

Abstract
The cancer stem cell hypothesis suggests that rare populations of tumor-initiating cells may be resistant to therapy, lead to tumor relapse and contribute to poor prognosis for cancer patients. We previously demonstrated the feasibility of p53 pathway restoration in p53-deficient tumor cell populations using small molecules including ellipticine or its derivatives. We now establish a single cell p53-regulated green fluorescent protein (EGFP)-reporter system in human DLD1 colon tumor cells expressing mutant p53 protein. We use these p53-EGFP reporter DLD1 cells to investigate the status of p53 transcriptional activity in putative colon cancer stem cell populations following exposure to p53 pathway-restoring drugs and/or classical chemotherapy. We demonstrate induction of p53-specific EGFP reporter fluorescence following overexpression of p53 family member p73 by an Adenovirus vector. We further show that p53-reporter activity is induced in DLD1 putative cancer stem cell side-populations analyzed by their Hoechst dye efflux properties following treatment with the p53 pathway restoring drug ellipticine. Combination of ellipticine with the cytotoxic agent 5-fluorouracil resulted in increased cytotoxicity as compared to either agent alone and this was associated with depletion of putative cancer stem cell populations as compared with 5-FU alone treatment. Our results support the feasibility of therapeutic targeting of mutant p53 in putative cancer stem cells as well as the potential to enhance cytotoxic chemotherapy.
Keywords: Cancer stem cell, p53, mutant p53, ellipticine, 5-fluorouracil, side-population, ALDH, flow cytometry, cytotoxicity
Introduction
Mutations in the p53 tumor suppressor gene occur commonly in human cancer and can contribute to disease progression and resistance to therapy.1–3 This is in part due to loss of activation of genes normally regulated by p53 that can limit tumor progression through cell cycle arrest,1,2 senescence,2 cell death1,2 or through remote cell autonomous effects of p53 on angiogenesis inhibition4 or death ligand activity.5 However, we hypothesize that p53 mutations may also contribute to tumor progression and resistance to therapy through the cancer stem cell phenotype by promoting the survival of such cells as well as the efflux of chemotherapeutic and other targeted agents from such cells. The cancer stem cell hypothesis suggests that rare populations of tumor-initiating cells may lead to tumor relapse, resistance to therapy and contribute to poor prognosis for cancer patients.6
We have previously attempted to target p53 pathway restoration in p53-mutant or p53-deficient tumor cell populations using small molecules that appear to restore transcriptional activity of p53 in such cells.7 Restoration of p53-pathway dependent transcriptional responses including upregulation of endogenous p53 target in tumor cells treated with small molecules was correlated with growth arrest or cell death induction and anti-tumor responses.7 Restoration of p53 pathway activity may occur through an altered conformation of mutant p53 protein, altered expression or activity of mutant p53 towards p53 family members or through stimulation of p53 family members such as p73.3,7 More recently we used ellipticine and various ellipticine derivatives as tool compounds to demonstrate p53 pathway restoration and involvement of p73 in the cytotoxicity towards tumor cells.8
In order to further investigate the effects of therapeutic compounds on p53 signaling at a single cell level, including in putative cancer stem cells, we now establish a single cell p53-regulated (EGFP)-reporter system in human DLD1 colon tumor cells expressing mutant p53 protein. We use these p53-EGFP reporter DLD1 cells to investigate the status of p53 transcriptional activity in putative colon cancer stem cell populations following exposure to p53 pathway-restoring small molecule ellipticine alone or in combination with the chemotherapeutic agent 5-FU. We demonstrate that a p53-specific EGFP reporter can be induced in putative cancer stem cell populations following treatment with the p53 pathway restoring drug ellipticine. We further show that combination of ellipticine with the cytotoxic agent 5-FU results in increased cytotoxicity as compared to either agent alone and that this is associated with depletion of putative cancer stem cell populations as compared with 5-FU alone treatment. Our results support the feasibility of therapeutic targeting of mutant p53 in putative cancer stem cells as well as the potential to enhance cytotoxic chemotherapy.
Materials and Methods
p53-EGFP reporter (PG13-EGFP)
The PG13-EGFP plasmid was generated by replacing the luciferase reporter of classical p53 reporter PG13-Luc by the EGFP cassette of pEGFP-N1. The EGFP cassette was obtained by PCR amplification using a primer that added a BamH I to the 3′ end so that the insert would have BamH1 sites on both ends. The primers used for amplification were EGFP-ORF-F: 5′—GCT-TCG-AAT-TCT-GCA-GTC-GAC-GGT-ACC-GC-3′ and EGFP-ORF-polyA-R: 5′-TCG-CGG-GAT-CCG-CCT-TAA-GAT-ACA-TTG-ATG-AG-3′. Following subcloning of the EGFP cassette to create PG13-EGFP, the correct orientation could be verified by XhoI/Not1 digestion releasing a fragment of 1290 bp.
p53 reporter-EGFP stable cell line
DLD1 cells were kept in DMEM 10% FBS at 37°C 5% CO2 and split every three to four days. To establish stable cell line, cells were seeded in 6-well plates and co-transfected with PG13-EGFP plasmid and puromycin resistance marker using lipofectamine 2000 the next day. 24 hours later, medium was replaced with puromycin (5 μg/ml for DLD1 and 50 μg/ml for SW480) containing medium and kept at 37°C for 3–4 days until the majority of cells started dying. The cells were then trypsinized and diluted to about 1 cell per microliter and 1 microliter was added to each well in 48-well plates containing medium. After 2–3 weeks, the wells containing single colony were trypsinized and split into three wells in 24-well plates. The next day, 10 and 20 μM of ellipticine was added to two of the three wells and cells were visualized under fluorescence microscopy. The clones that showed inducible EGFP signal were expanded.
DLD1 expression of EGFP in side population
P53-reporter (EGFP)-DLD1 cells were seeded in 6-well plates at 1 million cells per well. The next day, 20 μM of ellipticine was added to each well. At 24 hours later, cells were trypsinized and side population staining was performed according to methods described below.
Aldefluor assay
The Aldflluor assay was performed according to the manufacturer’s instructions (Stem Cell Technology). Briefly, cells were seeded in 6 well plates at 500,000 cells/well overnight. The next day, the cells were trypsinized and washed with assay buffer once, and then counted and resuspended at 1 million cells per 1ml of buffer. Five microliter of activated substrate was then added to 1 ml of cells. The cells were incubated at 37°C for 45 minutes before washing it once with assay buffer and followed by flow cytometry.
DLD1 drug treatment and cell counting
Cells were seeded in 24-well plates at 100,000 cells/well overnight. The next day, medium containing 0, 5, 10, 20, 40, 80 μM of 5-FU replaced the original medium. In some cases, 5, 10 or 20 μM of ellipticine were then added to appropriate wells. Cells were incubated at 37°C for four days, trypsinized and counted using a hemocytometer.
SW480 and DLD1 Side population identification and drug treatment
Cells were seeded in 5 cm plates at about 30–40% confluency overnight. The next day, media were replaced with media containing 0, 40 and 80 μM of 5-FU and 5 μM of ellipticine was added to appropriate plates. Cells were then incubated in the incubator at 37°C. After four days, the cells were trypsinized and washed with HBSS buffer and then resuspended in HBSS at 1 million cells/ml of buffer and divided into two tubes. Verapamil was added to one of the two tubes 10 minutes before adding Hoechst 33342 stain at 5 μg/ml. Cells were incubated at 37°C for 90 minutes followed by washing once with HBSS buffer, resuspending in 300 μl of HBSS buffer with 2 μg/ml of PI stain and flow cytometry.
HCT116 p53-null side population identification
Cells were processed in the same way as SW480 and DLD1 except that DMEM with 5% of FBS as medium was used when incubating cells at 37°C for 90 minutes.
Ad-p73 infections
Phoenix cells (obtained from Garry Nolan, Stanford University) were transfected with the PG13-EGFP vector described above and were subsequently infected with an adenovirus that expresses the p53 family member p73-beta (Ad-p73) as previously described.9 Ad-p73 was added to the culture medium at a multiplicity of infection (MOI) of ~50. The infected cells were cultured overnight and fluorescence of EGFP was visualized and captured by a laser microscope.
DR5 immunofluorescence
Cells were plated in wells of 8-well chamber slides and were stained for TRAIL DR5 expression after incubation in the presence of p53 conformation modifying small molecules such as CP-31398. Media was aspirated and cells were washed 3-times with PBS followed by the addition of 0.3 ml 37% formaldehyde for 5 minutes. Cells were washed 3-times with PBS for 5 minutes each wash. Non-specific binding was blocked by incubation of fixed cells for 1 hr at room temperature using 1% BSA in PBS. Cells were then washed 3-times with PBS for 5 minutes. A rabbit anti-human DR5 IgG antibody (Imgenex) was added at a 1:100 dilution and this was incubated overnight with gentle shaking at 4°C. Following removal of primary antibody, cells were washed 3-times with PBS for 5 minutes. A Cy3-labeled donkey anti-rabbit secondary antibody (Sigma) at a 1:400 dilution was added in 1% BSA and the wells were incubated for 1 hr in the dark at room temperature. Cells were washed 3 times with PBS for 5 minutes. Prior to fluorescence microscopy, Hoechst 33342 was added for 3 minutes at a concentration of 20μg/ml. Cells were washed 3-times with water prior to mounting slides for microscopy.
Results
Establishment of stable mutant p53-expressing DLD1 human colon cancer cells expressing a p53-responsive promoter driving an EGFP reporter
We initially introduced a p53-responsive promoter driving EGFP expression into Phoenix cells. We expressed the p53 family member p73 by an Adenovirus vector9 into the Phoenix cells carrying the p53-EGFP reporter and demonstrated a robust increase in EGFP expression in the Ad-p73 infected cells (Figure 1, upper panels). We then introduced the p53-EGFP reporter into DLD1 cells. We also demonstrated that treatment of DLD1 cells results in increased expression of the p53 target gene TRAIL receptor DR5 following exposure of the cells to the mutant p53 conformation-modifying drug CP-31398 (Figure 1, lower panels).10,11 Attempts to use pooled clones of p53-reporter EGFP DLD1 cells to demonstrate single cell upregulation in putative cancer stem cell populations revealed significant heterogeneity in EGFP expression that interfered with analysis of the populations. Thus we determined the need to isolate single cell DLD1 clones with p53-EGFP reporter expression that have fairly low basal EGFP expression and that could be induced to high EGFP expression upon exposure to small molecules that restore p53 pathway signaling in mutant p53-expressing DLD1 cells.
Figure 1. Upregulation of p53-dependent transcriptional activity or endogenous pro-apoptotic TRAIL receptor DR5 expression following p53 pathway restoration.

In the upper panels, a whole population of Phoenix cells expressing a p53-responsive EGFP reporter show increased EGFP reporter expression following infection by a p73-expressing Adenovirus (upper right panel vs. upper left panel). In the lower panels, mutant p53-expressing human colon cancer DLD1 cells were treated with 15 μg/ml of the mutant p53 conformation modifying drug CP-31398 for 12 hours followed by fluorescence microscopy for p53 target TRAIL receptor DR5 expression by immunofluorescence with a DAPI nuclear counterstain. The lower left panel shows DR5 expression in untreated DLD1 cells and the lower right panel shows DR5 expression in CP-31398 treated DLD1 cells.
DLD1 cells were therefore co-transfected with PG13-EGFP and a puromycin resistance marker. Single clones that showed inducible EGFP expression were isolated (example in Figure 2). One interesting and unexplained observation is that at 10 μM ellipticine, most EGFP was expressed in the cytoplasm while at 20 μM of ellipticine, EGFP signal was mostly in the nucleus of the p53-EGFP reporter DLD1 cells. All clones that were identified had a similar level of EGFP expression when ellipticine was added. Without ellipticine, none of the selected clones showed any observable EGFP signal under the fluorescence microscope.
Figure 2. Stable mutant p53-expressing human DLD1 PG13-EGFP colon cancer cell line increases EGFP expression in response to the p53 pathway restoring small molecule ellipticine.

Cells were seeded 20 hours before ellipticine was added at the concentration indicated at the top of each column to induce the expression of EGFP. The pictures in the middle row are green fluorescence only. The pictures in the bottom row are the bright field and the ones in the top row are combined.
Mutant p53-expressing DLD1 cells increased EGFP expression from a p53-responsive EGFP reporter in both non-side or side population cells exposed to the p53 pathway restoring small molecule ellipticine
There was a significant shift to higher fluorescence intensity both in side population and non-side population under flow cytometry when 20 μM of ellipticine was added (Figure 3). The shift was a little less in the side population cells as compared to the non-side population cells suggesting that p53 activity was activated in both populations although the activity in the side population may be a little lower. As there is usually higher expression of ABC transporters in side population, the lower p53 activity in side population may be due to the higher rate of drug efflux in side population cells as compared to non-side population cells. These results established the feasibility of restoring p53 signaling in dye-effluxing putative cancer stem cell SP cells.
Figure 3. DLD1 expression of p53-responsive EGFP reporter in side population (SP) putative cancer stem cells as compared to non-SP cells exposed to the p53 pathway restoring small molecule ellipticine.

Cells were treated with the indicated dose of ellipticine for 16 hr and then Hoechst 33342 dye was added as described in Materials and Methods followed by flow cytometry to analyze EGFP expression in untreated versus ellipticine-treated SP and non-SP DLD1 cells.
Use of the Aldelfuor assay as a second method in addition to SP to identify putative cancer stem cells in human colon cancer cell lines
A total of 5 human colon cancer cell lines, SW480, SW620, R6 (TRAIL-resistant clone of SW480 generated by exposure to TRAIL), HCT116 and HCT15, were assayed for the presence of aldehyde dehydrogenase (ALDH) activity12 using the Aldelfuor assay kit (Stem Cell Technology). For all of the 5 cell lines tested, a portion of the untreated cells demonstrated higher fluorescence intensity as compared to cells treated with the ALDH inhibitor DEAB (Figure 4, 5). The ALDH+ population was 2.8% for HCT15, 4.36% for SW620, 14.0% for HCT116, 3.38% for SW480, and 45.8% for R6, the TRAIL resistant cell line derived from SW480. This higher intensity population is continuous from the low intensity group of cells for HCT15 and HCT116 (Figure 5). By contrast, for SW480, SW620 and R6, the group of cells in the high fluorescence intensity region seems to be distinct from those in the low intensity region in the absence of DEAB (Figure 4). For R6, the cells in the high intensity region seemed to encompass the majority of the cells, while for HCT15 and HCT115, only a small percentage of cells shifted to lower intensity after DEAB. R6 also showed a greater degree of shift as compared with SW480 (Figure 4) suggesting that the TRAIL-resistant R613 may have more aldehyde dehydrogenase activity and thus more cancer stem cells than SW480. We have previously shown that SP cells of SW480 are responsive to TRAIL treatment.18
Figure 4. Some human colon tumor cells show a sub-population of ALDH-positive cells whose ALDH activity is blocked by the ALDH inhibitor DEAB.

This Aldelfuor assay is employed as a second means (in addition to SP) to identify putative colon cancer stem cells. Three colon cancer cell lines, SW480, SW620, R6, were assayed for the presence of aldehyde dehydrogenase activity. All three lines showed a distinct group of cells shifting to higher fluorescence intensity compared with the control cells with DEAB added. Individual gating was performed according to the profile of DEAB treated samples.
Figure 5. HCT116 and HCT15 cells show a continuum of ALDH positive cells whose activity is partially shifted by the ALDH inhibitor DEAB.

Two colon cancer cell lines, HCT116, HCT15 were assayed for the presence of ALDH activity. Both lines showed a shift to higher fluorescence intensity compared with DEAB treated control cells. The shift is continuous from the low intensity cell groups.
DLD1 cells show increased cytotoxicity to the combination of 5-FU and ellipticine as compared to treatment with either agent alone
DLD1 colon tumor cells were seeded on 24 well cell culture plates at about 50% confluency. The next day, 5-FU and ellipticine containing medium was used to replace the original medium and four days later, the dead cells were washed away and the cells still adhering to the plates were counted. Cell counts were performed with either 5-FU alone, ellipticine alone or the combination of both agents. The concentration of 5-FU added ranged from 5 μM to 80 μM and the concentration of ellipticine ranged from 5 μM to 20 μM. At most of the concentrations tested, the combined treatment resulted in increased cell death (Figure 6). While the difference in cell number between combined treatment and either treatment alone suggests an additive effect of the combined drugs, the design of the experiment does not specifically exclude potential synergistic effects given the level of observed toxicity with single agent treatment.
Figure 6. DLD1 cells show increased cytotoxicity to the combination of 5-FU and ellipticine as compared to treatment with either agent alone.
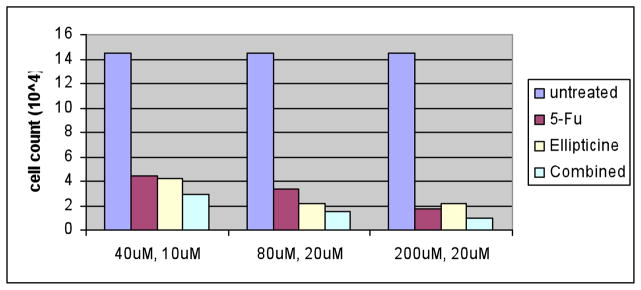
DLD1 cells were treated for 4 days with single agent or combination of 5-FU and ellipticine in 24 well plates. The concentrations in the x-axis are the concentrations of 5-FU and ellitpicine, respectively. Cell counts were determined by a hemocytometer and the numbers represent all the remaining cells in each particular well in the 24-well plate at the different treatment conditions.
Depletion of SW480 colon cancer side population cells after treatment with 5-FU in combination with ellipticine in contrast to enrichment of SP-cells after exposure to 5-FU alone
SW480 colon cancer cells were seeded on 5 cm cell culture plates at about 50% confluency. The next day, 5-FU (40 μM and 80 μM) and ellipticine (5 μM) containing medium was used to replace the original medium on cells. Four days later, the dead cells were washed away and the cells still adhering to the plates were trypsinized and stained with Hoechst 33342 dye and then analyzed by flow cytometry to identify the side populations. When cells were treated with 5-FU alone, the percentage of cells in the side population increased from 0.087% to 2.58% and 5.01% respectively (Figure 7). When cells were treated with 5 μM of ellipticine alone, the side population increased to 0.43% with a significantly altered fluorescence profile (Figure 7). In cells treated with both ellipticine and 5-FU, the ability of 5-FU to enrich the side population was abrogated and the percentage of the side population dropped to 0.34 and 0.25% for 40 and 80 μM 5-FU, respectively (Figure 7). The percentage of side population cells dropped by 7.6-fold when 40 μM of 5-FU was added with ellipticine and reduced 20-fold when 80 μM of 5-FU was added with ellipticine. One interesting observation was that when 5-FU was added alone, increasing the concentration of 5-FU increased the percentage of the side populations but when both 5-FU and ellipticine were added, increasing the concentration of 5-FU decreased the percentage of the side population. This may suggest that ellipticine has the ability to sensitize cells in the side population to 5-FU-induced apoptosis. Since ellipticine can rescue mutant p53 in SW480 cells and reduce the expression of ABC transporters, this result may fit into the hypothesis that functional p53 can increase the drug concentration in cells of the side population and increase cell death induced by cytotoxic drugs, such as 5-FU. The results also suggest that combining ellipticine with a cytotoxic drug such as 5-FU may be better at targeting the cells in the side population compared with using either drug alone.
Figure 7. Depletion of SW480 colon cancer side population cells after treatment with 5-FU in combination with ellipticine contrasts with enrichment of SP-cells after exposure to 5-FU alone.

SW480 cells were treated with ellipticine (5uM) and/or 5-FU at the doses indicated for 4 days in culture. Cells were collected, stained with Hoechst 33342 and analyzed for SP and non-SP cells −/+ verapamil by flow cytometry. Verapamil data is shown in the inset for each profile, and was used to properly gate the SP cells. The graph (lower panel) shows the quantified results for SP cells from the flow cytometry data (upper panel).
Depletion of DLD1 side population colon cancer cells after combined treatment with 5-FU plus ellipticine as compared to enrichment of the SP cells after treatment with 5-FU alone
DLD1 colon cancer cells were subjected to similar treatments as the SW480 cells above followed by side population analysis. The SP profiles were altered after cells were treated with 5-FU, ellipticine or both (Figure 8). There appeared to be an increased number of cells in the side population region after treatment with 5-FU (Figure 8, upper panels). More than 40% of cells were in the side population region after treatment with 5-FU. After treatment with ellipticine plus 5-FU there were fewer cells in the SP gate as compared to treatment with 5-FU alone. To test what caused the change in SP profile, each sample after drug treatment was also treated with verapamil, an inhibitor of ABC transporter proteins (Figure 9). In a second experiment, after treatment with 5-FU and ellipticine DLD1 cells showed decreased fluorescence in the SP gate. The observation that the side population profile after treatment with ellipticine is different from that after treatment with 5-FU may suggest that ellipticine can alter the degree of ABC transporter expression. Since the combination treatment of ellipticine and 5-FU caused the profile to look like the profile treated with ellipticine alone, the effect of ellipticine may be to abrogate the effect of 5-FU on increasing the SP.
Figure 8. Depletion of DLD1 side population colon cancer cells after combined treatment with 5-FU plus ellipticine as compared to enrichment of the SP cells after treatment with 5-FU alone.

DLD1 cells were treated with ellipticine and/or 5-FU at the doses indicated for 4 days in culture. SP analysis was performed as already described. The gated populations represent the SP as determined by verapamil incubation (not shown).
Figure 9. Decreased fluorescence of DLD1 side population cells treated with 5-FU and ellipticine.

DLD1 cells were treated with ellipticine and/or 5-FU at the doses indicated for 4 days in culture. SP analysis was performed as already described. The gated populations represent the SP as determined by verapamil incubation (lower profiles).
5-FU increases while the combination of 5-FU plus ellipticine decreases the side population of p53-null HCT116 colon cancer cells
To understand to role p53 plays in the effect of side population depletion caused by ellipticine, HCT116 p53-null cells were treated with 5-FU and ellipticine for 4 days followed by flow cytometry to identify the side population (Figure 10). As shown in our earlier experiments with DLD1 and SW480, 5-FU enriched the side population of p53-null HCT116 cells from 0.4% to 3.8% when 40 μM of 5-FU was used and to 7.7% when 80 μM of 5-FU was used to treat the cells. These results in p53-null cells suggest that the side population enriching effect of 5-FU may be in part independent of p53 activity (Figure 10). When ellipticine was added alone, the side population appeared to be enriched to 4.8% as compared with the 0.4% of the control cells. This may be attributed in part to cell death that was observed with the ellipticine-treated cells that may be more evident in the non-SP cells. The combination treatment of 5-FU plus ellipticine showed clear depletion of the side population as compared to 5-FU treatment alone. Because these were p53-null cells we conclude that this effect if unrelated to p53 itself but may be related to the effect of ellipticine on the p53 family member p73 as we have previously shown.8 Moreover, the combination treatment of 5-FU plus ellipticine resulted in an 8-fold and a 2-fold reduction in the percentage of the side population observed following treatment with 80 and 40 μM of 5-FU, respectively. This is a lesser reduction than observed in SW480 suggesting that p53 protein may still play a role in depletion of side population, in the presence of a mutant p53 conformation-modifying molecule such as ellipticine.
Figure 10. 5-FU increases while the combination of 5-FU plus ellipticine decreases the side population of p53-null HCT116 colon cancer cells.

The chart shows the percentage of side population cells present after each drug treatment for 4 days at the indicated doses.
Discussion
We demonstrate the feasibility of monitoring p53-dependent transcriptional activity at a single cell level in putative human cancer stem cell populations, the potential of p53 pathway-targeting agents to deplete such populations, as well as the potential for combining p53 pathway-targeting agents with classical cytotoxic chemotherapy to target mutant p53-expressing putative colon cancer stem cells. Combining a p53 pathway-restoring agent such as ellipticine with 5-FU appears to abrogate the effect of 5-FU to enrich putative cancer stem cell populations. The effect of putative cancer stem cell depletion caused by ellipticine was observed in both tumor cells with mutant p53 as well as in p53-null tumor cells.
The effect of p53 pathway restoration in side population cells needs to be further investigated to determine if p53 activity per se and restoration of the p53 signaling program may be the primary cause of putative cancer stem cell population depletion. The role of suppression the p-glycoprotein family needs to be further investigated as potentially mediating increased cytotoxicity when p53 pathway-restoring agents are combined with classical cytotoxic agents. Further studies need to be conducted to unravel the relationship between aldehyde dehydrogenase activity and side populations to further understand the biology and make-up of putative cancer stem cells.
One of the open questions in the field is whether targeting putative cancer stem cell populations in cancer therapy is potentially beneficial in terms of tumor response. A second question is whether directed drug screening against putative cancer stem cells is a strategy that may ultimately improve on therapeutic design against cancer. Our data documenting increased cytotoxicity when ellipticine is combined with 5-FU would support the idea that specific targeting of putative cancer stem cell populations should be further pursued, and that specific targeting of restoration of p53 function in such cancer stem cells may be useful in therapy. The availability of single cell p53-EGFP reporter lines with mutant p53 should be helpful in beginning to address the second question. With regard to targeting cancer stem cells, a recent study documented that the combination of metformin plus doxorubicin appears to have efficacy towards depletion of putative human breast cancer stem cell populations, although in the different breast cancer cell lines used the effects appeared in cells with either wild-type or mutant p53.14 Another study recently documented a potential benefit from targeting the epithelial-to-mesenchymal transition as a feature of breast cancer stem cell populations.15 There are efforts to target cancer stem cells using hedgehog16 and Notch17 inhibitors and targeting the TRAIL pathway18 may also result in killing putative cancer stem cells given the association between the c-Myc gene with both TRAIL sensitivity19,20 and the induced pleuripotent stem cell phenotype.21 Future efforts will further unravel the various pathways that are essential to target in putative cancer stem cells as well as the merits of various therapeutic combinations, and the impact of the tumor microenvironment including hypoxia22 on the efficacy of such directed therapies.
Acknowledgments
This work was supported in part by NIH Grant U54 CA105008 and by the Littlefield-AACR Grant in Metastatic Colon Cancer Research. W.S.E-D. is an American Cancer Society Research Professor.
Footnotes
This work was presented in part at the NCI Cancer Therapy Evaluation Program (CTEP) Workshop on Cancer Stem Cells on September 23, 2008 in Bethesda, Maryland and at the 14th International p53 Workshop in Shanghai, China on October 28, 2008.
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 3.Bassett EA, Wang W, Rastinejad F, El-Deiry WS. Structural and functional basis for therapeutic modulation of p53 signaling. Clin Cancer Res. 2008;14:6376–86. doi: 10.1158/1078-0432.CCR-08-1526. [DOI] [PubMed] [Google Scholar]
- 4.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene. 2003;22:7486–95. doi: 10.1038/sj.onc.1206949. [DOI] [PubMed] [Google Scholar]
- 5.Kuribayashi K, Krigsfeld G, Wang W, Xu J, Mayes PA, Dicker DT, Wu GS, El-Deiry WS. TNFSF10 (TRAIL), a p53 target gene that mediates p53-dependent cell death. Cancer Biol Ther. 2008;7:2034–8. doi: 10.4161/cbt.7.12.7460. [DOI] [PubMed] [Google Scholar]
- 6.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci USA. 2006;103:11003–8. doi: 10.1073/pnas.0604507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu C, Wang W, El-Deiry WS. Non-genotoxic anti-neoplastic effects of ellipticine derivative NSC176327 in p53-deficient human colon carcinoma cells involve stimulation of p73. Cancer Biol Ther. 2008;7:2039–46. doi: 10.4161/cbt.7.12.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das S, El-Deiry WS, Somasundaram K. Efficient growth inhibition of HPV 16 E6-expressing cells by an adenovirus-expressing p53 homologue p73beta. Oncogene. 2003;22:8394–402. doi: 10.1038/sj.onc.1206908. [DOI] [PubMed] [Google Scholar]
- 10.Wu GS, Burns TF, McDonald ER, 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, El-Deiry WS. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nature Genet. 1997;17:141–3. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 11.Takimoto R, Wang W, Dicker DT, Rastinejad F, Lyssikatos J, El-Deiry WS. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther. 2002;1:47–55. doi: 10.4161/cbt.1.1.41. [DOI] [PubMed] [Google Scholar]
- 12.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Z, McDonald ER, 3rd, Dicker DT, El-Deiry WS. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem. 2004;279:35829–39. doi: 10.1074/jbc.M405538200. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Altaba AR. Hedgehog-gli signaling regulates human glioma growth, cancer stem cell self-renewal and tumorigenicity. Curr Biol. 2007;17:165–72. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li Y-M, Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–52. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 18.Sussman RT, Ricci MS, Hart LS, Sun SY, El-Deiry WS. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL receptor DR4. Cancer Biol Ther. 2007;6:1490–5. doi: 10.4161/cbt.6.9.4905. [DOI] [PubMed] [Google Scholar]
- 19.Ricci MS, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, El-Deiry WS. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol. 2004;24:8541–55. doi: 10.1128/MCB.24.19.8541-8555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, Jin Z, Liu YY, Dicker DT, Chiao PJ, Flaherty KT, Smith CD, El-Deiry WS. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell. 2007;12:66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 22.Tavaluc RT, Hart LS, Dicker DT, El-Deiry WS. Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle. 2007;6:2554–62. doi: 10.4161/cc.6.20.4911. [DOI] [PubMed] [Google Scholar]