

Abstract
Synthetic efforts toward the rapid assembly of the AB ring system of the tetrapetalones is described. Key to this work was the use of [3+2] cycloaddition/oxidative extrusion methodology to furnish functionalized aryl enones. The Nazarov cyclization of these substrates was examined, and optimized to generate the AB ring carbon skeleton. Then, Pd-catalyzed cross-coupling were conducted, and conditions were identified that enabled installation of the requisite C14-N bond.
Keywords: Cross-coupling, Cyclization, Natural Products, Polycycles
Graphical Abstract

1. Introduction
In 2003, Hirota and co-workers isolated tetrapetalone A (1, Figure 1) from a Streptomyces culture broth, and this natural product was found to inhibit soybean lipoxygenase (SLO).1,2 Tetrapetalones B (2), C (3), and D (4) were also isolated from a Streptomyces culture extract in a separate report, and these natural products were also found to inhibit SLO.3 Since the catalytic domain of plant and mammalian lipoxygenases is highly conserved,4 the study of an SLO inhibitor like tetrapetalone A could lead to the identification of new human lipoxygenase (HLO) inhibitors. HLOs catalyze the first step in the conversion of arachidonic acid to the leukotrienes, which are signaling molecules implicated in several inflammatory diseases.5
Figure 1.

Structures of tetrapetalones A–D, derivatives, and ansaetherone.
The SLO inhibition of the tetrapetalones was moderate, but comparable to known SLO inhibitors kojic acid and nordihydroguaiaretic acid (NDGA) (Table 1).3,6 However, when tetrapetalone A was derivatized to tetrapetalone A-Me (5) via treatment of 1 with diazomethane, all SLO inhibitory activity was eliminated (entry 5)6. 5 was subjected to acidic hydrolysis to furnish tetrapetalone A-Me aglycon (6), and the absolute configuration of this material was determined by the preparation of Mosher esters.6 Hirota and co-workers also discovered the radical scavenger ansaetherone (7) present in a Streptomyces culture extract, and theorized that it could be an intermediate in the biosynthesis of the tetrapetalones.7
Table 1.
Inhibitory activities of 1–5 against soybean lipoxygenase
| Compound | IC50 (µM) |
|---|---|
| 1 | 190–460 |
| 2 | 320 |
| 3 | 360 |
| 4 | 240 |
| 5 | >1000 |
| Kojic acid (positive control) |
110–280 |
| NDGA (positive control) |
190–290 |
The tetrapetalone natural products possess a β-D-rhodinose moiety connected to a tetracyclic core with unprecedented complexity. Key features of this core include a para-quinol A ring, a five-membered B ring containing four contiguous stereogenic centers, a 7-membered C ring containing a bridging trisubstituted olefin, and a tetramic acid D ring. Furthermore, a study whereby Streptomyces were fed 13C-enriched building blocks revealed that the rhodinose moiety is derived from glucose, the A ring is derived from 3-amino-5-hydroxybenzoic acid, and the remainder of the molecule is constructed from propanoate and butanoate building blocks, probably by the action of polyketide synthase8.
To date, a total synthesis of tetrapetalone A has not been reported, though several laboratories have published synthetic approaches. The first report appeared in 2005 by Porco and coworkers, who sought to test a biosynthetic hypothesis: the tetracyclic core was formed through an oxidative transannular [4+3] cyclization of an ansa-bridged macrocycle.9a While the group initially reported the success of this strategy, a later disclosure indicated that while oxidation had been successful, the cycloaddition had not.9b In 2010, Sarpong and co-workers published an approach that culminated in the first synthesis of the tetracyclic skeleton of the tetrapetalone system.10 In brief, they constructed the AB ring system via Nazarov cyclization, ABD ring system via Paal-Knorr methodology, and the ABCD ring system via an oxidation/Friedel-Crafts acylation/oxidation cascade10. Two other approaches toward the tetrapetalone core have been published by Hong (2009)11 and Pettus (2014).12
In 2014, our group reported the first synthesis of tetrapetalone A-Me aglycon (6).13 Herein is an account of our early work on the project, detailing Nazarov cyclization studies targeting the indanone precursor to the AB ring system, and the installation of the C14-N bond of the A ring.
2. Results/Discussion
From the outset, we planned to construct the AB ring system using a polarized Nazarov cyclization on a substrate resembling aryl enone 8, to form bicycle 9 (Scheme 1).14 This strategy would allow us to convert the aromatic A ring into a para-quinol in a late-stage transformation, and the β-ketoester to a cyclopentane with the requisite C9 alkoxy group and C8 methyl group. Based on our earlier work on cyclizations of polarized aromatic systems,15 and Sarpong’s study of a closely related cyclization,16 we expected the cyclization to be possible, but perhaps difficult to achieve catalytically, and certainly difficult to achieve enantioselectively.
Scheme 1.

Outline of the polarized Nazarov cyclization in the context of AB ring construction
Over the course of our investigations on the tetrapetalone ring system, we prepared and cyclized a number of different aryl vinyl ketones. Aryl vinyl ketone 13 was easily prepared utilizing a Knovenagel condensation protocol previously reported in our group (Scheme 2).14 However, Knoevenagel condensation of α-methyl-trans-cinnamaldehyde with β-ketoester 12 failed.
Scheme 2.

Aryl enone synthesis via Knoevenagel condensation
We solved this problem by employing a different method previously explored in our group for the mild construction of aryl enones.14b,17 As shown in Scheme 3, alkynes of type 14 and nitrones of type 15 undergo [3+2] cycloaddition followed by oxidative chelotropic extrusion of nitrosomethane to furnish aryl enones of type 16. The results are shown in Table 2. For example, the protocol furnished aryl enone 16a, containing the requisite methyl group on C6, from alkyne 14a and nitrone 15a (entry 1). This substrate could not be prepared using the Knoevenagel condensation protocol in Scheme 2. The olefin geometry of aryl enone 16a was determined to be predominantly Z (>19:1 ratio), by examining the 3J coupling constants between the ketone carbon and the ester carbon with the C7 proton, which were obtained using selective proton-decoupled carbon NMR techniques.18 In an attempt to prepare the aryl enone unsubstituted at the alkene terminus, which we thought would be optimal for subsequent C ring formation, nitrone 15b was tested in the cycloaddition, but it decomposed under the elevated thermal conditions. Fortunately, nitrone 15c, featuring a vinyl trimethylsilyl group, underwent smooth cycloaddition/extrusion (entry 3). The trimethyl silyl group stabilizes the nitrone and serves as a protecting group for the desired 1,1-disubstituted alkene, as it is readily removed under acidic conditions (vide infra).
Scheme 3.

[3+2] cycloaddition/oxidative extrusion cascade for preparation of functionalized aryl enones
Table 2.
Synthesis of aryl enones 16a–e via [3+2] cycloaddition/oxidative extrusion cascade
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yield | dr |
| 1 | OTIPS (14a) | Ph (15a) | 80% (16a) | >19:1 (Z:E) |
| 2 | OTIPS (14a) | H(15b) | -- | -- |
| 3 | OTIPS (14a) | TMS(15c) | --a(16b) | 5:1 |
| 4 | Br(14b) | Ph(15a) | 85% (16c) | >20:1 |
| 5 | Br(14b) | TMS(15c) | 79% (16d) | 1.7:1 |
| 6 | Br(14b) | TBS(15d) | 77% (16e) | 7.1:1 |
See Experimental section
We also prepared aryl bromides 16c–e (entries 4–6) using the [3+2] cycloaddition/ cheletropic extrusion sequence. As in entries 1 and 3, nitrones 15a and 15c underwent the reaction cascade smoothly (entries 4 and 5). The more robust vinyl tert-butyldimethylsilane was also prepared, as alternative protection for the 1,1 disubstituted alkene (entry 6).
With aryl dienones 13 and 16a–e in hand, we could evaluate conditions for Nazarov cyclization (Table 3). Nazarov cyclization of 13 with catalytic Cu(OTf)2 gave the trans cyclopentanone as the only detectable product in 81% yield (entry 1). A similar result was obtained with aryl enone 16a using catalytic Cu(ClO4)2 (entry 2). When aryl enone 16b was subjected to Cu2+ catalysis, cleavage of the TMS group occurred, presumably due to trace Bronsted acid present in the reaction mixure (entry 3)19. This convenient desilylation facilitated the ring-closing metathesis studies that led to the formation of the C ring13. Indanone 18b was isolated (in crude form) as a 3:1 mixture of trans:cis isomers (see experimental section).
Table 3.
Synthesis of indanones via Nazarov cyclization
| # | Starting material | Conditions | Product | Yield; dr (trans:cis) |
|---|---|---|---|---|
| 1 |  |
5 mol % Cu(OTf)2, DCE, 60 °C |
 |
81%; >20:1 |
| 2 |  |
5 mol % Cu(ClO4)2, DCE, 45 °C |
 |
75%; >20:1 |
| 3 |  |
5 mol % Cu(ClO4)2, DCE, rt |
 |
--a; 3:1 |
| 4 |  |
cat. Sc(OTf)3/ Li(ClO4), DCE, 45 °C |
 |
81%; 3:1 |
| 5 |  |
cat. Sc(OTf)3/ Li(ClO4), DCE, 45 °C |
 |
-b |
| 6 | 16d | (TMS)NTf2 | 18d | -b |
| 7 | 16d | 1.1 eq. AlCl3, DCE, 0 °C |
18d | 71%; 3.6:1 |
| 8 | 16d | 1.1 eq. Sc(OTf)3, DCE, rt |
18d | 46%; 3.6:1 |
| 9 |  |
1.1 eq. AlCl3, DCE, 0 °C |
18d | ~70–75%; 3.6:1 |
See Experimental section
Decomposition
The inclusion of the electron-withdrawing bromide group on aryl enone substrates predictably reduced the reactivity of these substrates in the Nazarov cyclization reaction. For example, treatment of 16c with catalytic Sc(OTf) at 45 °C resulted in no reaction. It was only by using the Sc(OTf)3/Li(ClO4) catalyst combination (which we had used previously to cyclize other unreactive aryl enones)15 that an 81% yield of 18c could be obtained from 16c, as a 3:1 mixture of trans:cis isomers (entry 4). Surprisingly, subjection of 16d to these conditions, or to (TMS)NTf2,20 resulted in decomposition (entries 5 and 6). However, aryl enone 16d did cyclize upon treatment with either superstoichiometric AlCl3[16] (entry 7) or Sc(OTf)3 (entry 8), furnishing indanone 18d. As in entry 3, protodesilylation occurred under the reaction conditions.
We were curious to see whether the protodesilylation was contributing to the decomposition seen in the experiments in entries 5 and 6. Hence, we examined the Nazarov cyclization of aryl enone 16e, with the assumption that its vinyl tert-butyldimethylsilyl group would not be cleaved as readily as a vinyl trimethylsilyl group. Unfortunately, attempts to catalyze the cyclization of 16e with Sc(OTf)3/Li(ClO4), Cu(OTf)2/Li(ClO4), Al(OTf)3, and (TMS)NTf2 all resulted in decomposition. As in entry 7, superstoichiometric AlCl3 promoted the cyclization, affording indanone 18d. Thus, appending the bulkier TBS group on the vinyl terminus in place of the more labile TMS group did not appear to have any effect on the efficiency of the cyclization.
As shown in Scheme 4, indanone 18a was functionalized (at C8) and converted to the aryl triflate in preparation for the anticipated installation of the C17-N bond via Buchwald-Hartwig cross-coupling. Thus, 18a was decarbalkoxylated,21 and methylated to furnish indanone 19 (Scheme 4). This material was then globally deprotected with TBAF, chemoselectively protected at the less hindered hydroxyl group, and then treated with triflic anhydride and pyridine to furnish aryl triflate 20.
Scheme 4.

Transformation of 18a to aryl triflate 20
Aryl triflate 21 was synthesized following the reactions in Scheme 4, except the C8 methyl group was not installed. This material was then deprotected with TBAF and methylated to furnish aryl triflate 22 (Scheme 5).
Scheme 5.

Transformation of aryl triflate 21 to aryl triflate 22
As in Scheme 4, indanone 18c was subjected to decarbalkoxylation to furnish aryl bromide 23. Aryl bromide 25 was prepared in a similar fashion (Scheme 6). Indanone 18d was decarbalkoxylated, methylated, and reduced to furnish alcohol 24, which was protected as the triisopropylsilyl ether. Importantly, both the C8 methylation and the reduction in Scheme 6 were found to be diastereoselective, and the relative stereochemistry on the B ring was confirmed by single-crystal X-ray crystallography (Figure 2). This relative stereochemistry also matched the desired stereochemistry present in the B ring of tetrapetalone A.
Scheme 6.

Transformation of indanone 18d to aryl bromide 25
Figure 2.

ORTEP Diagram of Alcohol 24
We originally planned to install the requisite C14-N bond using the powerful and versatile Buchwald-Hartwig cross-coupling technology (Table 4).22 Initially, we tried the cross-coupling of aryl triflate 20 with lactam 26 (entry 1),23 with no success. We next attempted cross-coupling with tertiary amine 27, with a substitution pattern mapping onto C4 of tetrapetalone A. Our initial experiment again resulted in no reaction (entry 2), but use of the stronger base NaOtBu resulted in detectable amounts of cross-coupled products (entry 3). It was clear that the poor results of these initial studies were due to the considerable steric hindrance of both coupling partners: the ortho-substituted aryl triflate and the tertiary amine.
Table 4.
C14-N bond formation studies
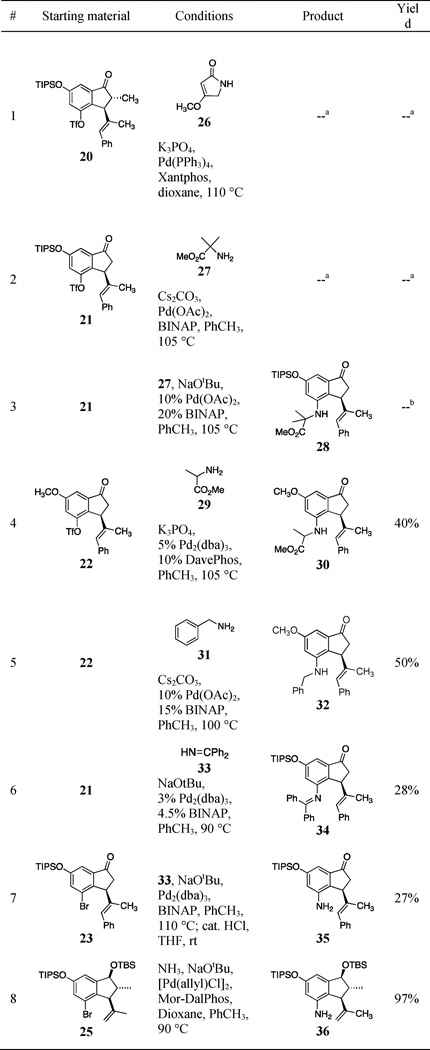 |
No reaction
Trace amount of product detected
To test this hypothesis, we attempted cross-coupling between aryl triflate 22 (containing the more robust methyl ether protecting group on C12) and the less sterically hindered amines 29 and 31 (entries 4 and 5). The result was a dramatic increase in the yield of the reaction. We also attempted the cross-coupling of the imine 3324 with aryl triflate 21, which delivered the imine derivative 34 in modest yield (entry 6). Imine 34 could be hydrolyzed to the aniline (35) in 56% yield using aqueous acid.
In many instances, phenol byproducts arising from hydrolysis of the triflyl group were detected in the reactions in entries 1–6. To avoid this, we prepared aryl bromides to test in the cross-coupling reactions. First, we attempted the cross coupling of sterically hindered amine 27 with aryl bromide 23. Unsurprisingly, no cross-coupled product was detected. We returned to the cross-coupling of imine 33, which furnished aniline 35 in 27% yield after hydrolysis (entry 7). The next coupling partner we investigated was ammonia, which has essentially no steric hindrance. Using the protocol of Stradiotto,25 we were able to cross-couple ammonia to aryl bromide 25 to furnish aniline 36 in 97% yield (entry 9). In a previous report, we successfully elaborated aniline 36 into tetrapetalone A-Me aglycon (6)13.
3. Conclusions
In summary, we have described our laboratory’s strategies toward the synthesis of the AB ring of the tetrapetalones. We used three key steps to construct the important architectural features of the AB ring system. We first used a [3+2] cycloaddition between strategically functionalized alkynoates and nitrones, followed by oxidative extrusion of nitrosomethane to furnish aryl enones. Then, we used Nazarov cyclization on these aryl enones to form the AB ring carbon skeleton. Last, we used Pd-catalyzed cross-coupling to install the requisite C14-N bond. Ongoing work in our laboratory is focused on the completion of the total synthesis of tetrapetalone A, as well as synthesis of other tetrapetalone natural products, and evaluation of their biological activity.
4. Experimental section
4.1. General
All reactions were carried out under an argon or N2 atmosphere in flame- or oven-dried glassware with magnetic stirring unless otherwise noted. Cold baths were prepared by mixing acetone/dry ice (−78 °C) or acetonitrile/dry ice (−40 °C). Reagents were used as obtained from commercial suppliers without further purification unless otherwise noted. Cu(ClO4)2 was dried in a vacuum oven at 40 °C for 24 h, then ground and stored in a glovebox prior to use. Tetrahydrofuran, diethyl ether, methylene chloride, 1,2-dichloroethane, and toluene were purchased from Fisher and dispensed using the Glass Contour solvent purification system. Celite 545 was purchased from EMD. ACS grade hexanes and ethyl acetate were used for column chromatography. Thin layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 glass-supported plates from EMD, and visualization was performed with a UV lamp followed by staining with potassium permanganate, p-anisaldehyde, ninhydrin, or ceric ammonium molybdate solution followed by heating. Column chromatography was carried out on EM Science 60 Å silica gel (230–400 mesh). Chloroform-d and Methanol-d4 was purchased from Cambridge Isotope Laboratories.
1H NMR spectra were recorded on a 400 MHz Bruker Avance spectrometer or a 500 MHz Bruker Avance spectrometer at room temperature. Chemical shifts are given in parts per million (ppm) from tetramethylsilane, referenced to solvent residual proton resonance (δ = 7.26 for CHCl3, δ = 3.31 for CH3OH). Some NMR spectra contain tetramethylsilane as an internal reference. NMR data are reported as: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, dq = doublet of quartets, br = broad), coupling constants (J) given in Hz, and integration. 13C NMR spectra were recorded on a 100 MHz Bruker Avance spectrometer or a 125 MHz Bruker Avance spectrometer at room temperature with proton decoupling. Chemical shifts are given in parts per million (ppm) from tetramethylsilane, referenced to solvent carbon resonance (δ = 77.16). Infrared spectra (IR) were recorded on a Shimadzu FTIR 8400S (ATR Mode) spectrometer or an ATI Mattson Genesis FT-IR spectrometer. High resolution mass spectra (HRMS) were measured by the Chemistry Instrumentation Center of the University of Buffalo, Buffalo, NY 14260 or the Mass Spectrometry Lab of the University of Illinois, Urbana, IL 61801. X-ray crystallography data were collected by the X-ray Crystallographic Facility of the University of Rochester, Rochester, NY 14627.
4.2. Preparation of Intermediates
β-Keto ester 12
In a 25 mL round-bottom flask, potassium hydride (30% in mineral oil, 273.0 mg, 2.0 mmol) and sodium hydride (60% in mineral oil, 324.4 mg, 8.1 mmol) were washed with dry hexanes, then THF (4 mL) was added, followed by dimethylcarbonate (0.7 mL, 8.1 mmol), and a solution of commercially-available 3’,5’-dimethoxy-acetophenone (730.7 mg, 4.1 mmol) in THF (5 mL). The reaction was stirred in a 65 C oil bath for 2 h, cooled, then quenched with water (20 mL). The layers were separated, and the aqueous layer was extracted with ether (3×10 mL). The organic phases were combined and dried over MgSO4, filtered, and concentrated. The crude material was purified by column chromatography (8:1 hexanes:ethyl acetate), yielding 619.7 mg (64%) of 12. 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 2.1 Hz, 1H), 6.65 (s, 1H), 3.95 (s, 2H), 3.80 (s, 6H), 3.73 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.16, 167.95, 161.03, 137.84, 106.34, 106.03, 55.66, 52.55, 45.85.
Aryl enone 13
In a 50 mL round-bottom flask, a solution of β-ketoester 12 (619.7 mg, 2.6 mmol) in benzene (12 mL) was combined with piperidine (30 µL, 0.3 mmol), acetic acid (90 µL, 1.6 mmol), and trans-cinnamaldehyde (0.4 mL, 2.9 mmol). The flask was fitted with a Dean-Stark trap, and refluxed for 16 h. The reaction was cooled, diluted with benzene, and washed with NH4Cl (50 mL), NaHCO3 (50 mL), and brine (25 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The crude material was purified by column chromatography (8:1 hexanes:ethyl acetate), yielding 677.3 mg (74%) of 13. Characterized as a mixture, E:Z =3:1, following the procedure of Ref. 18; E isomer: 1H NMR (400 MHz, CDCl3) δ 7.73 – 7.67 (m, 1H), 7.43 – 7.35 (m, 5H), 7.08 (d, J = 2.3 Hz, 2H), 7.02 (s, 1H), 6.76 (dd, J = 15.4, 11.8 Hz, 1H), 6.69 (t, J = 2.3 Hz, 1H), 3.84 (s, 6H), 3.74 (s, 3H). Z isomer: 1H NMR (400 MHz, CDCl3) δ 7.79 (dd, J = 15.5, 11.5 Hz, 1H), 7.57 (d, J = 1.8 Hz, 1H), 7.34 – 7.28 (m, 5H) 7.20 (d, J = 11.5 Hz, 1H), 6.96 (d, J = 2.3 Hz, 2H), 6.67 (t, J = 2.3 Hz, 1H), 3.84 (s, 6H), 3.77 (s, 3H).
Alkynoate 14a
Prepared according to Ref. 17. 1H NMR (400 MHz, CDCl3) δ 6.70 (d, J = 2.4 Hz, 2H), 6.52 (t, J = 2.4 Hz, 1H), 3.83 (s, 3H), 1.24 (m, 6H), 1.10 (d, J = 9.0 Hz, 36H); 13C NMR (100 MHz, CDCl3): δ 157.0, 154.5, 120.3, 117.5, 115.3, 86.6, 79.6, 52.7, 17.4, 12.6; IR (neat, cm−1): 2943.7, 2891.1, 2866.5, 2218.9, 1715.6, 1577.7, 1462.4, 1427.2, 1241.6.
Alkynoate 14b
Prepared according to Ref. 13. 1H NMR (400 MHz, CDCl3) δ 7.31 – 7.29 (m, 1H), 7.12 (t, J = 2.0 Hz, 1H), 7.01 – 6.97 (m, 1H), 3.83 (s, 3H), 1.31 – 1.18(m, 3H), 1.09 (d, J = 7.3 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 156.89, 154.26, 128.45, 126.25, 122.83, 122.67, 121.92, 84.63, 80.89, 53.04, 17.94, 12.69; IR (neat, cm−1): 2947, 2866, 2230, 1717, 1586, 1555, 1462, 1424, 1319, 1292, 1231, 1196, 1157, 976, 883, 853, 810, 733, 675, 648, 621; HRMS (ESI): Calculated for C19H28BrO3Si ([M+H]+): m/z 411.0991, found: 411.0990.
Nitrone 15a
Prepared according to Ref. 17. 1H NMR (400 MHz, CDCl3): δ 8.26 (s, 1H), 7.36 – 7.34 (m, 5H), 6.93 (s, 1H), 3.79 (s, 3H), 2.19 (s, 3H).
Nitrone 15b
Prepared according to Ref. 17. 1H NMR (400 MHz, CDCl3) δ 6.83 (s, 1H), 6.57 (s, 1H), 5.39 (s, 1H), 3.73 (s, 3H), 1.99 (s, 3H).
Nitrone 15c
Prepared according to Ref. 13. 1H NMR (500 MHz, CDCl3) δ 6.90 (s, 1H), 6.80 (s, 1H), 3.68 (s, 3H), 2.09 (s, 3H), 0.14 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 142.15, 138.07, 137.20, 54.46, 20.95, −0.20. IR (neat, cm−1): 2955, 2897, 1562, 1416, 1385, 1304, 1246, 1177, 1153, 1026, 961, 918, 833, 768, 748, 691, 644; HRMS (ESI): Calculated for C8H18NOSi ([M+H]+):m/z 172.1158, found: 172.1155.
Nitrone 15d
Prepared in analogy to 15c. 1H NMR (400 MHz, CDCl3) δ 6.87 (s, 1H), 6.86 (s, 1H), 3.71 (s, 3H), 2.14 (s, 3H), 0.91 (s, 9H), 0.13 (s, 6H).
Aryl enone 16a
In a dry 100 mL round-bottom flask was weighed the nitrone 15a (1.40 g, 8.0 mmol), 14a (3.6 g, 7.1 mmol), and toluene (35 mL), and the solution was heated at 80 °C for 16 h. The solution was concentrated, and CH2Cl2 (35 mL) was added, cooled to −20 °C, and m-CPBA (2.5 g, 11.6 mmol) was added and stirred for 30 min. The reaction was quenched with saturated Na2SO3 (50 mL), the layers were separated and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL), the organic phases were combined and washed with brine (50 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (8:1 hexanes:ethyl acetate), yielding 3.0 g (65%) of 16a. 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 0.8 Hz, 1H), 7.36 – 7.34 (m, 5H), 7.07 (d, J = 2.4 Hz, 2H), 6.98 (s, 1H), 6.65 (t, J = 2.0 Hz, 1H), 3.69 (s, 3H), 1.79 (s, 3H), 1.30 – 1.16 (m, 6H), 1.12 (d, J = 9.0 Hz, 36H); 13C NMR (100 MHz, CDCl3): δ 195.0, 165.8, 157.3, 147.8, 141.9, 138.9, 136.2, 133.3, 129.7, 129.2, 128.3, 128.0, 117.3, 113.7, 52.3, 17.8, 12.5; IR (neat, cm−1 ): 3023.7, 2944.7, 2892.3, 1724.2, 1713.6, 1678.6, 1672.0, 1587.8, 1442.8, 1333.8, 1249.2, 1197.6; HRMS (EI) calculated for C38H58O5Si2 650.3817, found 650.3817. Z, 3JC-H = 4.2 Hz for Cketone-H, 3JC-H= 7.5 Hz for Cester-H.
Aryl enone 16b
In a dry 100 mL round-bottom flask was weighed the nitrone 15c (0.33 g, 1.94 mmol), 14a (0.5 g, 1 mmol), and toluene (5 mL), and the solution was heated at 80 °C for 16 h. The solution was concentrated, and CH2Cl2 (35 mL) was added, cooled to −20 °C, and m-CPBA (0.38 g, 1.7 mmol) was added and stirred for 30 min. The reaction was quenched with saturated Na2SO3 (10 mL), the layers were separated and the aqueous phase was extracted with CH2Cl2 (2 × 10 mL), the organic phases were combined and washed with brine (10 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The crude material was used for the next step. 16b was characterized as two isomers (5:1) 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 0.8 Hz, 1H, maj. isomer), 7.04 (d, J = 2.0 Hz, 1H, maj. isomer), 6.89 (s, 1H, min. isomer), 6.80 (s, 1H, min. isomer), 6.61 (s, 1H, min. isomer), 6.65 (s, 1H, maj. isomer), 6.10 (s, 1H, maj. isomer), 5.97 (s, 1H, min. isomer), 3.83 (s, 3H, min. isomer), 3.67 (s, 3H, maj. isomer), 1.98 (s, 3H, min. isomer), 1.69 (s, 3H, maj. isomer), 1.26-1.21 (m, 6H, both isomers), 1.11-1.09 (m, 36H, both isomers), 0.16 (s, 9H, min. isomer), 0.09 (s, 9H, maj. isomer); 13C NMR (125 MHz, CDCl3) δ 194.9, 165.8, 157.3, 156.9, 149.6, 148.1, 146.9, 145.8, 145.3, 138.8, 129.0, 117.3, 115.8, 113.7, 52.3, 29.7, 19.7, 17.9, 17.8, 12.6, −0.5, −0.6; IR (neat) 2945, 2867, 1726, 1678, 1585, 1461, 1439, 1332, 1237, 1168, 1028, 998 cm−1; HRMS m/z calc. for C35H63O5Si3 (M + H+) 647.3978, found 647.3981.
Aryl enone 16c
To a 10 ml round-bottom flask was added 14b (0.617 g, 1.5 mmol), 15a (0.289 g, 1.65 mmol, 1.1 eq), and toluene (5 ml). The flask was purged with argon for 10 minutes and heated at 80 °C overnight. The reaction mixture was concentrated under reduced pressure. To a 10 ml round-bottom flask was added the crude material (0.75 g, 1.28 mmol) and CH2Cl2 (5 ml). The flask was cooled to 0 °C. m-CPBA (77%, 0.8625 g, 5 mmol, 3 eq) was added and the reaction mixture was stirred for 30 minutes. The reaction mixture was quenched with a saturated aqueous solution of Na2SO3, extracted with ether, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (9:1 hexanes:ethyl acetate) to afford 16c (0.67 g, 94%). 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 0.6 Hz, 1H), 7.69 (t, J = 1.5 Hz, 1H), 7.52 – 7.20 (m, 7H), 7.00 (s, 1H), 3.72 (s, 3H), 1.79 (d, J = 0.9 Hz, 3H), 1.35 – 1.17 (m, 3H), 1.08 (d, J = 7.3 Hz, 18H).
Aryl enone 16d
To a 25 ml round-bottom flask was added alkynoate 14b (0.950 g, 2.31 mmol), nitrone 15c (0.435 g, 2.54 mmol, 1.1 eq), and toluene (10 ml). The flask was heated at 80 °C overnight. The reaction mixture was concentrated under reduced pressure. To the crude material was added CH2Cl2 (10 ml). The flask was cooled to 0 °C. m-CPBA (77%, 1.55 g, 6.93 mmol, 3 eq) was added and the reaction mixture was stirred for one hour at 0 °C. The reaction mixture was quenched with a saturated aqueous solution of Na2SO3 and extracted three times with CH2Cl2. The combined organic extracts were added to a larger solution of saturated aqueous Na2SO3 and extracted three times with CH2Cl2. The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford aryl dienone 96 (1.12 g, 2.02 mmol, 88%) yield) as an inseparable mixture of geometric isomers (1.7:1) and vibrant yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.62 (s, 1H, maj. isomer), 7.49 (s, 1H maj. isomer), 7.39 (s, 1H, min. isomer), 7.29 (s, 1H, maj. isomer), 7.24 (t, J = 1.9 Hz, 1H, maj. isomer), 7.20 (t, J = 1.9 Hz, 1H, min. isomer), 7.08 (s, 1H, min. isomer), 6.86 (s, 1H, min. isomer), 6.09 (s, 1H, maj. isomer), 6.02 (s, 1H, min. isomer), 3.82 (s, 3H, min. isomer), 3.70 (s, 3H, maj. isomer), 1.97 (s, 3H, min. isomer), 1.68 (s, 3H, maj. isomer), 1.31 – 1.19 (m, 3H), 1.15 – 1.05 (m, 18H), 0.16 (s, 9H, mm. isomer), 0.08 (s, 9H, maj. isomer); 13C NMR (100 MHz, CDCl3) δ 193.81, 192.54, 167.28, 165.69, 157.38, 156.93, 150.29, 149.10, 146.88, 146.67, 146.42, 139.92, 139.55, 128.44, 128.31, 127.01, 124.63, 123.31, 122.86, 119.23, 119.12, 52.63, 52.58, 20.01, 18.70, 17.99, 17.95, 17.59, 12.70, −0.39, −0.47; IR (neat, cm−1): 2947, 2893, 2866, 1728, 1593, 1562, 1435, 1385, 1366, 1343, 1281, 1250, 1215, 1169, 1099, 1057, 995, 972, 941, 914, 880, 845, 752, 733, 687, 648; HRMS (ESI): Calculated for C26H42BrO4Si2 ([M+H]+): m/z 553.1805, found: 552.1814.
Aryl enone 16e
To a 50 ml round-bottom flask was added 15d (0.533 g, 2.5 mmol), 14b (1.028 g, 2.5 mmol), and toluene (25 ml). The flask was purged with argon for 10 minutes. The reaction mixture was heated at 80 °C overnight. The solution was concentrated under reduced pressure. To the crude material (1.4 g) was added CH2Cl2 (25 ml). The solution was cooled to 0°C. m-CPBA (77%, 1.00 g) was added, and the solution stirred for 30 minutes. The reaction was quenched with a saturated aqueous solution of Na2SO3, diluted and extracted with ether, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford 16e (1.147 g, 77%). 1H NMR (400 MHz, CDCl3) δ 7.63 (s, 1H), 7.51 (s, 1H), 7.32 – 7.28 (m, 1H), 7.23 (t, J = 1.9 Hz, 1H), 6.09 (s, 1H), 3.70 (s, 3H), 1.70 (s, 3H), 1.33 – 1.17 (m, 3H), 1.08 (d, J = 7.3 Hz, 18H), 0.83 (s, 9H), 0.03 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 193.74, 165.70, 157.36, 149.29, 147.51, 143.56, 139.41, 128.34, 128.28, 124.66, 123.30, 119.25, 52.66, 26.42, 20.35, 17.94, 17.23, 12.68, −4.65.
Indanone 17
In a 10 mL round-bottom flask, Cu(OTf)2 (51.5 mg, 0.1 mmol) was weighed, and a solution of 13 (467.3 mg, 1.3 mmol) and DCE (5 mL) was added. The reaction was stirred in an oil bath at 60 °C for 1.5 h, then filtered through a plug of silica gel and concentrated, yielding 372.6 mg (81%) of 17. 1H NMR (400 MHz, CDCl3): δ 7.38 – 7.25 (m, 5H), 6.78 (d, J = 2.0 Hz, 1H), 6.68 (d, J = 2.0 Hz, 1H), 6.50 (d, J = 15.6 Hz, 1H), 6.24 (dd, J = 8.0, 15.6 Hz, 1H), 4.53 (dd, J = 2.8, 8.0 Hz, 1H), 3.80 (s, 3H), 3.76 (s, 6H), 3.62 (d, J = 3.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 198.4, 168.7, 161.8, 157.9, 137.3, 137.0, 136.8, 131.1, 128.9, 128.4, 127.4, 126.2, 126.0, 106.6, 96.6, 60.9, 55.6, 52.7, 43.3; IR (neat, cm−1): 3003.4, 2950.9, 2941.2, 1738.2, 1708.8, 1611.4, 1493.8, 1306.2, 1203.0; HRMS (EI) calculated for C21H21O5 353.1384, found 353.1369.
Indanone 18a
In a dry 10 mL round-bottom flask was weighed Cu(ClO4)2 (7.0 mg, 0.03 mmol), and a solution of 16a (341 mg, 0.5 mmol) in DCE (5 mL) was added. The solution was stirred at 45 °C and monitored by NMR (aliquots were periodically removed from the reaction, filtered through Celite with CH2Cl2, and concentrated) for 8 h, then the reaction mixture was filtered through a plug of silica gel and flushed with CH2Cl2 then concentrated. The crude material was purified by column chromatography (10:1 petroleum ether:ether), yielding 272 mg (80%) of 18a. 1H NMR (400 MHz, CDCl3): δ 7.35 – 7.15 (m, 5H), 6.88 (d, J = 2.0 Hz, 1H), 6.66 (d, J = 2.0 Hz, 1H), 6.26 (s, 1H), 4.46 (d, J = 3.2 Hz, 1H), 3.78 (s, 3H), 3.49 (d, J = 3.2 Hz, 1H), 1.64 (s, 3H), 1.24 (m, 6H), 1.12 (d, J = 9.0 Hz, 36H); 13C NMR (100 MHz, CDCl3): δ 198.7, 169.2, 157.7, 154.5, 138.3, 138.2, 137.6, 137.4, 128.8, 127.9, 126.2, 117.3, 106.7, 61.0, 52.8, 50.0, 17.8, 12.5; IR (neat, cm−1): 2945.1, 2891.1, 2866.0, 1747.4, 1712.7, 1606.6, 1479.3, 1348.2, 1180.4; HRMS (EI) calculated for C38H58O5Si2 650.3817, found 650.3817.
Indanone 18b
In a dry 10 mL round-bottom flask was weighed Cu(ClO4)2 (13.0 mg, 0.05 mmol), and a solution of crude 16b from previous step in DCE (10 mL) was added. The solution was stirred at room temperature and monitored by NMR (aliquots were periodically removed from the reaction, filtered through Celite with CH2Cl2, and concentrated) for 2 h, then the reaction mixture was filtered through a plug of silica gel and flushed with CH2Cl2 concentrated. The crude material was used for the next step. 18b was characterized as two isomers (3:1) 1H NMR (400 MHz, CDCl3) δ 6.84 (d, 1H, J = 2.0 Hz, maj. isomer), 6.76 (d, 1H, J = 2.0 Hz, min. isomer), 6.63 (d, 1H, J = 2.0 Hz, maj. isomer), 6.42 (d, 1H, J = 2.0 Hz, min. isomer), 5.15 (s, 1H, min. isomer), 4.96 (s, 1H, min. isomer), 4.82 (s, 1H, maj. isomer), 4.70 (s, 1H, maj. isomer), 4.34 (s, 1H, min. isomer), 4.30 (d, 1H, J = 2.8 Hz, maj. isomer), 3.81 (s, 1H, maj. isomer), 3.79 (s, 2.25H), 3.69 (s, 0.25H), 3.41 (d, 0.75H, J = 3.2 Hz), 1.63 (s, 3H, maj. isomer), 1.58 (s, 3H, min. isomer), 1.31-1.22 (m, 6H, both isomers), 1.13-1.11 (m, 36H, both isomers); 13C NMR (125 MHz, CDCl3) δ 198.7, 169.3, 157.6, 156.9, 154.4, 153.0, 144.2, 141.4, 138.7, 138.5, 138.0, 117.3, 114.8, 112.6, 112.2, 106.6, 105.9, 105.0, 60.8, 52.7, 51.1, 50.3, 47.9, 29.7, 20.1, 17.9, 17.8, 13.3, 13.1, 12.6; IR (neat) 2945, 2892, 2867, 1720, 1605, 1478, 1348, 1310, 1248, 1176, 882, 766 cm−1; HRMS m/z calc. for C32H54O5NaSi2 (M + Na+) 597.3402, found 597.3402.
Indanone 18c
To a flame-dried 25 ml round-bottom flask in a glove box was added Sc(OTf)3 (0.03 g, 0.06 mmol, 0.05 eq) and Li(ClO4) (0.03 g, 0.18 mmol, 0.15 eq). The flask was removed from the glove box and dry DCE (12 ml) was added. 16c (0.67 g, 1.2 mmol) was added, and the reaction mixture was heated at 45 °C overnight. The reaction mixture was quenched with water, extracted with ether, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (9:1 hexane: ethyl acetate) to afford 18c as a 3:1 mixture of anti:syn isomers (0.546 g, 81%). For major (presumably anti) isomer: 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 2.2 Hz, 1H), 7.37 – 7.20 (m, 5H), 7.18 (d, J = 2.2 Hz, 1H), 6.38 (s, 1H), 4.47 (d, J = 3.0 Hz, 1H), 3.81 (s, 3H), 3.61 (d, J =3.0 Hz, 1H), 1.70 (d, J = 1.1 Hz, 3H), 1.35–1.23 (m, 3H), 1.11 (d, J = 7.2 Hz, 18H).
Indanone 18d
To a 25 ml round-bottom flask in a glove box was added AlCl3 (0.231 g, 1.73 mmol, 1.1 eq). The flask was removed from the glove box and DCE (15 ml) was added. The flask was cooled to 0 °C. Aryl enone 16d (0.869 g, 1.57 mmol) was added, and the reaction mixture turned deep red. The reaction mixture was stirred for 30 minutes. The reaction mixture was quenched with saturated aqueous potassium sodium tartrate, which turned the reaction mixture green, then yellow. The reaction mixture was extracted three times with Et2O. The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford 110 as a 3.6:1 mixture of anti:syn isomers (0.597 g, 1.24 mmol, 79% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 2.2 Hz, 1H, maj. isomer), 7.15 (d, J = 2.2 Hz, 1H, maj. isomer), 7.09 (d, J = 2.2 Hz, 1H, min. isomer), 7.06 (d, J = 2.2 Hz, 1H, min. isomer), 5.19 (s, 1H, min. isomer), 5.09 (s, 1H, min. isomer), 4.92 (s, 1H, maj. isomer), 4.69 (s, 1H, maj. isomer), 4.30 (s, 1H, min. isomer), 4.28 (d, J = 2.7 Hz, 1H, maj. isomer), 3.82 (s, 3H, min. isomer), 3.78 (s, 3H, maj. isomer), 3.50 (d, J = 2.7 Hz, 1H, maj. isomer), 2.34 (d, J = 4.5 Hz, 1H, min. isomer), 1.64 (s, 3H), 1.35 – 1.20 (m, 3H), 1.15 −1.05 (m, 18H); 13C NMR (100 MHz, CDCl3) δ 197.82, 168.72, 157.41, 157.30, 156.72, 146.72, 143.52, 139.76, 139.38, 138.48, 131.91, 125.57, 123.67, 122.40, 119.91, 117.07, 114.25, 113.05, 111.15, 107.44, 107.15, 61.23, 53.09, 52.98, 51.46, 50.48, 48.17, 20.53, 18.63, 17.96, 17.59, 17.39, 16.94, 13.00, 12.69. IR (neat, cm-1): 2947, 2866, 1721, 1651, 1586, 1555, 1458, 1377, 1292, 1219, 1157, 1134, 1069, 961, 883, 799, 729, 687, 648; HRMS (ESI): Calculated for C23H34BrO4Si ([M+H]+): m/z 481.1410, found: 481.1402.
Indanone 19
In a 200 mL round-bottom flask, indanone 18a (2.6 g, 4.0 mmol) was dissolved in toluene (45 mL), then water (1 mL) and DABCO (224.8 mg, 2.0 mmol) were added. The solution was refluxed for 12 h, and quenched with saturated aqueous NH4Cl (100 mL). The layers were separated and the organic phase was washed with water (2 × 100 mL) then brine (100 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (16:1 hexanes:ethyl acetate) to give 2.4 g (99%) of decarboxylated indanone. 1H NMR (400 MHz, CDCl3): δ 7.35 – 7.15 (m, 5H), 6.87 (d, J = 1.6 Hz, 1H), 6.63 (d, J = 2.0 Hz, 1H), 6.23 (s, 1H), 4.14 (dd, J = 2.0, 8.0 Hz, 1H), 3.02 (dd, J = 8.0, 19.2 Hz, 1H), 2.47 (dd, J = 2.0, 19.2 Hz, 1H), 1.70 (s, 3H), 1.24 (m, 6H), 1.07 (d, J = 7.4 Hz, 36H); 13C NMR (100 MHz, CDCl3): δ 206.1, 157.4, 154.6, 140.0, 139.3, 139.1, 138.0, 128.8, 127.9, 126.0, 125.6, 116.5, 105.9, 45.3, 44.6, 17.8, 15.5, 13.0, 12.6; IR (neat, cm−1): 2943.7, 2865.5, 1715.6, 1470.1, 1343.8, 1175.1; HRMS (EI) calculated for C36H57O3Si2 593.3841, found 593.3829.
In a 10 mL round-bottom flask, a solution of i-Pr2NH (0.2 mL, 1.0 mmol), n-BuLi (2.5 M solution in hexanes, 0.3 mL, 0.7 mmol), and THF (1 mL) was stirred at 0 °C for 15 minutes, then cooled to −40 °C. A solution of the decarboxylated material (300.0 mg, 0.5 mmol) in THF (2 mL) was added, and the reaction was stirred at −40 °C for 5.5 h, then MeI (0.2 mL, 3.2 mmol) was added and the reaction was stirred for an additional 4 h at −20 °C. The reaction was quenched while cold with saturated aqueous NH4Cl and the layers were separated. The organic phase was diluted with hexanes (10 mL) and washed with water (3 × 10 mL), then brine (10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (16:1 hexanes:ethyl acetate) to give 247.3 mg (81%) of 19.
1H NMR (400 MHz, CDCl3):δ 7.35 – 7.15 (m, 5H), 6.87 (d, J = 2.0 Hz, 1H), 6.62 (d, J = 2.0 Hz, 1H), 6.30 (s, 1H), 3.60 (d, J = 3.2 Hz, 1H), 2.43 (dt, J = 7.4, 3.2 Hz, 1H), 1.72 (s, 3H), 1.36 (d, J = 7.4 Hz, 3H), 1.24 (m, 6H), 1.07 (d, J = 7.4 Hz, 36H); 13C NMR (100 MHz, CDCl3): δ 208.6, 157.4, 154.7, 139.1, 138.7, 138.0, 137.6, 128.9, 127.9, 126.0, 125.7, 116.6, 106.2, 55.1, 49.9, 17.8, 15.9, 15.2, 13.1, 12.6; IR (neat, cm−1): 2943.2, 2866.0, 1715.6, 1604.7, 1470.6, 1341.9, 1174.6; HRMS (EI) calculated for C37H59O3Si2 607.3997, found 607.3998.

Indanone S1
In a 200 mL round-bottom flask, crude 18b (207 mg, 0.36 mmol) from previous steps was dissolved in toluene (4.1 mL), then water (0.41 mL) and DABCO (21 mg, 0.18 mmol) were added. The solution was refluxed for 12 h, and quenched with saturated aqueous NH4Cl (100 mL). The layers were separated and the organic phase was washed with water (2 × 100 mL) then brine (100 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude material was used for the next step.
In a 25 mL round-bottom flask, the crude material from previous step was dissolved in THF (3.1 mL) and the solution was cooled to 0 °C, then TBAF (1.0 M solution in THF, 0.6 mL, 0.6 mmol) was added and the reaction was stirred for 1 h. The solution was quenched with saturated aqueous NH4Cl and the layers were separated. The organic phase was diluted with diethyl ether (20 mL) and washed with water (3 × 10 mL), then brine (10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (1:1 hexanes: ethyl acetate) to give 66 mg (33%, yield over 4 steps from 14a) of S1. 1H NMR (400 MHz, CD3OD) δ 6.59 (d, 1H, J = 2.0 Hz), 5.56 (d, 1H, J = 2.4 Hz), 4.76 (s, 1H), 4.69 (s, 1H), 4.02 (dd, 1H, J = 2.0, 7.6 Hz), 2.94 (dd, 1H, J = 8.0, 19.2 Hz), 2.34 (dd, 1H, J = 2.0, 19.2 Hz), 1.60 (s,3H).
Aryl triflate 20
In a 25 mL round-bottom flask, diprotected 19 (402.0 mg, 0.7 mmol) was dissolved in THF (6 mL) and the solution was cooled to 0 °C, then TBAF (1.0 M solution in THF, 1.2 mL, 1.2 mmol) was added and the reaction was stirred for 1 h. The solution was quenched with saturated aqueous NH4Cl and the layers were separated. The organic phase was diluted with diethyl ether (20 mL) and washed with water (3×10 mL), then brine (10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (2:1 hexanes:ethyl acetate) to give 147.4 mg (76%) of diphenol. 1H NMR (400 MHz, CDCl3): δ 7.38 – 7.30 (m, 5H), 6.93 (d, J = 2.0 Hz, 1H), 6.72 (d, J =2.0 Hz, 1H), 6.71 (s, 1H), 3.67 (d, J =4.0 Hz, 1H), 2.60 (dt, J = 7.2, 4.0 Hz, 1H), 1.76 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H).
In a 25 mL round-bottom flask, diol from the previous step (240.0 mg, 0.8 mmol) was dissolved in CH2Cl2 (11 mL), and TEA (0.1 mL, 0.7 mmol) and TIPS-Cl (0.2 mL, 0.8 mmol) were added. The reaction was stirred for 16 h, then quenched with saturated aqueous NH4Cl and the layers were separated. The organic phase was diluted with CH2Cl2 (10 mL) and washed with water (10 mL), then brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (15:1 hexanes:ethyl acetate) to give 155.6 mg (42%) of monophenol. 1H NMR (400 MHz, CDCl3): δ 7.38 – 7.30 (m, 5H), 6.86 (d, J = 2.0 Hz, 1H), 6.75 (s, 1H), 6.68 (d, J = 2.0 Hz, 1H), 5.68 (s, 1H), 3.68 (d, J =4.4 Hz, 1H), 2.57 (dt, J = 7.2, 4.4 Hz, 1H), 1.78 (s, 3H), 1.37 (d, J = 7.2 Hz, 3H), 1.28 (m, 3H), 1.09 (d, J = 7.4 Hz, 18H); 13C NMR (100 MHz, CDCl3): δ 207.4, 158.1, 154.8, 138.6, 138.1, 136.7, 131.8, 128.9, 128.3, 128.2, 127.1, 114.0, 106.3, 55.0, 48.6, 17.7, 14.7, 14.2, 12.6; IR (neat, cm−1): 3204.8, 2943.2, 2865.5, 1680.9, 1470.1, 1345.7, 1177.5; HRMS (EI) calculated for C28H39O3Si 451.2663, found 451.2648.
In a 25 mL round-bottom flask, monophenol from the previous step (155.6 mg, 0.4 mmol) was dissolved in CH2Cl2 (2 mL) and cooled to 0 °C, then pyridine (0.08 mL, 1.0 mmol) and Tf2O (0.1 mL, 0.6 mmol) were added and the reaction was stirred in an ice bath for 2 h. The reaction was quenched with saturated aqueous NH4Cl and the layers were separated. The organic phase was diluted with hexanes (15 mL) and washed with brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (8:1 hexanes:ethyl acetate) to give 190.2 mg (93%) of aryl triflate 20. 1H NMR (400 MHz, CDCl3): δ 7.38 –7.30 (m, 6H), 7.05 (d, J = 2.4 Hz, 1H), 6.56 (s, 1H), 3.83 (d, J = 4.0 Hz, 1H), 2.58 (dt, J = 7.2, 4.0 Hz, 1H), 1.67 (s, 3H), 1.40 (d, J = 7.2 Hz, 3H), 1.27 (m, 3H), 1.12 (d, J = 7.2 Hz, 18H); 13C NMR (100 MHz, CDCl3): δ 205.8, 157.7, 147.3, 140.3, 138.0, 137.4, 134.9, 129.1, 128.9, 128.8, 128.0, 126.5, 119.9, 113.9, 55.0, 50.9, 49.1, 17.7, 14.8, 14.2, 12.5; IR (neat, cm−1): 2944.6, 2868.0, 1723.8, 1615.3, 1470.6, 1424.3, 1313.4, 1245.0, 1207.8, 1139.0, 992.3; HRMS (EI) calculated for C29H37O5SiSF3 582.2078, found 582.2077.
Aryl triflate 21
21 was prepared following the synthetic procedure for 20, except methylation was not carried out. 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.32 (t, J = 7.2 Hz, 2H), 7.28 −7.23 (m, 3H), 7.07 (d, J = 2.4 Hz, 1H), 6.53 (s, 1H), 4.39 (dd, J = 2.8, 8.0 Hz, 1H), 3.10 (dd, J = 8.4, 19.6 Hz, 1H), 2.63 (dd, J = 3.2, 19.6 Hz, 1H), 1.62 (s, 3H), 1.32 – 1.27 (m, 3H), 1.13 – 1.11 (m, 18H); 13C NMR (400 MHz, CDCl3) δ 203.7, 157.8, 147.3, 141.1, 139.7, 137.3, 135.7, 128.9, 128.5, 128.1, 126.6, 120.0, 113.7, 53.4, 45.5, 43.5, 17.7, 14.1, 12.5; IR (neat) 2946, 2868, 1725, 1617, 1476, 1426, 1315, 1245, 1217, 1141, 1064, 999, 942, 882 cm−1; HRMS m/z calc. for C28H35O5F3NaSSi (M + Na+) 591.1819, found 591.1824.
Aryl triflate 22
In a 25 mL round-bottom flask, 21 (569 mg, 1 mmol) was dissolved in THF (8.6 mL) and the solution was cooled to 0 °C, then TBAF (1.0 M solution in THF, 1.7 mL, 1.7 mmol) was added and the reaction was stirred for 1 h. The solution was quenched with saturated aqueous NH4Cl and the layers were separated. The organic phase was diluted with diethyl ether (20 mL) and washed with water (3 × 10 mL), then brine (10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (3:1 hexanes:ethyl acetate) to give 408 mg (99%) of phenol. 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.32 (m, 3H), 7.27 – 7.22 (m, 3H), 7.12 (m, 1H), 6.53 (s, 1H), 6.34 (b, 1H), 4.40 (dd, 1H, J = 2.8, 8.0 Hz), 3.13 (dd, 1H, J = 8.0, 19.6 Hz), 2.66 (dd, 1H, J = 2.8, 19.6 Hz), 1.63 (s, 3H); 13C NMR (400 MHz, CDCl3) δ 204.4, 157.5, 147.6, 141.1, 139.6, 137.2, 135.4, 128.8, 128.6, 128.1, 126.6, 116.3, 109.4, 77.2, 45.5, 43.5, 14.2.
TMSCHN2 (2.0 M in Et2O, 0.275 mL, 0.55 mmol) was added to a stirred solution of phenol from the previous step (206 mg, 0.5 mmol) and N, N-diisopropylethylamine (91 mg, 0.7 mmol) in methanol-acetonitrile (1:9, 2 mL) at room temperature. The mixture was stirred for 15 h at room temperature, and concentrated in vacuo. The crude was dissolved in Et2O (15 mL), washed with saturated aqueous NH4Cl (3 × 10 mL) and water (3 × 10 mL), then brine (10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (3:1 hexanes:ethyl acetate) to give 186 mg (82%) of 22. 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.21 (m, 5H), 7.11 (s, 1H), 6.53 (s, 1H), 4.40 (dd, J = 2.8, 8.0 Hz, 1H), 3.91 (s, 3H), 3.12 (dd, J = 8.0, 19.6 Hz, 1H), 2.65 (dd, J = 2.8, 19.6 Hz, 1H,), 1.63 (s, 3H); 13C NMR (400 MHz, CDCl3) δ 203.7, 161.2, 147.5, 141.3, 139.7, 137.3, 135.7, 128.9, 128.6, 128.1, 126.6, 116.4, 105.8, 77.2, 56.3, 45.5, 43.5, 14.2; IR (neat) 2917, 1720, 1486, 1425, 1296, 1244, 1212, 1140, 1071, 1036, 964, 861 cm−1; HRMS m/z calc. for C20H17O5F3S (M+) 426.0743, found 426.0743.
Aryl bromide 23
To a 25 ml round-bottom flask was added DABCO (0.142 g, 1.26 mmol, 1.43 eq), toluene (8 ml), H2O (0.8 ml), and 18c (0.491 g, 0.882 mmol). The flask was purged with argon for 10 minutes. The reaction mixture was refluxed overnight. The reaction mixture was diluted with water and ether, extracted with ether, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 ether:hexane) to afford aryl bromide 23 (0.371 g, 84%). 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 2.2 Hz, 1H), 7.34 – 7.20 (m, 5H), 7.19 (d, J = 2.2 Hz, 1H), 6.39 (s, 1H), 4.15 (dd, J = 8.0, 2.0 Hz, 1H), 3.08 (dd, J = 19.3, 8.0 Hz, 1H), 2.61 (dd, J = 19.3, 2.0 Hz, 1H), 1.63 (s, 3H), 1.40–1.20 (m, 3H), 1.11 (d, J = 7.3 Hz, 18H). 13C NMR (100 MHz, CDCl3) δ 204.98, 157.16, 147.42, 140.36, 137.89, 137.30, 131.25, 128.86, 128.23, 126.52, 122.57, 112.37, 44.44, 18.02, 17.89, 12.72.
Alcohol 24
To a 25 ml round-bottom flask was added 18d (0.475 g, 0.987 mmol), DABCO (0.055 g, 0.493 mmol, 0.5 eq), toluene (10 ml), and H2O (1 ml). The solution was heated at 80 °C overnight. The solution was quenched with saturated aqueous NH4Cl and extracted three times with Et2O. The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford decarboxylated indanone (0.382 g, 0.902 mmol 91% yield).
LDA was prepared according to the following: to a 50 ml round-bottom flask was added THF (15 ml) and diisopropylamine (1.37 ml, 9.74 mmol, 1.21 eq). The solution was cooled to −40 °C, and n-BuLi (3.66 ml, 2.42M in hexanes, 1.1 eq) was added. The solution was stirred at −40 °C for 40 minutes. Then, decarboxylated indanone from the previous step (3.41 g, 8.05 mmol), in THF (38 ml) was added dropwise. The solution was stirred and maintained at −40 °C for three hours. Then, Mel (24.9 ml, 400 mmol, 50 eq) was added to the solution, which was slowly raised to room temperature overnight. The reaction was quenched the next day with 1M aqueous HCl and extracted three times with Et2O. The combined organic fractions were washed with saturated NaHCO3, concentrated under reduced pressure, and taken to the next step without further purification.
The crude material from the previous step was dissolved in MeOH (80 ml) and cooled to 0 °C. NaBH4 (0.610 g, 16.1 mmol, 2 eq) was added. The reaction was stirred at 0 °C for 2 hours, then quenched with water. The solution was extracted three times with Et2O, and the combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford alcohol 24 (2.37 g, 5.39 mmol, 67% over two steps) as a crystalline solid. 1H NMR (400 MHz, CDCl3) δ 6.99 (s, 1H), 6.88 (s, 1H), 4.86 (s, 1H), 4.62 (s, 1H), 4.55 (dd, J = 8.5, 4.9 Hz, 1H), 3.19 (d, J = 5.4 Hz, 1H), 2.18 (d, J =8.5 Hz, 1H), 2.14 – 2.07 (m, 1H), 1.74 (s, 3H), 1.30–1.21 (m, 3H), 1.16 (d, J = 6.9 Hz, 3H), 1.10 (d, J = 7.4 Hz, 18H); 13C NMR (100 MHz, CDCl3) δ 156.59, 147.61, 147.12, 134.96, 124.24, 120.96, 115.16, 112.45, 82.41, 58.76, 49.23, 20.49, 17.99, 17.89, 12.70. IR (neat, cm−1): 3287 (br), 3075 (w), 2943 (s), 2866 (s), 1605 (s), 1555 (s), 1458 (s), 1412 (w), 1377 (w), 1285 (s), 1211 (m), 1177 (m), 1142 (s), 1057 (s), 999 (s), 880 (s), 845 (s), 814 (w), 760 (s), 733 (m), 683 (s), 644 (w); HRMS (ESI): Calculated for C22H35BrO2SiNa ([M+H+Na]+): m/z 461.1487, found: 461.1492.
Aryl bromide 25
To a 100 ml round-bottom flask was added alcohol 24 (2.54 g, 5.78 mmol), TBSCl (3.48 g, 23.1 mmol, 4 eq), imidazole (3.15 g, 46.2 mmol, 8 eq), and THF (57 ml). The solution was stirred at room temperature overnight. The reaction was quenched with saturated aqueous NH4Cl and extracted three times with Et2O. The combined organic fractions were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford aryl bromide 25 (2.90 g, 5.24 mmol, 91% yield) as a translucent oil. 1H NMR (400 MHz, CDCl3) δ 6.94 (d, J = 2.2 Hz, 1H), 6.74 (d, J = 2.2 Hz, 1H), 4.88 (s, 1H), 4.86 (s, 1H), 4.58 (d, J = 6.3 Hz, 1H), 3.20 (d, J =7.2 Hz, 1H), 2.08 – 1.97 (m, 1H), 1.62 (s, 3H), 1.31 – 1.19 (m, 3H), 1.15 (d, J = 6.9 Hz, 3H), 1.09 (d, J = 7.2 Hz, 18H), 0.93 (s, 9H), 0.17 (s, 3H), 0.14 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.27, 148.76, 145.74, 134.25, 123.99, 120.63, 114.67, 113.31, 82.12, 58.89, 49.43, 25.92, 19.39, 18.13, 18.03, 17.25, 12.78, −3.95, −4.12. IR (neat, cm-1): 2951, 2931, 2893, 2866, 1605, 1559, 1458, 1362, 1296, 1254, 1180, 1146, 1115, 1067, 999, 868, 833, 772, 733, 683, 671.
Amine 28
An oven-dried resealable Schlenk flask was evacuated and backfilled with argon. The flask was charged with Pd(OAc)2 (10 mol %), BINAP (20 mol %), and NaOtBu (1.4 equiv.) and evacuated and backfilled with argon. The flask was capped with a rubber septum, and toluene (2 mL/mmol triflate), the aryl triflate 21 (57 mg, 0.1 mmol, 1.0 equiv.), and the amine 27 (1.2 equiv.) were added through the septum. The septum was replaced with a Teflon screwcap, the flask was sealed, and the mixture was heated to 105 °C with stirring. The mixture was cooled to room temperature, diluted with ether, filtered through Celite, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel to furnish amine 28 (3 mg, 6%). 1H NMR (500 MHz, CDCl3) δ 7.36 – 7.20 (m, 5H), 6.83 (s, 1H), 6.65 (s, 1H), 6.36 (s, 1H), 4.16 (dd, J = 2.0, 8.0 Hz, 1H,), 3.82 (s, 3H), 3.00 (dd, J = 8.0, 19.0 Hz, 1H), 2.53 (dd, J = 2.0, 19.0 Hz, 1H), 1.64 (s, 3H), 1.32 – 1.28 (m, 3H), 1.26 (s, 6H), 1.13 – 1.11 (m, 18H); 13C NMR (400 MHz, CDCl3) δ 206.1, 158.2, 157.8, 139.7, 138.6, 138.2, 137.9, 128.8, 128.2, 128.0, 126.12, 126.09, 109.5, 104.4, 55.6, 45.2, 43.9, 29.7, 22.7, 17.9, 14.8, 12.7; IR (neat, cm−1) 2925, 2866, 1718, 1608, 1480, 1445, 1344, 1308, 1220, 1196, 1167, 992, 883.
Amine 30
An oven-dried resealable Schlenk flask was evacuated and backfilled with argon. The flask was charged with Pd2(dba)3 (5 mol %), DavePhos (10 mol %), and K3PO4 (1.4 equiv.) and evacuated and backfilled with argon. The flask was capped with a rubber septum, and toluene (2 mL/mmol triflate), the aryl triflate 22 (18 mg, 0.04 mmol, 1.0 equiv.), and the amine 29 (1.2 equiv.) were added through the septum. The septum was replaced with a Teflon screwcap, the flask was sealed, and the mixture was heated to 105 °C with stirring. The mixture was cooled to room temperature, diluted with ether, filtered through Celite, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel to furnish amine 30 as a 3:1 mixture of isomers (6 mg, 40%). 1H NMR (400 MHz,CDCl3) δ 7.36 – 7.24 (m, 5H, both isomers), 6.76 (s, 1H, both isomers), 6.66 (d, J = 2.4 Hz, 1H, both isomers), 6.26 (d, J = 2.0 Hz, 1H, both isomers), 4.93 (d, J = 8.8 Hz, 1H, maj. isomer), 4.77 (d, J = 8.0 Hz, 1H, min. isomer), 4.15 – 4.08 (m, 2H, both isomers), 3.82 (s, 3H, both isomers), 3.75 (s, 3H, min. isomer), 3.66 (s, 3H, maj. isomer), 3.03 (dd, J = 8.0, 19.2 Hz, 1H, both isomers), 2.57 (dd, J = 2.8, 19.2 Hz, 1H, both isomers), 1.67 (s, 3H, maj. isomer) 1.63 (s, 3H, min. isomer) 1.43 (d, J = 6.8 Hz, 3H, maj. isomer) 1.41 (d, J = 7.2 Hz, 3H, min. isomer); 13C NMR (400 MHz, CDCl3) δ 205.9, 174.2, 161.7, 145.9, 139.5, 138.9, 137.3, 135.2, 128.8, 128.7, 128.4, 128.2, 128.1, 126.7, 103.9, 103.8, 93.7, 93.6, 55.5, 52.4, 52.2, 51.5, 51.2, 46.2, 43.1, 29.7, 18.7, 18.6, 13.5, 13.4; IR (neat) 2923, 1743, 1707, 1612, 1514, 1449, 1363, 1204, 1170, 1087, 1049 cm−1; HRMS m/z calc. for C23H25O4NNa (M + Na+) 402.1676, found 402.1668.
Amine 32
An oven-dried resealable Schlenk flask was evacuated and backfilled with argon. The flask was charged with Pd(OAc)2 (10 mol %), BINAP (15 mol %), and Cs2CO3 (1.4 equiv.) and evacuated and backfilled with argon. The flask was capped with a rubber septum, and toluene (2 mL/mmol triflate), the aryl triflate 22 (18 mg, 0.04 mmol, 1.0 equiv.), and the amine 31 (1.2 equiv.) were added through the septum. The septum was replaced with a Teflon screwcap, the flask was sealed, and the mixture was heated to 100 °C with stirring. The mixture was cooled to room temperature, diluted with ether, filtered through Celite, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel to furnish amine 32 (8 mg, 50%). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.23 (m, 8H), 7.10 (m, 2H), 6.64 (d, J = 1.5 Hz, 1H), 6.61 (s, 1H), 6.39 (d, J = 1.5 Hz, 1H), 4.67 (s, 1H), 4.32 (d, J = 5.0 Hz, 2H), 4.08 (dd, J = 2.0, 7.5 Hz, 1H), 3.82 (s, 3H), 3.03 (dd, J = 7.5, 19.0 Hz, 1H), 2.57 (dd, J = 2.5, 19.0 Hz, 1H), 1.64 (s, 3H); 13C NMR (400 MHz, CDCl3) δ 206.0, 161.8, 147.0, 139.3, 138.8, 138.3, 137.0, 134.7, 128.8, 128.7, 128.2, 128.0, 127.5, 127.4, 126.7, 103.6, 93.0, 55.5, 47.9, 46.4, 43.2, 13.6; IR (neat) 2921, 2851, 1703, 1610, 1511, 1451, 1365, 1237, 1202, 1166, 1061 cm−1; HRMS m/z calc. for C26H25O2N (M+) 383.1880, found 383.1885.
Amine 34
An oven-dried resealable Schlenk flask was evacuated and backfilled with argon. The flask was charged with Pd2(dba)2 (3 mol %), BINAP (4.5 mol %), and NaOtBu (1.4 equiv.) and evacuated and backfilled with argon. The flask was capped with a rubber septum, and toluene (2 mL/mmol triflate), the aryl triflate 21 (69 mg, 0.121 mmol, 1.0 equiv.), and the amine 33 (1.2 equiv.) were added through the septum. The septum was replaced with a Teflon screwcap, the flask was sealed, and the mixture was heated to 90 °C with stirring. The mixture was cooled to room temperature, diluted with ether, filtered through Celite, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel to furnish amine 34 (20 mg, 28%). 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.2 Hz, 2H,), 7.50 – 7.45 (m, 1H), 7.32 – 7.21 (m, 7H), 7.19 – 7.16 (m, 1H), 7.12 – 7.05 (m, 2H), 7.08 – 7.01 (m, 2H), 6.89 (s, 1H), 6.43 – 6.48 (m, 1H), 6.36 (s, 1H), 3.89 (dd, J = 2.8, 8.0 Hz, 1H), 2.87 (dd, J = 8.0, 19.2 Hz, 1H), 2.52 (dd, J = 2.8, 19.2 Hz, 1H), 1.64 (s, 3H), 1.14 – 1.07 (m, 3H), 1.03 – 1.01 (m, 18H); 13C NMR (400 MHz, CDCl3) δ 206.1, 167.7, 156.5, 149.8, 139.5, 139.2, 138.8, 137.7, 135.8, 130.9, 129.6, 129.0, 128.9, 128.8, 128.1, 127.8, 127.4, 126.0, 118.5, 108.3, 77.2, 46.6, 43.6, 17.8, 13.9, 12.4; IR (neat) 2942, 2865, 1713, 1661, 1598, 1465, 1447, 1317, 1278, 1178, 1072, 1000, 941, 919, 883 cm−1; HRMS m/z calc. for C40H46O2NSi (M + H+) 600.3292, found 600.3306.
Amine 35
To a flame-dried 3 ml vial in a glove box was added Pd2(dba)3 (0.0046 g, 0.005 mmol, 0.05 eq), BINAP (0.011 g, 0.015 mmol, 0.15 eq), and NaOtBu (0.0115 g, 0.12 mmol, 1.2 eq). The vial was capped with a pierceable PFTE seal. The vial was removed from the glove box and dry toluene (0.6 ml) was added. The mixture was stirred for five minutes. 33 (0.025 ml, 0.15 mmol, 1.5 eq) and 23 (0.050 g, 0.1 mmol) were added by syringe, and the vial was heated at 110 °C behind a blast shield overnight. The reaction mixture was concentrated under reduced pressure. The crude residue was dissolved in THF (0.33 ml) in a 5 ml round-bottom flask. 1M aqueous HCl was added (0.033 ml) and the reaction was stirred for 15 minutes. The reaction was quenched with aqueous saturated NaHCO3, diluted and extracted with ether, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography (9:1 hexane: ethyl acetate) to afford aniline 35 (11.6 mg, 27%). 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.13 (m, 5H), 6.69 (d, J = 2.0 Hz, 1H), 6.64 (s, 1H), 6.45 (d, J = 2.0 Hz, 1H), 4.07 (dd, J = 7.8, 2.6 Hz, 1H), 3.99 (s, 2H), 3.01 (dd, J = 19.3, 7.8 Hz, 1H), 2.53 (dd, J = 19.3, 2.6 Hz, 1H), 1.67 (d, J = 0.9 Hz, 3H), 1.36 – 1.18 (m, 3H), 1.10 (d, J = 7.2 Hz, 18H). 13C NMR (101 MHz, CDCl3) δ 206.09, 157.66, 145.81, 139.68, 138.78, 137.29, 133.96, 128.90, 128.39, 127.69, 127.49, 126.84, 103.48, 103.28, 43.46, 18.09, 17.96, 12.75. IR (neat, cm− 1) 2943, 2865, 1701, 1619, 1482, 1351, 1236, 1184, 1068, 1013, 946, 883; HRMS m/z calc. for C27H38NO2Si (M + H+) 436.2666, found 436.2658.
Amine 36
To a 100 ml Schlenk bomb was added [Pd-(allyl)Cl]2 (0.0192 g, 0.0524 mmol, 0.01 eq), Mor-DalPhos (0.0974 g, 0.210 mmol, 0.04 eq), NaOtBu (1.01 g, 10.5 mmol, 2 eq), and toluene (52 ml). The solution was stirred for 10 minutes. Then, 25 (2.9 g, 5.24 mmol) was added, followed by NH3 (0.5 M solution in 1,4-dioxane, 31.4 ml, 3 eq.). The Schlenk bomb was sealed and heated at 110 °C for 1 hour. The reaction vessel was cooled, quenched with saturated aqueous NH4Cl, and extracted three times with Et2O. The combined extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (19:1 hexanes:ethyl acetate) to afford aniline 36 (2.48 g, 5.06 mmol, 97% yield). 1H NMR (400 MHz, CDCl3) δ 6.24 (d, J = 2.0 Hz, 1H), 6.05 (d, J = 2.0 Hz, 1H), 5.05 (d, J = 2.0 Hz, 1H), 4.94 (s, 1H), 4.56 (d, J = 7.4 Hz, 1H), 3.75 (s, 2H), 3.15 (d, J = 8.4 Hz, 1H), 2.10 – 1.98 (m, 1H), 1.64 (s, 3H), 1.32 – 1.16 (m, 3H), 1.14 (d, J = 6.8Hz, 3H), 1.09 (d, J = 7.0 Hz, 18H), 0.95 (s, 9H), 0.17 (s, 3H), 0.14 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.72, 147.94, 147.28, 144.22, 118.61, 113.66, 106.24, 105.50, 82.18, 56.32, 49.05, 26.01, 18.26, 18.20, 18.12, 16.32, 12.86, − 3.91, −4.00; IR (neat, cm−1): 2951, 2928, 2893, 2866, 1613, 1470, 1362, 1254, 1204, 1157, 1096, 1009, 837, 683; HRMS (ESI): Calculated for C28H52NO2Si2 ([M+H]+): m/z 490.3537, found: 490.3531.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIGMS R01 GM-079364) for funding this project, Dr. William W. Brennessel (University of Rochester) for performing X-ray crystallography, and Dr. Furong Sun (University of Illinois, Urbana-Champaign) and Dr. Alice Bergmann (University of Buffalo) for performing high-resolution mass spectrometry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
1H and 13C NMR spectra for compounds can be found in the Supplementary Data. Supplementary Data associated with this article can be found in the online version at doi:
References and notes
- 1.Komoda T, Sugiyama Y, Abe N, Imachi M, Hirota H, Hirota A. Tetrahedron Lett. 2003;44:1659–1661. [Google Scholar]
- 2.Komoda T, Sugiyama Y, Abe N, Imachi M, Hirota H, Koshino H, Hirota A. Tetrahedron Lett. 2003;44:7417–7419. [Google Scholar]
- 3.Komoda T, Kishi M, Abe N, Sugiyama Y, Hirota A. Biosci. Biotechnol. Biochem. 2004;68:903–908. doi: 10.1271/bbb.68.903. [DOI] [PubMed] [Google Scholar]
- 4.Prigge ST, Boyington JC, Gaffney BJ, Amzel LM. Proteins Struct. Funct. Genet. 1996;24:275–291. doi: 10.1002/(SICI)1097-0134(199603)24:3<275::AID-PROT1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 5.Salmon JA, Higgs GA. Br. Med. Bull. 1987;43:285–296. doi: 10.1093/oxfordjournals.bmb.a072183. [DOI] [PubMed] [Google Scholar]
- 6.Komoda T, Yoshida K, Abe N, Sugiyama Y, Imachi M, Hirota H, Koshino H, Hirota A. Biosci. Biotechnol. Biochem. 2004;6S:104–111. doi: 10.1271/bbb.68.104. [DOI] [PubMed] [Google Scholar]
- 7.Komoda T, Akasaka K, Hirota A. Biosci. Biotechnol. Biochem. 2008;72:2392–2397. doi: 10.1271/bbb.80282. [DOI] [PubMed] [Google Scholar]
- 8.Komoda T, Yasumasa S, Hirota A. Org. Biomol. Chem. 2007;5:1615–1620. doi: 10.1039/b618425a. [DOI] [PubMed] [Google Scholar]
- 9.(a) Wang X, Jr, Porco JA. Angew. Chem. Int. Ed. 2006;44:3067–3071. doi: 10.1002/anie.200500247. [DOI] [PubMed] [Google Scholar]; (b) Wang X, Porco JA., Jr Angew. Chem. Int. Ed. 2006;45:6607. [Google Scholar]
- 10.(a) Marcus AP, Sarpong R. Org. Lett. 2010;12:4560–4563. doi: 10.1021/ol1018536. [DOI] [PubMed] [Google Scholar]; (b) Marcus AP, Sarpong R. Org. Lett. 2014;16:3420. [Google Scholar]
- 11.Li C, Li X, Hong R. Org. Lett. 2009;11:4036–4039. doi: 10.1021/ol901349b. [DOI] [PubMed] [Google Scholar]
- 12.Weaver MG, Bai W-J, Jackson SK, Pettus TRR. Org. Lett. 2014;16:1294–1297. doi: 10.1021/ol4034447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlsen PN, Mann TJ, Hoveyda AH, Frontier AJ. Angew. Chem. Int. Ed. 2014;53:9334–9338. doi: 10.1002/anie.201404410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) He W, Sun X, Frontier AJ. J. Am. Chem. Soc. 2003;125:14278–14279. doi: 10.1021/ja037910b. [DOI] [PubMed] [Google Scholar]; (b) He W, Herrick IR, Atesin TA, Caruana PA, Kellenberger CA, Frontier AJ. J. Am. Chem. Soc. 2008;130:1003–1011. doi: 10.1021/ja077162g. [DOI] [PubMed] [Google Scholar]
- 15.Malona JA, Colbourne JM, Frontier AJ. Org. Lett. 2006;8:5661–5664. doi: 10.1021/ol062403v. [DOI] [PubMed] [Google Scholar]
- 16.Marcus AP, Lee AS, Davis RL, Tantillo DJ, Sarpong R. Angew. Chem. Int. Ed. 2008;47:6379–6383. doi: 10.1002/anie.200801542. [DOI] [PubMed] [Google Scholar]
- 17.Canterbury DP, Herrick IR, Um J, Houk KN, Frontier AJ. Tetrahedron. 2009;65:3165–3179. doi: 10.1016/j.tet.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kingsbury CA, Draney D, Sopchik A, Rissler W, Durham D. J. Org. Chem. 1976;41:3863–3868. [Google Scholar]
- 19.Vaidya T, Cheng R, Carlsen PN, Frontier AJ, Eisenberg R. Org. Lett. 2014;16:800–803. doi: 10.1021/ol403542k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathieu B, Ghosez L. Tetrahedron. 2002;58:8219–8226. [Google Scholar]
- 21.Jiricek J, Blechert S. J. Am. Chem. Soc. 2004;126:3534–3538. doi: 10.1021/ja0399021. [DOI] [PubMed] [Google Scholar]
- 22.For selected reviews, see:Yang BH, Buchwald SL. J. Organomet. Chem. 1999;576:125–146.Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-I, editor. New York: Wiley-Interscience; 2002. pp. 1051–1095.Wolfe JP, Wagaw S, Marcoux J-F, Buchwald SL. Ace. Chem. Res. 1998;31:805–818.Hartwig JF. Angew. Chem. Int. Ed. 1998;37:2046–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L.Kienle M, Dubbaka SR, Brade K, Knochel P. Eur. J. Org. Chem. 2007:4166–4176.Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338–6361. doi: 10.1002/anie.200800497.Marion N, Nolan SP. Ace. Chem. Res. 2008;41:1440–1449. doi: 10.1021/ar800020y.Hartwig JF. Ace. Chem. Res. 2011;41:1534–1544. doi: 10.1021/ar800098p.Surry DS, Buchwald SL. Chem. Sci. 2011;2:27–50. doi: 10.1039/C0SC00331J.
- 23.(a) Yin J, Buchwald SL. J. Am. Chem. Soc. 2003;124:6043–6048. doi: 10.1021/ja012610k. [DOI] [PubMed] [Google Scholar]; (b) Wallace DJ, Klauber DJ, Chen C-y, Volante RP. Org. Lett. 2003;5:4749–4752. doi: 10.1021/ol035959g. [DOI] [PubMed] [Google Scholar]
- 24.Wolfe JP, Ahman J, Sadighi JP, Singer RA, Buchwald SL. Tetrahedron. Lett. 1997;38:6367–6370. [Google Scholar]
- 25.Lundgren RJ, Peters BD, Alsabeh PG, Stradiotto M. Angew. Chem. Int. Ed. 2010;49:4071–4074. doi: 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.