

Abstract
In the past decade, the availability of powerful molecular techniques has accelerated the pace of discovery of several new primary immunodeficiencies (PID) and revealed the biologic basis of other established PID. These genetic advances, in turn, have facilitated more precise phenotyping of associated skin and systemic manifestations and provide a unique opportunity to better understand the complex human immunologic response. These continuing medical education articles will provide an update of recent advances about PID that may be encountered by dermatologists through their association with eczematous dermatitis, infectious, and non-infectious cutaneous manifestations. Part I will discuss new primary immunodeficiencies that have an eczematous dermatitis. Part II will focus on primary immunodeficiencies that greatly increase susceptibility to fungal infection and the noninfectious presentations of PID.
INTRODUCTION
Eczematous dermatitis is a common finding among several primary immunodeficiencies (PID) and may be the presenting clinical manifestation to the dermatologist. However, atopic dermatitis is also common in the general population, thus recognition of additional features of immunodeficiency can facilitate earlier diagnosis. In a series of 75 patients with severe dermatitis with no known underlying primary immunodeficiency, Aghamohammadi et al.1 identified 5 patients with hyper-immunoglobulin (Ig) E syndrome (HIES) and one patient with Wiskott-Aldrich syndrome (WAS) (mean age at diagnosis: 5 years old). This underscores the importance of eliciting a history of recurrent infections or family history suggestive of immunodeficiency in patients with severe atopic dermatitis. In this continuing medical education article we provide an update on primary immunodeficiencies associated with dermatitis.
HYPER-IGE SYNDROMES
The first HIES to be described was Job’s syndrome. This multisystem PID was initially described in 1966 as a disorder of recurrent cold abscesses, eczematous dermatitis, and lung disease.2, 3 Autosomal dominant hyper-IgE syndrome (AD-HIES) shares several clinical features with dedicator of cytokinesis 8 (DOCK8) deficiency, also known as autosomal recessive (AR)-HIES, but also has several key differences, which result in distinct phenotypes and prognoses. In addition, there are two other rare autosomal recessive diseases associated with HIES. The first is caused by mutations in phosphoglucomutase-3 (PGM3). The second disorder, associated with a mutation in tyrosine kinase 2 (Tyk2), was reported in one patient with elevated IgE, eczema, and susceptibility to viral, fungal, and bacterial infections including mycobacteria.4, 5 A subsequently reported patient with a mutation in Tyk2 also had a susceptibility to mycobacterial infections, but did not have HIES, making the link between Tyk2 mutation and AR-HIES uncertain.6, 7 Although elevated serum IgE and eczematous skin disease is a known presentation among the aforementioned HIES immunodeficiencies, Wiskott-Aldrich syndrome and Netherton syndrome may also present with similar skin and laboratory findings. Figure 1 reviews PIDs associated with eczematous dermatitis and distinctive features of each syndrome. Below we review in greater detail AD-HIES and DOCK8 deficiency, as well as recently described PGM3 deficiency.
Figure 1.

Clinical features of primary immunodeficiencies with eczematous dermatitis stratified by prevalence of allergies and asthma.
Autosomal dominant hyper-IgE syndrome
Key points
Early onset dermatitis
Cold abscesses and lung infections
Multisystem disease with skeletal and connective tissue abnormalities
In 2007, AD-HIES was found to be caused by dominant negative mutations in the signal transducer and activator of transcription 3 (STAT3) gene, a key transcription factor which regulates a diverse number of biologic processes including cell growth regulation and inflammation.8, 9
The majority of patients with AD-HIES develop a neonatal papulopustular eruption (Fig 2, A), often within the first week of life that typically starts on the face and scalp, but can generalize. The rash often changes into an eczematous morphology within the first year.10 Chronic dermatitis (Fig 2, B) in AD-HIES is strongly associated with Staphylococcus aureus skin colonization and infection. Control of S aureus through prophylactic systemic antimicrobials and topical antiseptics limits eczematous disease, but recurrences throughout life are common. Exacerbations of dermatitis are often due to resistant S aureus strains or poor antimicrobial adherence. Dilute sodium hypochlorite baths, as used in atopic dermatitis, may be effective, but further clinical study in this population is needed.11 Recommended therapy is a half-cup of household bleach in a full tub of water with exposure for 15 minutes three days weekly. For those who are not able or willing to use dilute bleach baths, chlorhexidine or sodium hypochlorite containing washes may be helpful.12 In contrast to DOCK8 and atopic patients with high serum IgE levels, anaphylaxis is rare and food allergies are not a major concern in AD-HIES, although the latter is more prevalent in AD-HIES than in the general population.13, 14
Figure 2.

Cutaneous findings in autosomal dominant hyper-IgE syndrome. A 3 week old infant with neonatal pustular eruption of the (A) face. B. A 7 year old boy with chronic dermatitis on the lower back. C. Characteristic facies with coarse facial features, broad nasal bridge, large bulbous nose, prominent skin pores and prognathism in a 44 year old female. D. High arched palate. E. Gorlin’s sign demonstrating hyperextensibility similar to that seen in Ehlers-Danlos syndrome. F. Joint hyperextensibility in a 6 year old boy.
S aureus is also the major pathogen responsible for recurrent cold skin abscesses and sinopulmonary infections in AD-HIES. Pulmonary infection results in abscess formation and pneumatocele development (Fig 3, A), which predispose patients to subsequent Pseudomonas, Aspergillus, and nontuberculous mycobacterial (NTM) infections and additional morbidity. Prophylactic anti-staphylococcal antibiotics are recommended to decrease risk of pneumonia and staphylococcal abscesses.3 Chronic mucocutaneous candidiasis (CMC) occurs in 83% of patients, and many patients require long-term antifungal treatment.3, 10 STAT3 is integral for the differentiation of Th17 cells. AD-HIES patients lack Th17 cells, thereby leading to impaired interleukin (IL)-17/IL-22 signaling and this high risk of CMC.15 STAT3 is also important for production of other proinflammatory cytokines and CD8+ T cell memory maintenance, which likely contributes to the risk of reactivation of varicella-zoster virus (VZV) and Epstein-Barr virus (EBV).16 Memory B cell differentiation is also impaired, leading to variable specific antibody production; therefore, some patients require chronic immune globulin replacement in addition to prophylactic antimicrobials.17
Figure 3.
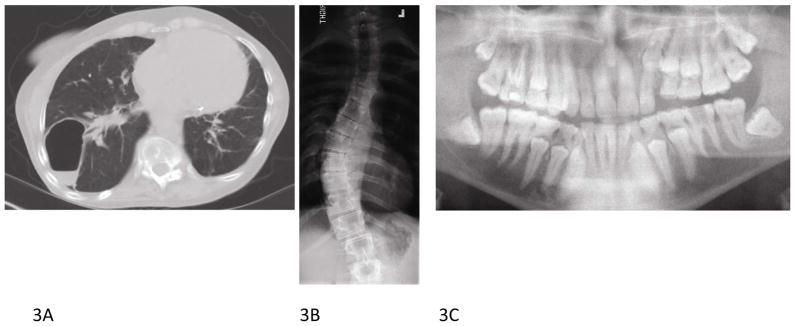
Autosomal dominant hyper-IgE syndrome radiographic findings. A. 42 year old female with right lung pneumatocele and (B) severe scoliosis. C. Multiple retained primary teeth visualized by panoramic radiograph in 21 year old male.
As the name implies, elevated serum IgE is seen in all AD-HIES patients, with peak IgE levels above 2000 IU/mL in 97% of patients and eosinophilia in 93%.3 However, IgE levels may diminish over time and may be within the normal range in adulthood. Craniofacial, musculoskeletal, and vascular abnormalities are also common and help distinguish AD-HIES from other PID associated with eczematous dermatitis and skin infection. A characteristic facial appearance emerges in childhood comprised of facial asymmetry, large bulbous nose, prominent chin, and prominent skin pores (Fig 2, C). Craniosynostosis, Chiari I malformations, retained primary teeth (Fig 3, C) and midline oropharyngeal anomalies are also common, particularly high-arched palate (Fig 2, D) and midline sagittal cleft of the tongue. Musculoskeletal abnormalities, including scoliosis (Fig 3, B), develop in approximately three quarters of patients, minimal trauma fractures occur in more than 50% patients, and joint hyperextensibility is also common (Fig 2, F).7, 18 Patients with AD-HIES have decreased bone mineral density (BMD) and increased osteoclast activity. Studies in STAT3 knock out mice confirm an important role for STAT3 in bone homeostasis19; however, a recent study did not demonstrate a relationship between BMD, as measured on dual-energy x-ray absorptiometry (DEXA) scan or serum markers of osteoclastic activity, and fracture risk in AD-HIES.19–21 Another new finding is that STAT3 is also involved in the regulation of matrix metalloproteinases (MMP) and plasma levels of MMP8 and MMP9 are elevated, while MMP3 is lower in patients with AD-HIES and may be responsible for impaired tissue remodeling.22
A newly identified systemic manifestation of AD-HIES is vascular anomalies, including lacunar infarcts in the brain, coronary aneurysms (37%), and coronary dilation and tortuosity (70%). The coronary abnormalities have been associated with risk of myocardial infarction.23–25 AD-HIES is also associated with an increased risk of malignancy, most commonly non-Hodgkin’s lymphoma.26 Life expectancy for patients with AD-HIES is the fifth to sixth decade. Mortality is most commonly from infection.7
DOCK8 deficiency
Key points
Autosomal recessive
Elevated IgE and multiple allergies
Eczematous dermatitis
Severe human papillomavirus (HPV), molluscum contagiosum virus (MCV), and herpes viral infections
In 2009, mutations in DOCK8 were identified as the genetic basis of the majority of cases of AR-HIES, a syndrome now commonly referred to as DOCK8 deficiency.27, 28 Both DOCK8 deficiency and AD-HIES are characterized by elevated serum IgE, eosinophilia, eczematous dermatitis, and recurrent sinopulmonary and staphylococcal skin infections. Unlike AD-HIES, DOCK8 deficiency is also characterized by severe cutaneous viral infections due to herpes simplex virus (HSV) (Fig 4, A), HPV, MCV, and varicella-zoster virus (VZV). Widespread HPV and MCV infection (Fig 4, B, C, and D) may be disfiguring and resistant to standard treatments. Interferon (IFN)3 has been used to treat the mucocutaneous viral infections, with improvement in HPV infection and, in one case, HSV infection.29, 30 Patients are at elevated risk of squamous cell carcinoma at sites of HPV infection (Fig 4, E), and cutaneous T cell lymphoma has also been reported.28, 31 Malignancy is a frequent cause of death in the second and third decade of life.7, 32
Figure 4.

The spectrum of cutaneous findings in DOCK8 immunodeficiency. A. Severe herpetic stomatitis in a 7 year old girl. B. Generalized molluscum contagiosum with verrucous HPV infection on the distal fingers in a 22 year old female. C. Generalized verrucosis of the arm, extensive pink to red brown thin papules resembling epidermodysplasia verruciformis in a 25 year old male (D), and squamous cell carcinoma on the face (E). Biopsy of the chest revealed coarse keratohyaline granules and abundant pale gray cytoplasm similar to findings seen in epidermodysplasia verruciformis. F. Extensive dermatitis with excoriations and post-auricular crusting (G) in a 5 year old with DOCK8 immunodeficiency.
DOCK8 deficiency is associated with impaired natural killer (NK) cell development and survival, which likely contributes to the profound susceptibility to cutaneous viral infections.33 In addition, CD4+ and CD8+ T cells are frequently reduced and often diminish further with age. Further, plasmocytoid dendritic cells are profoundly decreased. This cell population is critical for production of IFN3 in response to the cutaneous viral infections through the toll like receptor (TLR)-9 signaling pathway.29, 30 Consistent with an elevated risk of recurrent sinopulmonary infections, memory B cells are often reduced in number, and, although total serum IgG may be within normal range, specific antibody production is frequently impaired.17, 34, 35 Mucocutaneous candidiasis is less common in DOCK8 deficiency as compared to AD-HIES. Differentiation of Th17 cells and IL-17 production is impaired, albeit to a lesser degree than seen in AD-HIES.36 The mechanism is not yet understood, but may be related to general T cell deficiency.
Patients with DOCK8 deficiency present with variable severity of dermatitis (Fig 4, F and G). Signs begin in the first several months of life with a classic atopic dermatitis distribution and appearance.31 In contrast, most patients with AD-HIES present with a neonatal pustular eruption that eventually progresses to an eczematous dermatitis.10 Additionally, patients with DOCK8 deficiency frequently have multiple food allergies and asthma. Treatment of the atopic dermatitis is often difficult due to concurrent cutaneous viral infections and a predilection for bacterial infections that can worsen with the use of topical or systemic corticosteroids. Skin superinfection with Staphylococcus aureus is frequent and antiseptic measures, including dilute sodium hypochlorite baths and systemic antibiotics, are recommended based on studies in the general atopic dermatitis population as well as our experience with dermatitis in the PID setting.11, 37
Given the risk of malignancy (squamous cell carcinoma and lymphoma) and the reduced life expectancy associated with DOCK8 deficiency, allogeneic hematopoietic stem cell transplant (HSCT) is the treatment of choice.38–40 Cutaneous viral infections and dermatitis have shown dramatic improvement in the first 6 months after transplant.31, 40
PGM3 deficiency
Key points
Autosomal recessive
Elevated IgE levels with dermatitis, multiple allergies, and asthma
Neurologic abnormalities
PGM3 deficiency was described in 2014 as a novel autosomal recessive PID associated with atopic dermatitis, recurrent infections, and elevated IgE.41, 42 PGM3 is a protein that catalyzes the conversion of N-acetlyglucosamine-6-phosphate (GlcNAc-6-P) into GlcNAc-1-P in the synthesis of uridine diphosphate (UDP)-GlcNAc, a critical component of the glycosylation pathway, thereby affecting a wide range of diverse proteins. Atopic dermatitis was a universal feature in all 17 patients described.41, 42 Similar to DOCK8 deficiency, patients with PGM3 mutations frequently have other prominent atopic features, including asthma and allergies. These patients tended to have susceptibility to viral infections, including cutaneous HSV and MCV, as seen in DOCK8 deficiency. Much like in AD-HIES, patients with PGM3 deficiency have sinopulmonary infections, with bronchiectasis and pneumatocele development, skin and soft tissue bacterial infections. Mucocutaneous candidiasis can also develop, although it is not a consistent feature. One unique cutaneous finding in PGM3 deficiency is leukocytoclastic vasculitis seen in numerous patients. Distinguishing features of AD-HIES, DOCK8 deficiency, and PGM3 deficiency are shown in Table I. In contrast to AD-HIES and DOCK8 deficiency, neurologic impairment is a prominent feature in patients with PGM3 deficiency, and include developmental delay and low IQ (88%), ataxia (88%), dysarthria (63%), myoclonus (63%), sensorineural hearing loss (50%), and electroencephalography (EEG) abnormalities (38%).41 Hypomyelination is seen on brain magnetic resonance imaging (MRI). Common hematologic manifestations include cytopenias, mostly lymphopenia and neutropenia.
Table 1.
Comparison of hyper-immunoglobulin E syndromes
| STAT3 deficiency (Job’s syndrome) | DOCK8 deficiency | PGM3 deficiency | |
|---|---|---|---|
| GENETIC FEATURES | |||
| Inheritance | Autosomal dominant | Autosomal recessive | Autosomal recessive |
| Gene | STAT3 | DOCK8 | PGM3 |
| Protein | Signal transducer and activator of transcription 3 | Dedicator of cytokinesis 8 | Phosphoglucomutase 3 |
| Function | Mediates cellular responses to interleukins, stem cell factor, and other growth factors | Activates Rho GTPases, cytoskeletal reorganization, cell migration, phagocytosis | Enzyme catalyzing conversion of GlcNAc-6-P into GlcNAc-1-P |
| IMMUNOLOGIC FEATURES | |||
| Eosinophila | Common | Common | Common |
| Allergies | Rare | Common | Common |
| Asthma | Rare | Common | Common |
| Sinopulmonary infection | Common | Common | Common |
| Bronchiectasis | Common | Rare | Less common |
| DERMATOLOGIC FEATURES | |||
| Newborn rash | Cephalic papulopustular eruption | - | - |
| Eczematous dermatitis | Common | Highly variable | Highly variable |
| Bacterial skin abscesses | Common | Less common | Common |
| Leukocytoclastic vasculitis | - | - | Common |
| Mucocutaneous viral infection | Rare | Very common, severe | Occasional reports |
| Mucocutaneous candidiasis | Common | Less common | Less common |
| OTHER FEATURES | |||
| Malignancy | SCC (rare) | SCC (vulvar, facial, anal) | - |
| Characteristic facial appearance | Coarse features, asymmetry, broad nasal root, hypertelorism, prognathism | - | Narrow palpebral fissures |
| Oral findings | Retained primary dentition, high-arched palate | - | High-arched palate |
| Joint hyperextensibility | Very common | Rare | Rare |
| Minimal trauma fractures | Very common | Rare | - |
| Scoliosis | Very common | - | Rare |
| Neurologic symptoms | - | - | Conductive hearing loss, ataxia, myoclonus |
DOCK8 – dedicator of cytokinesis 8, PGM3 - phosphoglucomutase 3, STAT3 – signal transducer and activator of transcription 3 GlcNAc-1-P – N-acetylglucosamine-1-phosphate; GlcNAc-6-P – N-acetylglucosamine -6-phosphate
IMMUNE DYSREGULATION, POLYENDOCRINOPATHY AND ENTEROPATHY, X-LINKED (IPEX) SYNDROME
Key points
X-linked recessive
Early onset dermatitis
Multi-organ autoimmune disease due to loss of peripheral tolerance
Immune dysregulation, polyendocrinopathy and enteropathy, X-linked syndrome, is an X-linked recessive condition due to loss of function mutations in forkhead box protein 3 (FOXP3). It is characterized by a decreased or absent T regulatory (Treg) cell population and multi-organ autoimmune disease. There is considerable phenotypic variation in patients with IPEX syndrome without an obvious genotype-phenotype correlation.43 Affected males develop autoimmune enteropathy in infancy that can be life threatening. Patients may also develop insulin-dependent diabetes mellitus (IDDM) and thyroid dysfunction early in life.44 Antibodies to organ specific antigens have been demonstrated in patients with IPEX, explaining autoimmune manifestations such as IDDM, thyroiditis, cytopenias, hepatitis, and nephritis.45 The most common cutaneous finding is eczematous dermatitis.46 Other less frequent presentations include psoriasiform dermatitis, erythroderma, urticaria, pemphigoid nodularis, cheilitis, onychodystrophy, and alopecia universalis.44, 47, 48 Elevated IgE and eosinophilia are common, as are food allergies. Infections are likely due to both the immune suppressive drugs employed to manage the autoimmune disease and impaired skin barrier and gut epithelium. Severe disease may lead to mortality in early childhood. HSCT is currently the only curative treatment, although gene therapy is being investigated.49, 50
MAMMALIAN STERILE 20-LIKE 1(MST1) DEFICIENCY
Key Points
Autosomal recessive
Bacterial, viral, and candidal cutaneous infections
Structural cardiac anomalies
Mammalian sterile 20-like 1 deficiency, previously known as serine/threonine protein kinase 4 (STK4) deficiency, is an autosomal recessive PID associated with bacterial and viral infections (HSV, HPV, MCV, EBV) as well as mucocutaneous candidiasis. MST1 encodes a serine-threonine kinase that is ubiquitously expressed but has increased levels in cells of hematopoietic origin.51 First reported in 2012, four consanguineous affected families with MST1 deficiency have now been described.51–53 Systemic findings include structural cardiac anomalies (atrial septal defects and patent foramen ovale) and valvular disease.52, 53 Eczematous dermatitis has been reported, but is poorly characterized.53 Additionally, multiple autoantibodies including antinuclear, anticardiolipin, and antineutrophil cytoplasmic antibodies, and autoimmune hemolytic anemia has been described.51–53 Mst1 and Mst2 were recently found to be important regulators of Foxp3 expression and Treg development, providing a biologic rationale for the autoimmune manifestations of this condition.54, 55 Affected patients have a peripheral neutropenia with normal bone marrow maturation, as well as T and B cell lymphopenia. The primary therapeutic intervention for this condition is infection control. Three patients with MST1 deficiency have undergone HSCT, but 2 died within 6 months due to graft-versus-host disease and infectious complications.53
CONCLUSION
Genetic advances have identified several novel primary immunodeficiencies and allowed better characterization of their cutaneous phenotypic presentation. In Part 1, we reviewed the clinical characteristics, genetic basis, and immunologic abnormalities in PID associated with eczematous dermatitis, including several newly described syndromes. In Part 2, we will provide an update on other recently described PID that are not associated with eczematous dermatitis.
Learning objectives.
After completing this Journal CME activity, the learner should be able to differentiate primary immunodeficiencies that present with eczematous dermatitis based on infectious and non-infectious manifestations; identify the numerous systemic manifestations of autosomal dominant hyper-IgE syndrome;
Acknowledgments
Funding/Support: This study was supported by the Intramural Research Program of the National Institutes of Health (NIH), Center for Cancer Research, National Cancer Institute.
ABBREVIATION AND ACRONYM LIST
- AD
autosomal dominant
- AD-HIES
autosomal dominant hyper-IgE syndrome
- AR
autosomal recessive
- BMD
bone mineral density
- CMC
chronic mucocutaneous candidiasis
- DEXA
dual-energy x-ray absorptiometry
- DOCK8
dedicator of cytokinesis 8
- EBV
Epstein-Barr virus
- EEG
electroencephalography
- FOXP3
forkhead box protein 3
- GlcNAc-6-P
N-acetlyglucosamine-6-phosphate
- HIES
hyper-IgE syndrome
- HPV
human papillomavirus
- HSCT
hematopoietic stem cell transplant
- HSV
herpes simplex virus
- IDDM
insulin-dependent diabetes mellitus
- Ig
immunoglobulin
- IFN
interferon
- IL
interleukin
- IPEX
immune dysregulation, polyendocrinopathy and enteropathy, X-linked syndrome
- MCV
molluscum contagiosum virus
- MMP
metalloproteinase
- MRI
magnetic resonance imaging
- MST1
mammalian sterile 20-like 1
- NK
natural killer
- NTM
nontuberculous mycobacterial
- PGM3
phosphoglucomutase 3
- PID
primary immunodeficiency
- STAT3
signal transducer and activator of transcription 3
- STK4
serine/threonine protein kinase 4
- Treg
regulatory T
- TLR
toll-like receptor
- TYK2
tyrosine kinase 2
- UDP
uridine diphosphate
- VZV
varicella-zoster virus
- WAS
Wiskott-Aldrich syndrome
- WHIM
warts, hypogammaglobulinemia, immunodeficiency and myelokathexis
- WILD
warts, immunodeficiency, primary lymphedema, and anogenital dysplasia
Footnotes
Public disclosure statement: The authors have no conflict of interest to declare
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Dominique C. Pichard, National Institutes of Health, National Cancer Institute, Bethesda, MD.
Alexandra F. Freeman, National Institutes of Health, National Institute of Allergy and Infectious Disease. Bethesda, MD.
Edward W. Cowen, National Institutes of Health, National Cancer Institute, Bethesda, MD.
References
- 1.Aghamohammadi A, Moghaddam ZG, Abolhassani H, Hallaji Z, Mortazavi H, Pourhamdi S, et al. Investigation of underlying primary immunodeficiencies in patients with severe atopic dermatitis. Allergol Immunopathol (Madr) 2014;42:336–41. doi: 10.1016/j.aller.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet. 1966;1:1013–5. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- 3.Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Ann N Y Acad Sci. 2012;1250:25–32. doi: 10.1111/j.1749-6632.2011.06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–55. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Minegishi Y, Karasuyama H. Hyperimmunoglobulin E syndrome and tyrosine kinase 2 deficiency. Curr Opin Allergy Clin Immunol. 2007;7:506–9. doi: 10.1097/ACI.0b013e3282f1baea. [DOI] [PubMed] [Google Scholar]
- 6.Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, Kreins AY, Grant AV, Abel L, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012;160:1055–7. doi: 10.1016/j.jpeds.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freeman AF, Holland SM. Clinical manifestations of hyper IgE syndromes. Dis Markers. 2010;29:123–30. doi: 10.3233/DMA-2010-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 9.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 10.Eberting CL, Davis J, Puck JM, Holland SM, Turner ML. Dermatitis and the newborn rash of hyper-IgE syndrome. Arch Dermatol. 2004;140:1119–25. doi: 10.1001/archderm.140.9.1119. [DOI] [PubMed] [Google Scholar]
- 11.Huang JT, Abrams M, Tlougan B, Rademaker A, Paller AS. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:e808–14. doi: 10.1542/peds.2008-2217. [DOI] [PubMed] [Google Scholar]
- 12.Ryan C, Shaw RE, Cockerell CJ, Hand S, Ghali FE. Novel sodium hypochlorite cleanser shows clinical response and excellent acceptability in the treatment of atopic dermatitis. Pediatr Dermatol. 2013;30:308–15. doi: 10.1111/pde.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegel AM, Stone KD, Cruse G, Lawrence MG, Olivera A, Jung MY, et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J Allergy Clin Immunol. 2013;132:1388–96. doi: 10.1016/j.jaci.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boos AC, Hagl B, Schlesinger A, Halm BE, Ballenberger N, Pinarci M, et al. Atopic dermatitis, STAT3- and DOCK8-hyper-IgE syndromes differ in IgE-based sensitization pattern. Allergy. 2014;69:943–53. doi: 10.1111/all.12416. [DOI] [PubMed] [Google Scholar]
- 15.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35:806–18. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207:155–71. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 19.Li J. JAK-STAT and bone metabolism. JAKSTAT. 2013;2:e23930. doi: 10.4161/jkst.23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sowerwine KJ, Shaw PA, Gu W, Ling JC, Collins MT, Darnell DN, et al. Bone Density and Fractures in Autosomal Dominant Hyper IgE Syndrome. J Clin Immunol. 2014;34:260–4. doi: 10.1007/s10875-013-9982-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araya N, Inose H, Kato T, Saito M, Sumiya S, Yamada T, et al. Spinal deformity caused by hyperimmunoglobulin E syndrome. J Neurosurg Spine. 2014;21:292–5. doi: 10.3171/2014.4.SPINE13629. [DOI] [PubMed] [Google Scholar]
- 22.Sekhsaria V, Dodd LE, Hsu AP, Heimall JR, Freeman AF, Ding L, et al. Plasma metalloproteinase levels are dysregulated in signal transducer and activator of transcription 3 mutated hyper-IgE syndrome. J Allergy Clin Immunol. 2011;128:1124–7. doi: 10.1016/j.jaci.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman AF, Kleiner DE, Nadiminti H, Davis J, Quezado M, Anderson V, et al. Causes of death in hyper-IgE syndrome. J Allergy Clin Immunol. 2007;119:1234–40. doi: 10.1016/j.jaci.2006.12.666. [DOI] [PubMed] [Google Scholar]
- 24.Freeman AF, Collura-Burke CJ, Patronas NJ, Ilcus LS, Darnell D, Davis J, et al. Brain abnormalities in patients with hyperimmunoglobulin E syndrome. Pediatrics. 2007;119:e1121–5. doi: 10.1542/peds.2006-2649. [DOI] [PubMed] [Google Scholar]
- 25.Freeman AF, Avila EM, Shaw PA, Davis J, Hsu AP, Welch P, et al. Coronary artery abnormalities in Hyper-IgE syndrome. J Clin Immunol. 2011;31:338–45. doi: 10.1007/s10875-011-9515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leonard GD, Posadas E, Herrmann PC, Anderson VL, Jaffe ES, Holland SM, et al. Non-Hodgkin’s lymphoma in Job’s syndrome: a case report and literature review. Leuk Lymphoma. 2004;45:2521–5. doi: 10.1080/10428190400004463. [DOI] [PubMed] [Google Scholar]
- 27.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–302.e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keles S, Jabara HH, Reisli I, McDonald DR, Barlan I, Hanna-Wakim R, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with IFN-3 2b therapy. J Allergy Clin Immunol. 2014;133:1753–5.e3. doi: 10.1016/j.jaci.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Zahrani D, Raddadi A, Massaad M, Keles S, Jabara HH, Chatila TA, et al. Successful interferon-alpha 2b therapy for unremitting warts in a patient with DOCK8 deficiency. Clin Immunol. 2014;153:104–8. doi: 10.1016/j.clim.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148:79–84. doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr. 2004;144:93–9. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 33.Crawford G, Enders A, Gileadi U, Stankovic S, Zhang Q, Lambe T, et al. DOCK8 is critical for the survival and function of NKT cells. Blood. 2013;122:2052–61. doi: 10.1182/blood-2013-02-482331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caracciolo S, Moratto D, Giacomelli M, Negri S, Lougaris V, Porta F, et al. Expansion of CCR4+ activated T cells is associated with memory B cell reduction in DOCK8-deficient patients. Clin Immunol. 2014 doi: 10.1016/j.clim.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Speckmann C, Enders A, Woellner C, Thiel D, Rensing-Ehl A, Schlesier M, et al. Reduced memory B cells in patients with hyper IgE syndrome. Clin Immunol. 2008;129:448–54. doi: 10.1016/j.clim.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 36.McDonald DR. TH17 deficiency in human disease. J Allergy Clin Immunol. 2012;129:1429–35. doi: 10.1016/j.jaci.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bath-Hextall FJ, Birnie AJ, Ravenscroft JC, Williams HC. Interventions to reduce Staphylococcus aureus in the management of atopic eczema: an updated Cochrane review. Br J Dermatol. 2010;163:12–26. doi: 10.1111/j.1365-2133.2010.09743.x. [DOI] [PubMed] [Google Scholar]
- 38.Boztug H, Karitnig-Weiß C, Ausserer B, Renner ED, Albert MH, Sawalle-Belohradsky J, et al. Clinical and immunological correction of DOCK8 deficiency by allogeneic hematopoietic stem cell transplantation following a reduced toxicity conditioning regimen. Pediatr Hematol Oncol. 2012;29:585–94. doi: 10.3109/08880018.2012.714844. [DOI] [PubMed] [Google Scholar]
- 39.Gatz SA, Benninghoff U, Schütz C, Schulz A, Hönig M, Pannicke U, et al. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation. Bone Marrow Transplant. 2011;46:552–6. doi: 10.1038/bmt.2010.169. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh S, Schuster FR, Adams O, Babor F, Borkhardt A, Comoli P, et al. Haploidentical stem cell transplantation in DOCK8 deficiency - Successful control of pre-existing severe viremia with a TCRaβ/CD19-depleted graft and antiviral treatment. Clin Immunol. 2014;152:111–4. doi: 10.1016/j.clim.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014;133:1400–9.e5. doi: 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol. 2014;133:1410–9.e13. doi: 10.1016/j.jaci.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baris S, Schulze I, Ozen A, Aydiner EK, Altuncu E, Karasu GT, et al. Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol. 2014;34:601–6. doi: 10.1007/s10875-014-0059-7. [DOI] [PubMed] [Google Scholar]
- 44.Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211. doi: 10.3389/fimmu.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuda M, Torgerson TR, Selmi C, Gambineri E, Carneiro-Sampaio M, Mannurita SC, et al. The spectrum of autoantibodies in IPEX syndrome is broad and includes anti-mitochondrial autoantibodies. J Autoimmun. 2010;35:265–8. doi: 10.1016/j.jaut.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 46.Martin-Santiago A, Hervas JA, Hervas D, Rosell A, Caimari M, de Carlos JC, et al. Diagnostic value of the skin lesions in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Dermatol. 2013;30:e221–2. doi: 10.1111/pde.12126. [DOI] [PubMed] [Google Scholar]
- 47.Moraes-Vasconcelos D, Costa-Carvalho BT, Torgerson TR, Ochs HD. Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J Clin Immunol. 2008;28(Suppl 1):S11–9. doi: 10.1007/s10875-008-9176-5. [DOI] [PubMed] [Google Scholar]
- 48.McGinness JL, Bivens MM, Greer KE, Patterson JW, Saulsbury FT. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55:143–8. doi: 10.1016/j.jaad.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 49.Passerini L, Santoni de Sio FR, Porteus MH, Bacchetta R. Gene/Cell Therapy Approaches for Immune Dysregulation Polyendocrinopathy Enteropathy X-linked Syndrome. Curr Gene Ther. 2014;14:422–8. doi: 10.2174/1566523214666141001123828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horino S, Sasahara Y, Sato M, Niizuma H, Kumaki S, Abukawa D, et al. Selective expansion of donor-derived regulatory T cells after allogeneic bone marrow transplantation in a patient with IPEX syndrome. Pediatr Transplant. 2014;18:E25–30. doi: 10.1111/petr.12184. [DOI] [PubMed] [Google Scholar]
- 51.Crequer A, Picard C, Patin E, D’Amico A, Abhyankar A, Munzer M, et al. Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS One. 2012;7:e44010. doi: 10.1371/journal.pone.0044010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schäffer AA, et al. The phenotype of human STK4 deficiency. Blood. 2012;119:3450–7. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nehme NT, Pachlopnik Schmid J, Debeurme F, André-Schmutz I, Lim A, Nitschke P, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood. 2012;119:3458–68. doi: 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du X, Shi H, Li J, Dong Y, Liang J, Ye J, et al. Mst1/Mst2 Regulate Development and Function of Regulatory T Cells through Modulation of Foxo1/Foxo3 Stability in Autoimmune Disease. J Immunol. 2014;192:1525–35. doi: 10.4049/jimmunol.1301060. [DOI] [PubMed] [Google Scholar]
- 55.Tomiyama T, Ueda Y, Katakai T, Kondo N, Okazaki K, Kinashi T. Antigen-specific suppression and immunological synapse formation by regulatory T cells require the Mst1 kinase. PLoS One. 2013;8:e73874. doi: 10.1371/journal.pone.0073874. [DOI] [PMC free article] [PubMed] [Google Scholar]