regressions (8/10 tumors) compared to ONC201 (4/10) or sorafenib (0/10) alone following two weeks of this treatment (Figure 3E). The combination was well tolerated with no overt signs of toxicity or loss of body weight (Figure S9B).
Sorafenib and KSR-1 affect ONC201 response by regulating FLIP, IAP, and Bcl-2 proteins
We investigated the effects of ONC201 and sorafenib, alone and in combination, on surface TRAIL and DR5 that together act as apoptosis initiators. ONC201 consistently upregulated surface TRAIL and DR5 as previously reported (1) and sorafenib surprisingly upregulated TRAIL in two of the three cancer cell lines. Both surface TRAIL and DR5 were cooperatively upregulated by the combination of sorafenib and ONC201 in HepG2 cells, an observation that was not evident in the other tested hepatocellular carcinoma cells that did not exhibit synergy (Figure 4A). Cooperative induction of TRAIL and DR5 was also observed in tumor specimens from the HepG2 in vivo experiment with sorafenib and ONC201 (Figure S10). Investigating effects of combinatorial sorafenib and ONC201 therapy on Akt and ERK kinase activity in a cellular context with respect to their mutual substrate Foxo3a revealed a cooperative inhibitory effect on Akt phosphorylation site of Foxo3a (Figure 4B). The ERK phosphorylation site of Foxo3a was ablated by sorafenib monoagent therapy, likely due to the direct inhibition of Raf by sorafenib.
Figure 4. Sorafenib and KSR1 regulate FLIP, IAP, and Bcl-2 proteins.

A) Surface TRAIL and surface DR5 by flow cytometry following treatment with ONC201 (72 hours, 10 µM, n =3). *P< 0.05 by student’s two-tailed t test compared to all other conditions. (B) Western blot analysis of HepG2 cells treated with ONC201 (5 µM), sorafenib (40 µM), or the combination (72 hours). (C) Western blot analysis of HepG2 or HCT116 cell treated with sorafenib (40 µM) for 12 hours. (D) Western blot analysis of HCT116 cells with or with siRNA-mediated knockdown of KSR1. (E) Effect of FLIP, Mcl-1, or KSR1 siRNA-mediated knockdown on sensitivity to ONC201 in HCT116 and cells (72 hours, n=3). *P < 0.05 compared to control siRNA under the same treatment conditions by Student’s two-tailed t test. Confirmation of knockdown by Western blot is shown in the left panel.
We also investigated the effects of sorafenib on apoptosis regulators that we previously described as modulators of TRAIL sensitivity (3). Among IAP and Bcl-2 family members, sorafenib downregulates expression of survivin and XIAP in HepG2 cells (Figure 4C). Sorafenib also negatively regulated Mcl-1, Bax, Bak, cIAP1, and survivin to a moderate extent in HCT116 cells that undergo apoptosis in response to ONC201 treatment (1). By comparison, KSR1 knockdown resulted in downregulation of the anti-apoptotic proteins FLIP, Mcl-2, Bcl-2, cIAP1, cIAP2, and survivin in HCT116 cells that was even more dramatic in most cases than that observed with sorafenib (Figure 4D).
Based on these regulatory relationships and previously established relationships with TRAIL sensitivity we investigated the effects of knocking down the caspase-8 inhibitor FLIP or the Bcl-2 family protein Mcl-1 on ONC201 sensitivity in HCT116 cells (7). We found that knockdown of either of these proteins enhanced the response to ONC201 (Figure 4E). In addition, we found that these genetic manipulations also sensitized HepG2 cells to ONC201 (Figure S11). Together, these results suggest that FLIP, IAP, and Bcl-2 proteins may modulate sensitivity to ONC201 and that sensitivity to ONC201 can be increased through regulation of these proteins by simultaneous sorafenib combination treatment (Figure S12).
Discussion
The siRNA screen in the present studies identified several kinases that negatively regulate ONC201 response and may be evaluated as predictive markers in nascent clinical studies. It is worth noting that these markers are predictive of enhanced or blunted responses to ONC201, rather than complete versus complete absence of responses. In addition to serving as biomarkers, these kinases may be pharmacologically engaged directly or indirectly with other therapies to potentially augment ONC201 efficacy. To this effect, a number of FDA-approved anti-cancer therapies were identified in this study that combine with ONC201 to synergistically exert antitumor effects. The combination of sorafenib and ONC201 appears to be promising given the prevalence of cures and it was well tolerated in mouse models. One limitation of this study is that hepatocellular carcinoma xenografts, including HepG2 xenografts, are not very aggressive in mouse models, although the responses were impressive.
Synergy between sorafenib and TRAIL has been extensively reported in a variety of tumor types including hepatocellular carcinoma with sorafenib-mediated downregulation of Mcl-1 and FLIP being the most commonly proposed mechanism of TRAIL sensitization (3, 9–12). The mechanism of sorafenib-mediated regulation of these proteins is less clear, as reports have proposed a number of mechanisms including inhibition of transcription through Stat3 or NFKB and alternative mechanisms such as inhibition of translation. These findings suggest that other therapies that synergize with TRAIL, such as quinacrine, are worth investigating for combinatorial activity with ONC201 (13). By a similar notion, these findings also suggest that sorafenib may sensitize other TRAIL-based agents such as TRAIL-receptor agonists that have been developed including antibody- and protein scaffold-based approaches (14–16).
One limitation of these studies is the somewhat ambiguous biology of KSR1, which was previously reported as a regulator of Ras signaling. Despite selecting this lead based on this canonical molecular biology, we did not observe effects on MAPK or Akt signaling, but instead discovered a novel relationship between KSR1 and regulators of apoptosis. In support of our observations, a previous study has reported that KSR1 sensitizes endometrial carcinoma cells to TRAIL through downregulation of FLIP that did not involve transcription or protein degradation (17). Further study will be needed to elucidate this mechanism of regulation that could involve many established regulators of FLIP, IAP, and Bcl-2 stability and expression such as JNK (18) or NFKB (3). Nevertheless, the involvement of these apoptotic regulators in ONC201 sensitizations suggests that combining ONC201 with direct antagonists of these proteins under clinical development, such as XIAP or Bcl-2 inhibitors, may be an effective anti-cancer strategy.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by Oncoceutics and a Pennsylvania Department of Health grant awarded to Oncoceutics. W.S.E.-D. is an American Cancer Society Research Professor.
Footnotes
Conflicts of Interests: Joshua E. Allen and Wafik S. El-Deiry are shareholders of Oncoceutics, which is commercially developing ONC201. Joshua E. Allen is an employee of Oncoceutics.
Authors' Contributions
Conception and design: J.E. Allen, W.S. E.-D.,
Development of methodology: J.E. Allen, W.S. E.-D., J.L. Fritz, A. Beck
Acquisition of data: V.V. Prabhu, M. Talekar, P. van den Huevel, B. Lim, D.T. Dicker, J.L. Fritz, A. Beck
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J.E. Allen, D.T. Dicker, W.S. E.-D.
Writing, review, and/or revision of the manuscript: V.V. Prabhu, M. Talekar, P. van den Huevel, B. Lim, D.T. Dicker, J.L. Fritz, A. Beck, W. S. El-Deiry
Study supervision: W. S. El-Deiry
References
- 1.Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction and potent anti-tumor effects. Science Translational Medicine. 2013 doi: 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burns TF, El-Deiry WS. Identification of Inhibitors of TRAIL-induced Death (ITIDs) in the TRAIL-sensitive Colon Carcinoma Cell Line SW480 Using a Genetic Approach. Journal of Biological Chemistry. 2001 Oct 12;276(41):37879–37886. doi: 10.1074/jbc.M103516200. 2001. [DOI] [PubMed] [Google Scholar]
- 3.Ricci MS, Kim S-H, Ogi K, Plastaras JP, Ling J, Wang W, et al. Reduction of TRAIL-Induced Mcl-1 and cIAP2 by c-Myc or Sorafenib Sensitizes Resistant Human Cancer Cells to TRAIL-Induced Death. Cancer cell. 2007;12(1):66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Therrien M, Michaud NR, Rubin GM, Morrison DK. KSR modulates signal propagation within the MAPK cascade. Genes & Development. 1996 Nov 1;10(21):2684–2695. doi: 10.1101/gad.10.21.2684. 1996. [DOI] [PubMed] [Google Scholar]
- 5.Menke C, Bin L, Thorburn J, Behbakht K, Ford HL, Thorburn A. Distinct TRAIL Resistance Mechanisms Can Be Overcome by Proteasome Inhibition but not Generally by Synergizing Agents. Cancer Research. 2011 Mar 1;71(5):1883–1892. doi: 10.1158/0008-5472.CAN-10-2252. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engesaeter B, Engebraaten O, Florenes VA, Maelandsmo GM. Dacarbazine and the Agonistic TRAIL Receptor-2 Antibody Lexatumumab Induce Synergistic Anti-cancer Effects in Melanoma. PLoS ONE. 2012;7(9):e45492. doi: 10.1371/journal.pone.0045492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biology & Therapy. 2005;4(2):139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 8.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. New England Journal of Medicine. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen K-F, Tai W-T, Liu T-H, Huang H-P, Lin Y-C, Shiau C-W, et al. Sorafenib Overcomes TRAIL Resistance of Hepatocellular Carcinoma Cells through the Inhibition of STAT3. Clinical Cancer Research. 2010 Nov 1;16(21):5189–5199. doi: 10.1158/1078-0432.CCR-09-3389. 2010. [DOI] [PubMed] [Google Scholar]
- 10.Huang S, Sinicrope FA. Sorafenib Inhibits STAT3 Activation to Enhance TRAIL-Mediated Apoptosis in Human Pancreatic Cancer Cells. Molecular Cancer Therapeutics. 2010 Mar 1;9(3):742–750. doi: 10.1158/1535-7163.MCT-09-1004. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nojiri K, Sugimoto K, Shiraki K, Tameda M, Inagaki Y, Ogura S, et al. Sorafenib and TRAIL have synergistic effect on hepatocellular carcinoma. International Journal of Oncology. 2012;42(1):101–108. doi: 10.3892/ijo.2012.1676. [DOI] [PubMed] [Google Scholar]
- 12.Rosato RR, Almenara JA, Coe S, Grant S. The Multikinase Inhibitor Sorafenib Potentiates TRAIL Lethality in Human Leukemia Cells in Association with Mcl-1 and cFLIPL Down-regulation. Cancer Res. 2007 Oct 1;67(19):9490–9500. doi: 10.1158/0008-5472.CAN-07-0598. 2007. [DOI] [PubMed] [Google Scholar]
- 13.Wang W, Gallant J-N, Katz SI, Dolloff NG, Smith CD, Abdulghani J, et al. Quinacrine sensitizes hepatocellular carcinoma cells to TRAIL and chemotherapeutic agents. Cancer Biology & Therapy. 2011;12(3):229–238. doi: 10.4161/cbt.12.3.17033. [DOI] [PubMed] [Google Scholar]
- 14.Swers JS, Grinberg L, Wang L, Feng H, Lekstrom K, Carrasco R, et al. Multivalent Scaffold Proteins As Superagonists of TRAIL Receptor 2-Induced Apoptosis. Molecular Cancer Therapeutics. 2013 May 3; doi: 10.1158/1535-7163.MCT-12-1107. 2013. [DOI] [PubMed] [Google Scholar]
- 15.Allen JE, Ferrini R, Dicker DT, Batzer G, Chen E, Oltean DI, et al. Targeting TRAIL death receptor 4 with trivalent DR4 Atrimer complexes. Molecular Cancer Therapeutics. 2012 Jul 16; doi: 10.1158/1535-7163.MCT-12-0366. 2012. [DOI] [PubMed] [Google Scholar]
- 16.Abdulghani J, El-Deiry WS. TRAIL receptor signaling and therapeutics. Expert Opinion on Therapeutic Targets. 2010;14(10):1091–1108. doi: 10.1517/14728222.2010.519701. [DOI] [PubMed] [Google Scholar]
- 17.Llobet D, Eritja N, Domingo M, Bergada L, Mirantes C, Santacana M, et al. KSR1 Is Overexpressed in Endometrial Carcinoma and Regulates Proliferation and TRAIL-Induced Apoptosis by Modulating FLIP Levels. The American Journal of Pathology. 2011;178(4):1529–1543. doi: 10.1016/j.ajpath.2010.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen JE, El-Deiry WS. Oxaliplatin Uses JNK to Restore TRAIL Sensitivity in Cancer Cells Through Bcl-xL Inactivation. Gastroenterology. 2011;141(2):430–434. doi: 10.1053/j.gastro.2011.06.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.