

The current era of research in antiangiogenic therapy for cancer began in earnest in 1971 with the publication of Folkman's imaginative hypothesis,1 but 33 years would elapse before the first drug developed as an inhibitor of angiogenesis was approved by the Food and Drug Administration (FDA).2,3 This approval was based on the survival benefit observed in a randomized phase 3 trial of first-line treatment of metastatic colorectal cancer; in that trial, bevacizumab, a humanized monoclonal antibody directed against vascular endothelial growth factor (VEGF), was combined with conventional chemotherapy.4 Bevacizumab therapy also increased overall survival in the first-line treatment of advanced non–small-cell lung cancer when used in combination with standard chemotherapy.5 Two other antiangiogenic drugs, sorafenib and sunitinib, have also been approved by the FDA; these are oral small-molecule-receptor tyrosine kinase inhibitors (RTKIs). They target multiple receptor tyrosine kinases, including VEGF receptors and platelet-derived growth factor (PDGF) receptors.6 Sorafenib and sunitinib have been beneficial in the treatment of metastatic renal-cell cancer when used alone.7,8 Sorafenib monotherapy is also active in the treatment of hepatocellular carcinoma9 and was recently approved by the FDA for this indication.
The survival benefits of these treatments are relatively modest (usually measured in months), with the possible exception of the benefits for patients with renal-cell carcinoma. These treatments are also costly10 and have toxic side effects.11,12 These concerns raise the following questions with respect to improving antiangiogenic therapy: How do such drugs work, and how does bevacizumab increase the efficacy of chemotherapy? Several theories have been postulated,13-16 including the theory that antiangiogenic drugs improve chemotherapy by causing “vessel normalization” in tumors (see Appendix 1 of the Supplementary Appendix, available with the full text of this article at www.nejm.org). How do tumors become resistant to antiangiogenic drugs? Are there clinically useful markers that can predict the efficacy of this class of drug? Are there promising surrogate pharmacodynamic biomarkers that will help to determine the best dose of a particular agent? Will antiangiogenic RTKIs such as sunitinib or sorafenib consistently enhance the efficacy of chemotherapy? What accounts for the side effects of these agents?11,12
Many recent discoveries have the potential not only to answer some of these questions but also to indicate new therapeutic targets and treatment strategies. The purpose of this review is to summarize a number of these discoveries, made mainly over the past 5 years, and to point out their potential clinical impact.
The VEGF and VEGF-Receptor Family in Tumor Angiogenesis
Much attention has been focused on the VEGF family of growth factors and the receptor tyrosine kinases that mediate their proangiogenic effects17,18 (Fig. 1). This family of structurally related molecules includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF).17,18 The major mediator of tumor angiogenesis is VEGF-A, usually referred to as VEGF. VEGF signals mainly through VEGF receptor 2 (VEGFR-2), which is expressed at elevated levels by endothelial cells engaged in angiogenesis and by circulating bone marrow–derived endothelial progenitor cells. The role of VEGF receptor 1 (VEGFR-1) is a mystery with respect to VEGF-mediated angiogenesis. It binds VEGF with approximately 10 times the affinity of VEGFR-2 binding, but its signal-transducing properties are extremely weak.19
Figure 1. The Family of VEGF Molecules and Receptors.
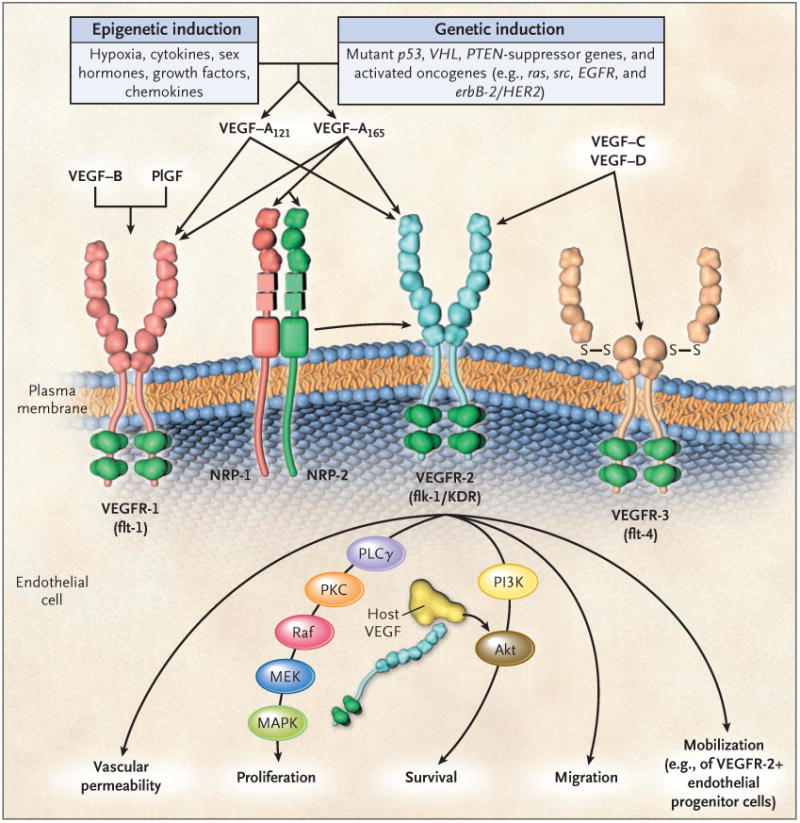
The major mediator of tumor angiogenesis is vascular endothelial growth factor A (VEGF-A, also called VEGF), specifically the circulating isoforms of VEGF — VEGF121 and VEGF165. These isoforms signal through VEGF receptor 2 (VEGFR-2), the major VEGF signaling receptor that mediates sprouting angiogenesis (called kinase-insert domain–containing receptor [KDR] in humans and fetal liver kinase 1 [flk-1] in mice). The role of VEGFR-1 in sprouting angiogenesis is much less clear. VEGF is expressed in most types of human cancer, and increased expression in tumors is often associated with a less favorable prognosis. Induction of or an increase in VEGF expression in tumors can be caused by numerous environmental (epigenetic) factors such as hypoxia, low pH, inflammatory cytokines (e.g., interleukin-6), growth factors (e.g., basic fibroblast growth factor), sex hormones (both androgens and estrogens), and chemokines (e.g., stromal-cell–derived factor 1). Other causes include genetic inductive changes such as activation of numerous different oncogenes or loss or mutational inactivation of a variety of tumor-suppressor genes. The binding of VEGF to VEGFR-2 leads to a cascade of different signaling pathways,19 two examples of which are shown, resulting in the up-regulation of genes involved in mediating the proliferation and migration of endothelial cells and promoting their survival and vascular permeability. For example, the binding of VEGF to VEGFR-2 leads to dimerization of the receptor, followed by intracellular activation of the PLCγ–PKC–Raf kinase–MEK–mitogen-activated protein kinase (MAPK) pathway and subsequent initiation of DNA synthesis and cell growth, whereas activation of the phosphatidylinositol 3′–kinase (PI3K)–Akt pathway leads to increased endothelial-cell survival. Activation of src can lead to actin cytoskeleton changes and induction of cell migration. VEGF receptors are located on the endothelial-cell surface; however, intracellular (“intracrine”)–signaling VEGF receptors (VEGFR-2) may be present as well, and they are involved in promoting the survival of endothelial cells. The detailed structure of the intracellular VEGFR-2 in endothelial cells is not yet known, but it is shown as the full-length receptor that is normally bound to the cell surface. Binding of VEGF-C to VEGFR-3 mediates lymphangiogenesis. VEGF165 can bind to neuropilin (NRP) receptors, which can act as coreceptors with VEGFR-2 (horizontal arrow) to regulate angiogenesis. EGFR denotes epidermal growth factor receptor, flt-1 fms-like tyrosine kinase 1, PlGF placental growth factor, PTEN phosphatase and tensin homologue, S–S disulfide bond, and VHL von Hippel–Lindau.
Most types of human cancer cells express VEGF, often at elevated levels; this is a likely consequence of the numerous and diverse genetic and epigenetic ways in which VEGF can be induced (Fig. 1).20 Hypoxia, a characteristic of solid tumors,21 is an important inducer of VEGF. Its effect is mediated through the hypoxia-inducible transcription factors 1α and 2α.21
It is commonly held that VEGF action is attributable to a paracrine mechanism by tumor cells — that is, tumor cells produce VEGF but cannot respond to it directly since they do not have cell-surface VEGF receptors. In contrast, endothelial cells engaged in angiogenesis express numerous VEGF receptors, but they produce very little or no detectable VEGF. It is now clear, however, that VEGF in amounts sufficient to drive tumor angiogenesis originates from various host cells in the body such as platelets and muscle cells22; such cells also include tumor-associated stromal cells.23,24 These findings explain, at least in part, why elevated VEGF levels in blood or even tumor tissue do not predict a benefit in patients receiving drugs that target the VEGF–VEGFR-2 pathway.25 The observation that tumor cells of many types,26-28 including those of hematologic tumors,26,27 express VEGF receptors (especially VEGFR-1) and also produce VEGF indicates that VEGF may sometimes act as a direct (cell-autonomous) autocrine growth factor for tumor cells. Furthermore, in some cases the VEGF receptors may be expressed not on the surface of the tumor cell but rather within the cell, where they promote cell survival by an “intracrine” mechanism; this has been shown for VEGFR-1 in breast-cancer cells.29 Hematopoietic stem cells also express both intracellular VEGFR-1 and VEGF, which can in some instances promote the growth and survival of these cells.30
Results in mice bearing a mutant VEGF gene only in vascular endothelial cells suggest that very low levels of autocrine-acting VEGF mediate endothelial-cell survival and vascular homeostasis by signaling through intracellular VEGFR-2.31 Such mice have severe cardiac defects and are subject to gastrointestinal perforations and thrombotic events; these adverse effects are sometimes observed in patients treated with bevacizumab.11,12
These observations have potential clinical implications with respect to the use of small-molecule antiangiogenic RTKIs or monoclonal antibodies.30,31 The RTKIs, by virtue of their ability to penetrate cells, could cause certain toxic side effects,31 such as myelosuppression (which can also be caused by targeting other receptor tyrosine kinases such as c-kit and fms-like tyrosine kinase 3), but they may also be more effective than antibodies when used to treat tumors with functional intracellular autocrine VEGF receptors.
There is growing evidence of the role, under certain circumstances, of neuropilins in tumor angiogenesis.32,33 These transmembrane receptors lack tyrosine kinase activity, bind semaphorin 3A, and are normally involved in axon guidance. However, neuropilins also bind VEGF165, a splice variant of VEGF-A, and thereby modulate angiogenesis32,33; they can act as coreceptors for VEGFR-2. Consequently, neuropilins are emerging as potentially promising antiangiogenic targets.34
Another advance is the development of monoclonal antibodies to PlGF,35 a growth factor that binds to VEGFR-1. These antibodies have few side effects in mice because PlGF, unlike VEGF, is expressed minimally or not at all by most normal cells and tissues.35 Moreover, anti-PlGF antibodies can act in concert with antibodies targeting the VEGF pathway of tumor angiogenesis.35
Circulating VEGF and a soluble form of VEGFR-2 have been used as surrogate markers of antiangiogenic drug activity.36,37 The measurement of these molecules in blood has been used as a preclinical means of establishing the optimal biologic dose of drugs that target VEGFR-2, including antibodies37 and small-molecule RTKIs.36 Such drugs can cause dose-dependent alterations in the levels of circulating VEGF or soluble VEGFR-2, and in mice these levels correlate with antitumor activity.36,37 The assessment of circulating VEGF in a complex with a VEGF antagonist such as the soluble “decoy” receptor drug called the “VEGF trap” (aflibercept) is also a promising approach for predicting the blockade of angiogenesis.38
Another receptor tyrosine kinase signaling pathway is mediated by tie-2, a receptor tyrosine kinase expressed principally on the vascular endothelium. There are two major ligands for tie-2, angiopoietin-1 (ang-1) and angiopoietin-2 (ang-2).39,40 Ang-1 acts as an agonist, whereas ang-2 acts as an antagonist, but it can promote angiogenesis, especially in cooperation with VEGF. These angiopoietins act in concert with VEGF to stabilize and mature new capillaries. Blockade of this tie-2 pathway has been more difficult than blockade of the VEGF pathway, in part because of the complexity of agonistic and antagonistic ligands for the same receptor and the problems in finding effective and specific drugs against tie-2 or the angiopoietins. However, antibodies and peptidelike antibodies (“peptibodies”) against ang-2 have recently been developed; these can block tumor angiogenesis and tumor growth in preclinical models.40
Research on this signaling system has also highlighted the importance of the pericyte, an accessory cell that is closely associated with blood vessels. Not only endothelial cells but also pericytes secrete ang-1. Moreover, pericytes express PDGF receptors, which may be relevant to the antiangiogenic effects of certain small-molecule RTKIs that target PDGF receptors.41
The Notch–Deltalike Ligand 4 Signaling Pathway
Studies have implicated what appears to be a pivotal new angiogenesis signaling pathway, notch–deltalike ligand (Dll) 442-46 (Fig. 2). Notch cellsurface receptors (i.e., notch 1, 2, 3, and 4) are expressed by various cell types and are generally involved in cell fate, differentiation, and proliferation. These receptors interact with transmembrane ligands (jagged 1, jagged 2, and Dll1, Dll3, and Dll4) on adjacent cells. Vascular endothelial cells express notch 1 and notch 4 receptors and the jagged 1, Dll1, and Dll4 ligands; among these, Dll4 is expressed exclusively by endothelial cells. Experiments involving gene disruption in mice have shown that notch–Dll4 signaling is essential for vascular development in the embryo — knockout of only one Dll4 allele is lethal to the embryo46; VEGF haploinsufficiency has a similar effect.48,49
Figure 2. The Endothelial-Cell–Associated Deltalike Ligand 4–Notch Signaling Pathway in Angiogenesis.
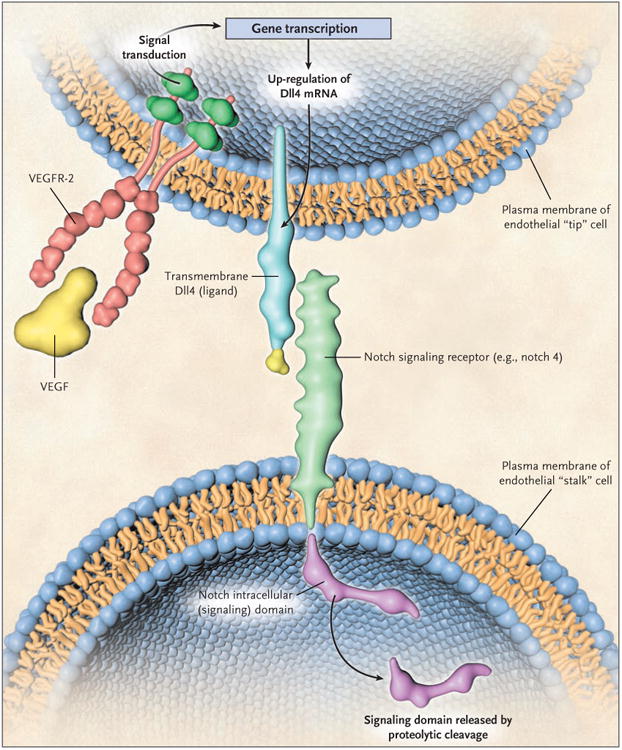
In mammals there are multiple deltalike ligands (Dlls), one of which, Dll4, is highly expressed in the vascular endothelial cells involved in angiogenesis. It tends to be expressed at higher levels in the “tip” cells of sprouting vessels and is induced by vascular endothelial growth factor (VEGF). Dll4 binds to notch receptors, two of which (notch 1 and notch 4) are expressed in the vascular endothelial cells composing the “stalk” component of a growing capillary sprout, adjacent to the tip cells. The interaction of Dll4 and notch receptors through the contact of adjacent endothelial cells leads to a series of proteolytic events whereby a notch intracellular signaling domain is cleaved and released by a γ-secretase; the domain then translocates to the nucleus. There it interacts with transcription factors and induces the expression of various target genes. The induction of Dll4–notch signaling is thought to act as a damping mechanism to prevent excessive angiogenesis and to promote the orderly development of new blood vessels. Blockade of Dll4–notch signaling interferes with this negative feedback mechanism, resulting in an increased density of vascular sprouts and branches, but these blood vessels are highly abnormal and do not perfuse blood adequately, leading to major increases in tumor hypoxia. The combined use of an anti-VEGF drug and a Dll4-targeting drug can be more effective than either drug used alone, and tumors that are resistant to an anti-VEGF drug can be treated with a Dll4-targeting drug. Thurston et al.47 describe more molecular details of Dll–notch receptor signaling and the domain structure of the receptor. VEGFR-2 denotes vascular endothelial growth factor receptor 2.
This finding may suggest that the notch–Dll4 signaling system is a major stimulator of angiogenesis and could be an appealing drug target, since Dll4 is up-regulated in the tumor vasculature,42 in part by VEGF.44 Paradoxically, drugs that target Dll4, including neutralizing antibodies, actually increase tumor angiogenesis, but most of the newly formed blood vessels are abnormal and functionally compromised in ways that drastically reduce blood flow. As a result, tumor hypoxia is increased by up to seven times the normal level,43 thereby retarding tumor growth. Apparently, angiogenesis induced by VEGF up-regulates Dll4 in the endothelial cells of developing blood vessels and in doing so allows Dll4 to act as a negative feedback mechanism to prevent an excess of chronic, functional angiogenesis.44,47
Another mechanism that inhibits excessive angiogenesis involves vasohibin, a protein in activated endothelial cells that is induced by stimulators of angiogenesis such as VEGF.50 Vasohibin is thus a specialized member of a large and growing number of endogenous angiogenesis inhibitors such as angiostatin, endostatin, thrombospondin-1, and tumstatin.2,51
The therapeutic potential of anti-Dll4 drugs is an issue of considerable interest. The question of whether they need to be combined with chemotherapy, radiation therapy, or other antiangiogenic drugs is important, because a therapy that induces such profound vessel dysfunction in tumors and elevated levels of tumor hypoxia could be counterproductive when added to radiation therapy or chemotherapy. On the other hand, the increase in proliferating endothelial cells induced by anti-Dll4 drugs could result in a more potent vascular targeting effect mediated by chemotherapy and perhaps radiation therapy.
Angiogenesis and Circulating Bone Marrow–Derived Cells
Many cell types can be mobilized from the bone marrow and can home to sites of new blood-vessel formation, where they amplify the angiogenic process52 (Fig. 3). These cells include various hematopoietic-cell (CD45+) populations, many of which are monocytic or myeloid cells that express such endothelial-cell markers as VE-cadherin, VEGFR-1, VEGFR-2, and tie-2.53-57 They also express chemokine receptors such as CXC chemokine receptor 4 (CXCR4), which binds stromal-cell–derived factor 1 (SDF-1, also called CXCL12), a chemokine that attracts lymphocytes and certain other cell types. Neutrophils58 and macrophages59 can also have proangiogenic properties. In addition, there is a nonhematopoietic (CD45−) bone marrow cell population, the circulating endothelial progenitor cells. In contrast to perivascular cells, which function through paracrine mechanisms such as local secretion of VEGF,57 circulating endothelial progenitors are thought to merge with the wall of a growing blood vessel, where they differentiate into endothelial cells60 (Fig. 3).
Figure 3. Circulating Bone Marrow-Derived Cell Populations That Stimulate or Amplify Tumor Angiogenesis.
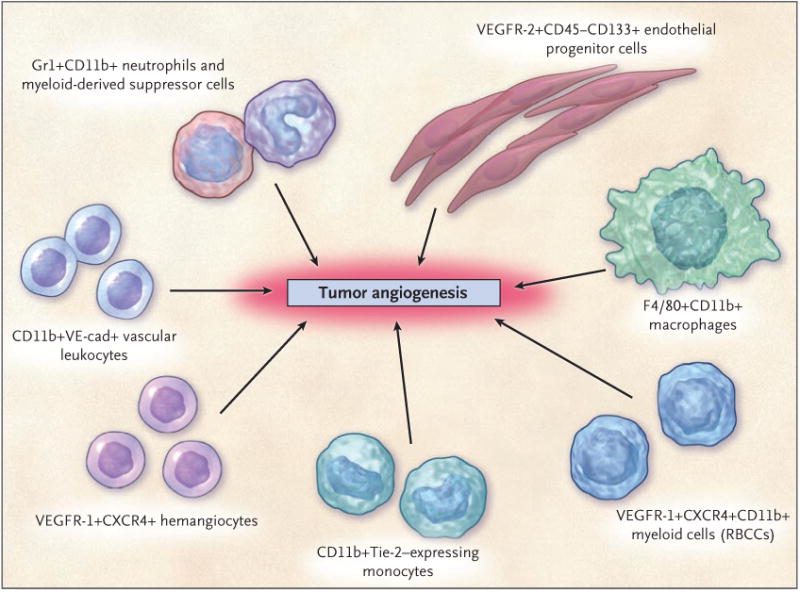
The various hematopoietic (CD45+) cell types appear to have a perivascular location with respect to the tumor neovasculature, whereas the CD45− endothelial progenitor cells can become incorporated into the lumen of a growing blood vessel and differentiate into mature endothelial cells. In recent preclinical studies, neutrophils have also been shown to contribute to the induction of tumor angiogenesis.53 F4/80 is a pan macrophage cell-surface marker. CXCR4 denotes CXC chemokine receptor 4, RBCCs recruited bone marrow–derived circulating cells, VE-cad vascular endothelial-cell cadherin (an adhesion molecule), and VEGFR vascular endothelial growth factor receptor. (Adapted from a figure provided by Dr. Michele dePalma, San Raffaele Scientific Institute, Milan.)
The study of circulating endothelial progenitors has generated considerable interest and controversy.52 The various cell-surface markers used to describe such cells (which can be expressed by other cell types) and the methods used to detect them probably contribute to the widely divergent reports of the levels of incorporation of these cells into new blood vessels52; these levels range from highs of 20 to 50% or more61,62 to lows of 5% or less,52,63 the lower levels being more common. These differences have cast doubt on the general role of circulating endothelial progenitors in tumor angiogenesis, but in virtually all studies of these cells in tumors, untreated tumors were analyzed, or, as in one clinical study, the treatment was completed long before the tumors were analyzed.63 In contrast, in preclinical studies, acute and marked mobilization of the progenitors from the bone marrow has been shown to occur immediately after treatment with vascular disrupting agents (VDAs).64 These potent drugs, mostly microtubule inhibitors, can rapidly shut down the abnormal tumor vasculature, thereby causing massive tumor hypoxia and necrosis.65,66 Invariably, a viable rim of tumor tissue remains.65,66 Within hours after VDA treatment, the number of circulating endothelial progenitors increases, and they invade and colonize the viable rim of the tumor, thereby contributing to the rapid regrowth of the tumor.64 However, this process can be blocked by an anti–VEGFR-2 antibody.64 In addition to their role in VDA treatment, the contribution of circulating endothelial progenitors may be critical during the very early stages of the development of a tumor, but the population of these cells in the vasculature is progressively diluted by differentiated endothelial cells.67 Furthermore, the incorporation of relatively few endothelial progenitor cells (e.g., approximately 12%) into new blood vessels can have major promoting effects on functional tumor growth such as the progression of microscopic metastatic lesions to macroscopic ones.68
The contribution of circulating bone marrow–derived cells to angiogenesis has a number of clinical implications. For example, a chemotherapy drug such as cyclophosphamide, administered at maximum tolerated doses, can mobilize circulating endothelial progenitors,69,70 which could conceivably contribute to regrowth of the tumor. Conversely, the closely spaced, regular administration of relatively less toxic doses of chemotherapy drugs, with no prolonged breaks — metronomic chemotherapy71 — can prevent mobilization of circulating endothelial progenitors and can act against not only the endothelial progenitors72 but also differentiated endothelial cells in the tumor neovasculature.73,74 Several clinical trials of metronomic chemotherapy are ongoing (Appendix 2 of the Supplementary Appendix).75
The use of hematopoietic growth factors in patients with cancer who receive high-dose or dose-dense chemotherapy merits consideration here because recombinant granulocyte colony-stimulating factor (G-CSF) can mobilize not only endothelial-cell progenitors,76 but also CD11b+ granulocyte differentiation antigen (Gr1)+ myeloid suppressor cells that can promote angiogenesis77,78 (Fig. 3). In preclinical studies, the number of mobilized progenitors can be used as a surrogate pharmacodynamic marker to help establish the optimal dose of metronomic chemotherapy,72,79 and enumeration of apoptotic circulating endothelial cells has been used to predict antiangiogenic drug activity induced by metronomic chemotherapy.80 However, these methods remain to be validated in larger prospective clinical studies. Finally, since many of the cells shown in Figure 3 express VEGFR-1 and CXCR4, there are prospects for using drugs that target these receptors to inhibit angiogenesis mediated by relevant bone marrow–derived cells.81,82
Resistance to Antiangiogenic Drugs
Intrinsic resistance and acquired resistance to antiangiogenic drugs are clinically significant problems. Preclinical studies have begun to shed light on the mechanisms of such resistance.83 With respect to intrinsic resistance, it has been shown that a tumor cell line from mice that is resistant to anti-VEGF antibodies becomes colonized by bone marrow–derived cells (CD11b+Gr1+ myeloid-suppressor cells) when transplanted into mice.84 Moreover, when normally sensitive tumor cells are mixed with these cells that are resistant to anti-VEGF antibodies and transplanted into other mice, the transplanted tumors resist anti-VEGF antibodies.84 Intrinsic resistance can also occur as a result of tumor cells using existing blood vessels in vasculature-rich organs such as the lungs,85 or simply as a result of the absence of VEGF or VEGF receptors in metastatic tumors growing in certain organ sites when drugs that target the VEGF pathway are used.86
Acquired resistance to anti–VEGFR -2 antibodies can be caused by the redundancy of angiogenesis stimulators. An example is the up-regulation of the angiogenesis stimulator basic fibroblast growth factor (bFGF) within the tumor after treatment with anti–VEGFR-2 antibody therapy; this effect is probably caused by the elevated levels of hypoxia induced by the drug treatment.87 Clinically, treatment with bevacizumab increases circulating PlGF, which could cause drug resistance.88 In humans, sunitinib can induce high levels of circulating PlGF and VEGF that revert to normal levels during drug-free periods7; these results can be reproduced in mice, but the presence of a tumor is not necessary for the effect.36 G-CSF and SDF-1, which can mobilize circulating endothelial progenitors52,76 and possibly other proangiogenic accessory cells,57 are also up-regulated in healthy mice treated with sunitinib. Moreover, it is possible that two or more such induced growth factors89 could act in a synergistic manner to promote tumor angiogenesis, as has been shown for bFGF and an isoform of PDGF.89 Such effects could contribute to rapid vascular regrowth in tumors after discontinuation of certain antiangiogenic treatments.90
Acquired resistance can also develop through the selection and overgrowth of mouse tumor cells with mutations in genes such as Tp53, which cause relative resistance to hypoxia.91 These variants may be less dependent on the oxygen supplied by the newly formed blood vessels than are tumor cells without the mutations.91 Rapid vascular remodeling of tumor-associated vessels as a consequence of antiangiogenic therapy is another cause of resistance.92 The mature remodeled vessels are resistant to antiangiogenic drugs, which usually target relatively immature vessels.93 These aforementioned mechanisms suggest various possible strategies to delay or possibly even reverse acquired resistance.87
Angiogenesis and Cancer Stem Cells
Studies have identified in tumors a minor population of cells with the characteristics of “tumor-initiating” cancer stem cells.94-96 These cells are thought to drive tumor growth and to constitute the seeds of resistance to treatment. The cancer stem-cell hypothesis is based largely on the results of transplantation of selectively enriched populations of cells into immunodeficient mice. Transplantation of extremely small numbers of such putative human cancer stem cells results in a high rate of tumor “takes,” whereas transplantation of much larger numbers of cancer cells lacking stem-cell characteristics does not.94,95 It has been suggested that conventional chemotherapy and other types of drugs attack the latter cells but not the cancer stem cells.96 The potent tumorigenic properties of cancer stem cells suggest that they may be strongly proangiogenic, and there is some evidence of this feature,97 which may help to explain some of the effects of tumor-inhibiting antiangiogenic drugs.
In addition, putative cancer stem cells in brain tumors reside in close proximity to blood vessels in a “vascular niche.”98 Treatment of orthotopic transplanted gliomas in mice with antibodies to VEGF disrupts the vascular niche and targets the stem-cell population.98 This population expresses high levels of VEGF and thus would be expected to be sensitive to anti-VEGF treatment.98 Low-dose metronomic chemotherapy may target the population of cancer stem cells or cells that are like cancer stem cells,99 especially when it is combined with an antiangiogenic drug such as anti–VEGFR-2 antibodies.74
Summary
The increasing use of antiangiogenic drugs for the treatment of cancer has emerged from decades of extensive basic and clinical research. The clinical benefits of such drugs, however, are relatively modest. Improvements are likely to come from a more thorough understanding of the molecular and cellular mechanisms governing tumor angiogenesis and the response to antiangiogenic therapies. A number of recent advances promise to bring about such improvements. These include new findings in the VEGF and the VEGF-receptor family, discovery of the notch–Dll4 signaling pathway in tumor angiogenesis, elucidation of the proangiogenic role of circulating bone marrow–derived cells, identification of the mechanisms of resistance to antiangiogenic drugs, and observations that suggest a role of angiogenesis in the survival and growth of cancer stem cells.
Many of these discoveries and others suggest strategies for improving the clinical benefits of antiangiogenic therapy. These strategies include the development of better preclinical models to study the biology of tumor angiogenesis and antiangiogenic therapies (see Appendix 3 of the Supplementary Appendix). Such improvements will also be critical in the use of long-term antiangiogenic therapy in the adjuvant setting in patients with early-stage disease. With respect to the treatment of metastatic disease, the magnitude and diversity of targets for antiangiogenic approaches100 suggest numerous possibilities for antiangiogenic drug combinations that should be much more effective than monotherapy in treating cancer.
Supplementary Material
Acknowledgments
Supported by grants from the National Cancer Institute of Canada, the Canadian Institutes of Health Research, the Ontario Institute for Cancer Research, and the National Institutes of Health, and by a Tier 1 Canada research chair in tumor biology, angiogenesis, and antiangiogenic therapy.
Dr. Kerbel reports serving as a consultant for ImClone Systems and Taiho Pharmaceuticals, as a scientific advisory board member for Attenuon, Adnexus Therapeutics, and Oxigene, and on speakers bureaus or ad hoc advisory boards for Pfizer, Roche, AstraZeneca, Novartis, Genentech, and Bristol-Myers Squibb; receiving grant support from ImClone Systems, Taiho Pharmaceuticals, and Oxigene; and having equity interests in Adnexus Therapeutics. No other potential conflict of interest relevant to this article was reported.
This article is dedicated to the memory of Dr. Judah Folkman, mentor, friend, and colleague, whose visionary hypothesis and numerous pioneering discoveries opened up and advanced the field of tumor angiogenesis and antiangiogenic therapy.
I thank Cassandra Cheng for excellent secretarial assistance and colleagues in the Kerbel laboratory for a critical review of an earlier version of the manuscript.
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Idem. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 4.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 5.Sandler A, Gray R, Perry MC, et al. Paclitaxel–carboplatin alone or with bevacizumab for non–small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. Erratum, N Engl J Med 2007;356:318. [DOI] [PubMed] [Google Scholar]
- 6.Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–45. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 7.Motzer RJ, Michaelson MD, Redman BG, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 8.Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–34. doi: 10.1056/NEJMoa060655. Erratum, N Engl J Med 2007;357: 203. [DOI] [PubMed] [Google Scholar]
- 9.Llovet J, Ricci S, Mazzaferro V, et al. Randomized phase III trial of sorafenib versus placebo in patients with advanced hepatocellular carcinoma (HCC) Proc Am Soc Clin Oncol. 2007;25(1):18S. abstract. [Google Scholar]
- 10.Berenson A. A cancer drug shows promise, at a price that many can't pay. New York Times; Feb 15, 2006. [PubMed] [Google Scholar]
- 11.Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors: a review. Eur J Cancer. 2006;42:3127–39. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 12.Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007;7:475–85. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 14.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312:1171–5. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
- 15.Blagosklonny MV. How Avastin potentiates chemotherapeutic drugs: action and reaction in antiangiogenic therapy. Cancer Biol Ther. 2005;4:1307–10. doi: 10.4161/cbt.4.12.2315. [DOI] [PubMed] [Google Scholar]
- 16.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 17.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 18.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 19.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–60. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–39. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 21.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 22.Kut C, Mac GF, Popel AS. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br J Cancer. 2007;97:978–85. doi: 10.1038/sj.bjc.6603923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukumura D, Xavier R, Sugiura T, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–25. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 24.Liang WC, Wu X, Peale FV, et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem. 2006;281:951–61. doi: 10.1074/jbc.M508199200. [DOI] [PubMed] [Google Scholar]
- 25.Jubb AM, Oates AJ, Holden S, Koeppen H. Predicting benefit from anti-angiogenic agents in malignancy. Nat Rev Cancer. 2006;6:626–35. doi: 10.1038/nrc1946. [DOI] [PubMed] [Google Scholar]
- 26.Kessler T, Fehrmann F, Bieker R, Berdel WE, Mesters RM. Vascular endothelial growth factor and its receptor as drug targets in hematological malignancies. Curr Drug Targets. 2007;8:257–68. doi: 10.2174/138945007779940089. [DOI] [PubMed] [Google Scholar]
- 27.Dong X, Han ZC, Yang R. Angiogenesis and antiangiogenic therapy in hematologic malignancies. Crit Rev Oncol Hematol. 2007;62:105–18. doi: 10.1016/j.critrevonc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Dallas NA, Fan F, Gray MJ, et al. Functional significance of vascular endothelial growth factor receptors on gastrointestinal cancer cells. Cancer Metastasis Rev. 2007;26:433–41. doi: 10.1007/s10555-007-9070-2. [DOI] [PubMed] [Google Scholar]
- 29.Lee TH, Seng S, Sekine M, et al. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med. 2007;4(6):e186. doi: 10.1371/journal.pmed.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerber HP, Malik AK, Solar GP, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002;417:954–8. doi: 10.1038/nature00821. [DOI] [PubMed] [Google Scholar]
- 31.Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellis LM. The role of neuropilins in cancer. Mol Cancer Ther. 2006;5:1099–107. doi: 10.1158/1535-7163.MCT-05-0538. [DOI] [PubMed] [Google Scholar]
- 33.Bielenberg DR, Klagsbrun M. Targeting endothelial and tumor cells with semaphorins. Cancer Metastasis Rev. 2007;26:421–31. doi: 10.1007/s10555-007-9097-4. [DOI] [PubMed] [Google Scholar]
- 34.Pan Q, Chanthery Y, Liang WC, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Fischer C, Jonckx B, Mazzone M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–75. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 36.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with anti-tumor efficacy. Proc Natl Acad Sci U S A. 2007;104:17069–74. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bocci G, Man SM, Green SK, et al. Increased plasma vascular endothelial growth factor (VEGF) as a surrogate marker for optimal therapeutic dosing of VEGF receptor-2 monoclonal antibodies. Cancer Res. 2004;64:6616–25. doi: 10.1158/0008-5472.CAN-04-0401. [DOI] [PubMed] [Google Scholar]
- 38.Rudge JS, Holash J, Hylton D, et al. VEGF Trap complex formation measures production rates of VEGF, providing a biomarker for predicting efficacious angiogenic blockade. Proc Natl Acad Sci U S A. 2007;104:18363–70. doi: 10.1073/pnas.0708865104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D. Signaling vascular morphogenesis and maintenance. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- 40.Oliner J, Min H, Leal J, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004;6:507–16. doi: 10.1016/j.ccr.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 41.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–95. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sainson RC, Harris AL. Anti-Dll4 therapy: can we block tumour growth by increasing angiogenesis? Trends Mol Med. 2007;13:389–95. doi: 10.1016/j.molmed.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 43.Noguera-Troise I, Daly C, Papadopoulos NJ, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–7. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 44.Lobov IB, Renard RA, Papadopoulos N, et al. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A. 2007;104:3219–24. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ridgway J, Zhang G, Wu Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–7. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- 46.Gale NW, Dominguez MG, Noguera I, et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–54. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7:327–31. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- 48.Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 49.Ferrara N, Carver-Moore K, Chen H, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 50.Watanabe K, Hasegawa Y, Yamashita H, et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J Clin Invest. 2004;114:898–907. doi: 10.1172/JCI21152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–79. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 52.Bertolini F, Shaked Y, Mancuso P, Kerbel RS. The multifaceted circulating endothelial cell in cancer: from promiscuity to surrogate marker and target identification. Nat Rev Cancer. 2006;6:835–45. doi: 10.1038/nrc1971. [DOI] [PubMed] [Google Scholar]
- 53.Okazaki T, Ebihara S, Asada M, Kanda A, Sasaki H, Yamaya M. Granulocyte colony-stimulating factor promotes tumor angiogenesis via increasing circulating endothelial progenitor cells and Gr1+CD11b+ cells in cancer animal models. Int Immunol. 2006;18:1–9. doi: 10.1093/intimm/dxh334. [DOI] [PubMed] [Google Scholar]
- 54.De Palma M, Venneri MA, Galli R, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–26. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 55.Venneri MA, De Palma M, Ponzoni M, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109:5276–85. doi: 10.1182/blood-2006-10-053504. [DOI] [PubMed] [Google Scholar]
- 56.Conejo-Garcia JR, Buckanovich RJ, Benencia F, et al. Vascular leukocytes contribute to tumor vascularization. Blood. 2005;105:679–81. doi: 10.1182/blood-2004-05-1906. [DOI] [PubMed] [Google Scholar]
- 57.Grunewald M, Avraham I, Dor Y, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–89. doi: 10.1016/j.cell.2005.10.036. Erratum, Cell 2006;126:811. [DOI] [PubMed] [Google Scholar]
- 58.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–8. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67:5064–6. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- 60.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 61.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–9. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 62.Spring H, Schüler T, Arnold B, Hämmerling GJ, Ganss R. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci U S A. 2005;102:18111–6. doi: 10.1073/pnas.0507158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peters BA, Diaz LA, Polyak K, et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11:261–2. doi: 10.1038/nm1200. [DOI] [PubMed] [Google Scholar]
- 64.Shaked Y, Ciarrocchi A, Franco M, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313:1785–7. doi: 10.1126/science.1127592. [DOI] [PubMed] [Google Scholar]
- 65.Siemann DW, Chaplin DJ, Horsman MR. Vascular-targeting therapies for treatment of malignant disease. Cancer. 2004;100:2491–9. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]
- 66.Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–35. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- 67.Nolan DJ, Ciarrocchi A, Mellick AS, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21:1546–58. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–8. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 69.Bertolini F, Paul S, Mancuso P, et al. Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res. 2003;63:4342–6. [PubMed] [Google Scholar]
- 70.Fürstenberger G, von Moos R, Lucas R, et al. Circulating endothelial cells and angiogenic serum factors during neoadjuvant chemotherapy of primary breast cancer. Br J Cancer. 2006;94:524–31. doi: 10.1038/sj.bjc.6602952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4:423–36. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- 72.Shaked Y, Emmenegger U, Man S, et al. Optimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activity. Blood. 2005;106:3058–61. doi: 10.1182/blood-2005-04-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Browder T, Butterfield CE, Kräling BM, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–86. [PubMed] [Google Scholar]
- 74.Klement G, Baruchel S, Rak J, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24. doi: 10.1172/JCI8829. Errata, J Clin Invest 2006;116: 2827, 3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garcia AA, Hirte H, Fleming G, et al. Phase II clinical trial of bevacizumab and low dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital Phase II Consortia. J Clin Oncol. 2008;26:76–82. doi: 10.1200/JCO.2007.12.1939. [DOI] [PubMed] [Google Scholar]
- 76.Natori T, Sata M, Washida M, Hirata Y, Nagai R, Makuuchi M. G-CSF stimulates angiogenesis and promotes tumor growth: potential contribution of bone marrow-derived endothelial progenitor cells. Biochem Biophys Res Commun. 2002;297:1058–61. doi: 10.1016/s0006-291x(02)02335-5. [DOI] [PubMed] [Google Scholar]
- 77.Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 78.Shojaei F, Wu X, Zhong C, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–31. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 79.Shaked Y, Bertolini F, Man S, et al. Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis: implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell. 2005;7:101–11. doi: 10.1016/j.ccr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 80.Mancuso P, Colleoni M, Calleri A, et al. Circulating endothelial cell kinetics and viability predict survival in breast cancer patients receiving metronomic chemotherapy. Blood. 2006;108:452–9. doi: 10.1182/blood-2005-11-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28:299–307. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin DK, Shido K, Kopp HG, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–67. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller KD, Sweeney CJ, Sledge GW., Jr The Snark is a Boojum: the continuing problem of drug resistance in the antiangiogenic era. Ann Oncol. 2003;14:20–8. doi: 10.1093/annonc/mdg033. [DOI] [PubMed] [Google Scholar]
- 84.Shojaei F, Wu X, Malik AK, et al. Tumor resistance to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 85.Leenders WP, Küsters B, Verrijp K, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004;10:6222–30. doi: 10.1158/1078-0432.CCR-04-0823. [DOI] [PubMed] [Google Scholar]
- 86.Karashima T, Inoue K, Fukata S, et al. Blockade of the vascular endothelial growth factor-receptor 2 pathway inhibits the growth of human renal cell carcinoma, RBM1-IT4, in the kidney but not in the bone of nude mice. Int J Oncol. 2007;30:937–45. [PubMed] [Google Scholar]
- 87.Casanovas O, Hicklin D, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 88.Willett CG, Boucher Y, Duda DG, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136–9. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- 89.Nissen LJ, Cao R, Hedlund EM, et al. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest. 2007;117:2766–77. doi: 10.1172/JCI32479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mancuso MR, Davis R, Norberg SM, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–21. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002;295:1526–8. doi: 10.1126/science.1068327. [DOI] [PubMed] [Google Scholar]
- 92.Glade Bender J, Cooney EM, Kandel JJ, Yamashiro DJ. Vascular remodeling and clinical resistance to antiangiogenic cancer therapy. Drug Resist Updat. 2004;7:289–300. doi: 10.1016/j.drup.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 93.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–65. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 95.Al Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. Erratum, Proc Natl Acad Sci U S A 2003;100:6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marx J. Mutant stem cells may seed cancer. Science. 2003;301:1308–10. doi: 10.1126/science.301.5638.1308. [DOI] [PubMed] [Google Scholar]
- 97.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–8. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 98.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 99.Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560–4. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- 100.Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007;11:539–54. doi: 10.1016/j.ccr.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.