

In this series we draw attention to medicines that have entered the European market with an entirely new mechanism of action. Publication is not to be confused with endorsement of use in clinical practice. Copyright of the images belongs to Leiden University, but use of the images (also available at http://coo.lumc.nl/trc and in the app stores) is free.
Introduction
Homozygous familial hypercholesterolaemia (HoFH) is an inherited disease which causes accelerated atherosclerosis. HoFH commonly arises from a loss-of-function mutation in both alleles of the low density lipoprotein (LDL) receptor 1. This mutation inhibits LDL uptake from the circulation, thus resulting in increased plasma concentrations of total cholesterol and LDL (and increased cardiovascular risk) in patients with HoFH 1,2. Statins, the most commonly used LDL lowering agents, are generally not sufficiently effective in patients with HoFH. This is because patients with HoFH often have minimal LDL receptor activity, and this is not fully counteracted by statin-induced up-regulation of the LDL receptor 1.
Mechanism
Lomitapide lowers cholesterol through a different pathway, i.e. through the inhibition of the microsomal triglyceride transfer protein (MTP) (Figure1) 1. MTP is involved in the loading of triglyceride onto apolipoprotein B100, which is a part of the assembly process of very low density lipoprotein (VLDL). After excretion by hepatocytes, VLDL is converted to LDL. By blocking the assembly of VLDL, lomitapide reduces VLDL release and VLDL-mediated triglyceride secretion. This leads to a reduction of LDL concentrations in plasma 1.
Figure 1.
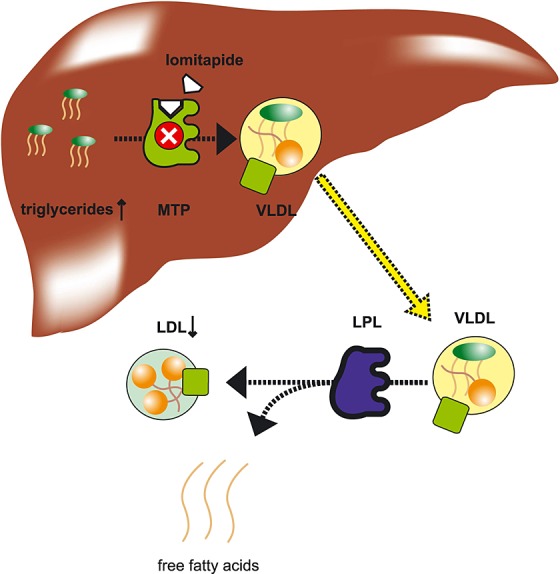
Mechanism of action of lomitapide. MTP is involved in the production of VLDL. After production in the liver, VLDL is released into the plasma. The enzyme lipoprotein lipase (LPL) catalyzes the breakdown of triglycerides from the VLDL into free fatty acids. As a result, VLDL is transformed into LDL. By inhibiting MTP, lomitapide reduces the production and release of VLDL, and LDL levels in plasma. A side effect of lomitapide is the accumulation of triglycerides in the liver, which is probably due to the inhibition of VLDL-mediated release of triglycerides from the liver.  apolipoprotein B100,
apolipoprotein B100,  triglycerides,
triglycerides,  cholesterol ester
cholesterol ester
Indication
Lomitapide has been approved by both the FDA and EMA for the treatment of adult HoFH patients as an adjunct to a low fat diet and other lipid-lowering therapies, with or without LDL apheresis 3,4.
Clinical application
In three clinical trials, lomitapide lowered plasma LDL concentrations effectively by up to 51% compared with baseline values 5. Lomitapide treatment is started with a daily oral dose of 5 mg, taken 2 or more hours after the evening meal 5. The dose may be gradually increased after 2 weeks, based upon tolerability and response, up to a maximum daily dose of 60 mg. Lomitapide is both a substrate and inhibitor for CYP3A4 metabolism 5. Co-administration with moderate to strong inhibitors of CYP3A4 is therefore contraindicated. The regular monitoring of hepatic function can mitigate the risks posed by the hepatic effects of lomitapide.
Adverse effects
Gastrointestinal side effects are very common in patients treated with lomitapide, with an incidence of about 90% 4. Diarrhoea, nausea, dyspepsia and vomiting were reported in over 30% of the patients, while pain, discomfort and bloating of the abdomen, constipation and flatulence were observed less frequently (±20%) 4. Elevated liver aminotransferase levels are a serious side effect reported in 10 out of 29 patients in a phase 3 trial. The frequency and severity of these adverse reactions are reduced when patients are on a low fat diet 4. The inhibition of VLDL-mediated triglyceride secretion can lead to hepatic accumulation of triglycerides 1.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–32. doi: 10.1161/CIRCULATIONAHA.113.001292. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, de Ferranti SD, Durrington P. Inherited disorders of LDL-cholesterol metabolism. [updated Nov 25 2014; cited Dec 16 2014]. Available at http://www.uptodate.com/contents/inherited-disorders-of-ldl-cholesterol-metabolism.
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. 2013. Lojuxta: EPAR – Product Information Sep [updated Dec 12 2014; cited Dec 15 2014]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002578/WC500148549.pdf.
- Rosenson RS, de Ferranti SD, Durrington P. Treatment of drug-resistant hypercholesterolemia. [updated Nov 7 2014; cited Dec 16 2014]. Available at http://www.uptodate.com/contents/treatment-of-drug-resistant-hypercholesterolemia.