

Abstract
The viral population, including bacteriophages, is an important component of the human microbiota, yet is poorly understood. We aim to determine whether bacteriophages modulate the composition of the bacterial populations, thus potentially playing a role in health or disease. We investigated the diversity and host interactions of the bacteriophages of Propionibacterium acnes, a major human skin commensal implicated in acne pathogenesis. By sequencing 48 P. acnes phages isolated from acne patients and healthy individuals and by analyzing the P. acnes phage populations in healthy skin metagenomes, we revealed that P. acnes phage populations in the skin microbial community are often dominated by one strain. We also found phage strains shared among both related and unrelated individuals, suggesting that a pool of common phages exists in the human population and that transmission of phages may occur between individuals. To better understand the bacterium–phage interactions in the skin microbiota, we determined the outcomes of 74 genetically defined Propionibacterium strains challenged by 15 sequenced phages. Depending on the Propionibacterium lineage, phage infection can result in lysis, pseudolysogeny, or resistance. In type II P. acnes strains, we found that encoding matching clustered regularly interspaced short palindromic repeat spacers is insufficient to confer phage resistance. Overall, our findings suggest that the prey–predator relationship between bacteria and phages may have a role in modulating the composition of the microbiota. Our study also suggests that the microbiome structure of an individual may be an important factor in the design of phage-based therapy.
Introduction
The human skin is inhabited by hundreds of microbial species, including bacteria, fungi and viruses. The homeostasis of this ecosystem is important to its function as a barrier against infection and colonization of pathogens on the skin surface. Bacteriophages are important components of the human microbiota. They are a reservoir of diversity-generating elements (Rohwer and Thurber, 2009) and regulate both the abundance and diversity of their bacterial hosts by predation (Suttle et al., 1990; Waterbury and Valois, 1993; Rohwer, 2003; Rodriguez-Valera et al., 2009). Although the skin bacterial community has been studied by several groups (Gao et al., 2007; Costello et al., 2009; Grice et al., 2009; Kong et al., 2012; The Human Microbiome Project Consortium, 2012; Blaser et al., 2013; Fitz-Gibbon et al., 2013; Nakatsuji et al., 2013), relatively few studies have characterized the skin viral community (Foulongne et al., 2012; Ma et al., 2014; Wylie et al., 2014). In particular, the composition and dynamics of bacteriophages and their interactions with bacterial hosts on the skin are not well understood.
The microbial community in the skin pilosebaceous unit is dominated by Propionibacterium acnes, which accounts for nearly 90% of the microbiota (Fitz-Gibbon et al., 2013). Although P. acnes is a major skin commensal, it has been considered a pathogenic factor for acne vulgaris (Leyden, 2001; Bojar and Holland, 2004), one of the most common skin diseases affecting over 80% of adolescents and young adults (White, 1998; Bergler-Czop and Brzezińska-Wcisło, 2013). Our previous 16S ribosomal RNA metagenomic study demonstrated that P. acnes strain population structure in pilosebaceous units differs significantly between acne patients and healthy individuals (Fitz-Gibbon et al., 2013). Certain strains are highly associated with the disease (Lomholt and Kilian, 2010; McDowell et al., 2012; Fitz-Gibbon et al., 2013; Tomida et al., 2013), while some strains are enriched in healthy skin (Fitz-Gibbon et al., 2013).
In parallel, P. acnes phages are dominant bacteriophages in the pilosebaceous unit (Fitz-Gibbon et al., 2013). It has been known for over 50 years that P. acnes phages exist on the human skin (Brzin, 1964). They have the morphology of siphoviruses, consisting of a ~50 nm icosahedral head and a ~150 nm flexible tail (Farrar et al., 2007). Zierdt et al., (1968) isolated phage 174 from spontaneous plaques of a P. acnes isolate. Phage 174 was able to lyse nearly all P. acnes strains tested in the study. Subsequently, more P. acnes phages were isolated, which exhibited lytic as well as pseudolysogenic behavior (Lood and Collin, 2011). However, in the past decades, the study of P. acnes phages had been limited to the development of phage typing systems to distinguish different serotypes of P. acnes (Jong et al., 1975; Webster and Cummins, 1978). Recent sequencing of 14 P. acnes phages (Farrar et al., 2007; Lood and Collin, 2011; Marinelli et al., 2012) suggested that they have limited genetic diversity with over 85% nucleotide identity in the genome. All sequenced genomes are similar in size and structure with 45–47 genes encoded in two oppositely transcribed regions named the left arm and right arm (Farrar et al., 2007; Lood and Collin, 2011; Marinelli et al., 2012).
Much is to be learned about whether bacteriophages drive the diversity and dynamics of the skin bacterial community. The ratio between P. acnes phages and P. acnes was ~1:20 in pilosebaceous units, based on a pooled healthy skin sample that we analyzed previously (Fitz-Gibbon et al., 2013), but can vary in a large range among individuals and over time. P. acnes phages do not encode integrases in their genomes (Farrar et al., 2007; Lood and Collin, 2011), suggesting their inability to stably integrate into the host chromosome. They can kill the host bacteria through cell lysis or can enter a pseudolysogenic state in the host strain (Lood and Collin, 2011), in which the phage DNA persists in infected cells without lysing the host or integrating into its genome. Whether P. acnes phages modulate the relative abundances of different P. acnes strains by selectively killing specific strains of P. acnes and thus play a role in skin health and disease is unknown.
On the other hand, bacterial hosts can influence phage populations through antiviral mechanisms, such as the restriction modification mechanism and the bacterial adaptive immune system utilizing clustered regularly interspaced short palindromic repeat (CRISPR) sequence arrays (Horvath and Barrangou, 2010). In our effort to characterize the strain diversity of P. acnes in the pilosebaceous unit, we discovered that all sequenced type II P. acnes strains harbor Type I-E CRISPR and CRISPR-associated (Cas) proteins (Fitz-Gibbon et al., 2013; Tomida et al., 2013). Marinelli et al. (2012) suggested that the CRISPR mechanism explains the resistance of certain P. acnes strains to phage infection, yet noting that some of their observations were inconsistent with this theory.
To better understand how bacteriophages modulate the bacterial composition of the skin microbiota and their potential roles in skin health and disease, in this study, we determined the diversity of P. acnes phages and their interactions with bacterial hosts in the skin of acne patients and healthy individuals. We sequenced the genomes of 48 P. acnes phages isolated from 37 individuals and investigated whether certain phage strains dominate the skin microbiota. By analyzing the skin metagenome data from the Human Microbiome Project (HMP), we further verified our conclusions from analyzing sequenced phage isolates. We also challenged a panel of 74 genetically defined Propionibacterium strains against 15 of the sequenced phages to determine the outcome and mechanisms of their interactions.
Materials and methods
Phage isolation and DNA extraction
Skin follicle samples were previously collected from the nose of acne patients and individuals with healthy skin as reported in the study by Fitz-Gibbon et al., (2013). To best represent the diversity of populations and history of medical care, the subjects were recruited from private practice, managed care and public hospital settings, as well as outside of dermatology clinics in Southern California. Written informed consent was provided by all study subjects.
The follicle contents collected on the surface of the nose strip were mashed using a sterile loop (Fisherbrand, Pittsburgh, PA, USA), and plated onto a blood agar plate (Teknova Brucella Agar Plate with Hemin and Vitamin K, Teknova, Hollister, CA, USA). The sample plates were incubated at 37 °C for 5–7 days anaerobically using the AnaeroPack System (Mitsubishi Gas Chemical Company, Tokyo, Japan) (Fitz-Gibbon et al., 2013).
Phage plaques observed on the culture plates were isolated by puncturing the agar with a sterile pipette tip and resuspending each tip in 50 μl SM buffer (0.1 M sodium chloride, 8 mM magnesium sulfate heptahydrate, 1M Tris-HCl, pH 7.5, 2% gelatin and 1 mM calcium chloride). Each phage resuspension was spread onto A-media plates (12 g l−1 pancreatic digest of casein, 12 g l−1 yeast extract, 22.2 mM D-glucose, 29.4 mM g l−1 potassium phosphate monobasic, 8 mM magnesium sulfate heptahydrate and 20 g l−1 agar) with top agar containing P. acnes strain ATCC6919. After incubation at 37 °C for 2 days, single plaques were selected and propagated using the same host strain, medium and incubation conditions. Suspensions of each phage isolate were prepared by eluting plates with 8 ml SM buffer at room temperature, filtering with a 0.22-μm PES filter (Millipore, Billerica, MA, USA) to remove bacterial cells, and storing at 4 °C. Phage titers were determined by plaque assay.
Phage DNA extraction was performed using the Lambda Mini Kit (Qiagen, Valencia, CA, USA) with the following modifications. Phage particles were precipitated in Buffer L2 by centrifugation at 20 000 g at 4 °C for 1 h. Extracted DNA was precipitated overnight at −20 °C before centrifugation.
Phage electron microscopy
Copper grids (400 mesh formvar per carbon film) (Ted Pella, Redding, CA, USA) were glow discharged. Phage cultures were applied, followed by a wash with 0.22-μm-filtered water. The samples were stained with 1% uranyl acetate and examined under a JEOL JEM-1200EX electron microscope (JEOL, Peabody, MA, USA) with an accelerating voltage of 80 kV.
Phage genome sequencing, assembly and annotation
Phage genomes were sequenced in multiplex using the Roche GS FLX Titanium (Roche, Branford, CT, USA) or the Illumina MiSeq (Illumina, San Diego, CA, USA) platform. Sequence reads were initially assembled using MIRA 3.2.1 (Chevreux et al., 1999), and the resulting contigs were manually finished in Consed 23.0 (Gordon et al., 1998). Some phage genomes required additional PCRs and amplicon sequencing to fill the gaps between contigs. Fully assembled phage genomes were annotated using Genemark.hmm (Lukashin and Borodovsky, 1998) and Glimmer v3.02 (Delcher et al., 1999) with manual corrections. All phage genome sequences have been deposited in GenBank under BioProject PRJNA173665 with accession numbers JX570702-JX570714, KJ578758-KJ578792.
Genome analysis and phylogenetic tree construction
Sequences present in all 62 phage genomes were defined as core regions of the phage genome. To identify these core regions, we first generated alignments between the PA6 genome and each of the other 61 phage genomes using Nucmer (Kurtz et al., 2004). This yielded 61 sets of starting and ending coordinates describing intervals within the PA6 genome that align with a given phage genome. We then calculated the core regions for all phages by determining the overlapping intervals between all of the 61 coordinate sets. The core region sequences were concatenated for the subsequent multiple sequence alignments. Single nucleotide polymorphisms (SNPs) in the core regions were identified by using the ‘show-snps' option of Nucmer with the default setting. In addition, the set of non-synonymous SNPs was obtained by masking the third codon positions in the coding regions. Using MEGA5 (Tamura et al., 2011), phylogenetic trees were constructed by the Neighbor Joining method from p-distances based on all SNP sites in the core regions or only the non-synonymous SNPs. Bootstrapping was performed on 1000 replicates.
Analysis of nucleotide polymorphism within single phage strains
The genetic variation within a phage strain was measured by the number of SNPs found in the sequencing data from a clonal phage population. The SNPs were identified as sites in a strain's genome assembly that were covered by ⩾30 reads with Phred quality score ⩾30 and with ⩾10% of these reads differing from the consensus sequence.
Analysis of metagenomic shotgun sequencing data of the skin microbiota
Phage diversity was analyzed in the metagenomic shotgun sequencing data from 27 retroauricular crease samples collected in the HMP (The Human Microbiome Project Consortium, 2012). The SRA accessions of these samples are SRS013261, SRS024598, SRS013258, SRS024596, SRS019016, SRS019015, SRS019033, SRS019063, SRS019064, SRS019081, SRS024655, SRS024620, SRS020263, SRS020261, SRS017851, SRS017849, SRS057083, SRS024482, SRS045606, SRS058221, SRS018978, SRS058182, SRS016944, SRS046688, SRS015381, SRS052988 and SRS019116. Access to the phenotype data for this study (phs000228.v3.p1) was obtained from dbGaP. MIRA (Chevreux et al., 1999) was used to identify phage reads in each data set by mapping against the P. acnes phage PA6 genome. Parameters similar to the default were used: ⩾20 nt overlap and ⩾60% identity. To estimate the number of P. acnes phage strains in each data set, we first performed a de novo assembly using the extracted phage reads from the metagenomic data. We then aligned all the phage reads to the resulting contigs to identify SNPs in the core genome regions. The assemblies and alignments were manually inspected using Consed (Gordon et al., 1998). The same criteria used for nucleotide polymorphism identification in single phage strains were applied as described above.
Analysis of phage genes under diversification
Multiple sequence alignments of Group VI and Group VIII phages and their related phages (PHL037M02 and PHL073M02) were generated using MAFFT (Katoh et al., 2002). The positions of all mismatches and gaps were recorded. Sites of discrepancy were plotted in Artemis (Rutherford et al., 2000).
Propionibacterium culture
P. acnes, P. humerusii, P. granulosum and P. avidum strains were cultured under anaerobic conditions in Clostridial Reinforced medium (Oxoid, Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for 4–6 days. Propionibacterium cultures were used to prepare top agar overlays for phage culture on A-media plates.
Phage resistance test
The resistance/susceptibility of Propionibacterium strains against phages was determined using a modified cross-streak assay. Fifteen of the 48 newly sequenced phages were randomly chosen for the analysis. Two sets of phages that each belongs to the same group, PHL010M04 and PHL066M04 in Group VIII and PHL115M02, PHL085N00, and PHL085M01 in Group VI, were included. The bacterial strains were streaked across in A-media plates, along with ATCC6919 on the same plate as a control. Approximately 5 μl of 106 PFU ml−1 phage suspension was spotted onto each bacterial streak. The plates were incubated at 37 °C anaerobically for 2 days. At least five replicates of each cross-streak experiment were performed to determine whether the strains were susceptible or resistant.
For the strains that showed resistance in the modified cross-streak experiment, we further quantitatively determined the resistance by assaying the efficiency of plaquing of the phages relative to P. acnes strain ATCC6919, calculated as the following:
 |
We considered a 100-fold or greater increase in efficiency of plaquing to be evidence of resistance.
The plaques on cross-streak plates were visually inspected by one person and scored for turbidity based on the re-growth of the bacteria after plaque formation using the following scale: 0=clear, 1=little to no re-growth, 2=mild re-growth, 3=moderate re-growth and 4=heavy re-growth. The average plaque turbidity score of all the strains from the same P. acnes clade was calculated for each of the tested phages and was compared among different clades.
Pseudolysogeny characterization
PCR was performed on phage suspensions using the primers annealing to the ends of the phage genomes (Forward 5′-CCGAAGCCGACCACATCACACC-3′, Reverse 5′-TCATCCAACACCTGCTGCTGCC-3′) to determine whether phage genomes are circularized. The PCR was run under the following conditions: initial denaturation at 94 °C for 5 min, 35 cycles of denaturation at 94 °C for 45 s, annealing at 53 °C for 35 s and extension at 72 °C for 1 min, with a final extension at 72 °C for 10 min.
P. acnes ATCC6919 cultures, which were re-grown after lytic infection with specific phages, were tested for superinfection immunity by passaging sequentially two to four times without further phage infection. Phage resistance was assayed using the same cross-streak method described above. The presence of phage DNA in re-grown cultures was determined by PCR using the primers targeting the phage gp11 gene (Forward 5′-GGCTGGAACACGTAAAGCG-3′, Reverse 5′-CACGATCGATCAACTCAACC-3′). The PCR was run under the following conditions: initial denaturation at 94 °C for 5 min, 35 cycles of denaturation at 95 °C for 45 s, annealing at 58 °C for 35 s and extension 72 °C for 1 min, with a final extension at 72 °C for 10 min.
CRISPR analysis
CRISPR spacer sequences were previously identified in P. acnes genomes (Fitz-Gibbon et al., 2013; Tomida et al., 2013). The spacer sequences were aligned against all phage genomes using BLASTn. Protospacers with up to two mismatches were identified.
Results
Phage isolation and genome features
In an effort to determine the diversity of the skin microbiota, we previously collected 203 skin samples from 179 individuals, including 94 samples from acne patients and 109 samples from healthy individuals with clear skin (Li, 2010; Fitz-Gibbon et al., 2013). Twenty-four individuals were sampled twice over a 4–6 month period. When we cultured the skin samples for bacteria, we observed phage plaques in 49 of these samples: 14 from acne patients and 35 from healthy individuals. P. acnes phages were found more frequently in samples from healthy individuals than from acne patients. Our rate of phage detection (24% of investigated samples) is similar to those previously reported, ranging from 26 to 30% (Marples et al., 1973; Puhvel and Amirian, 1979). The phages that we isolated have the morphology of siphoviruses as previously described (Farrar et al., 2007). A representative electron micrograph is shown in Supplementary Figure S1. Among the phage isolates obtained from these samples, we selected 21 phages from acne patients and 27 from healthy individuals for whole genome sequencing using 454 or MiSeq platforms (Supplementary Table S1). In some samples, multiple phage plaques were isolated and selected for sequencing. All phage genomes were assembled, completed and annotated (Supplementary Figure S2). A representative phage genome is shown in Figure 1.
Figure 1.
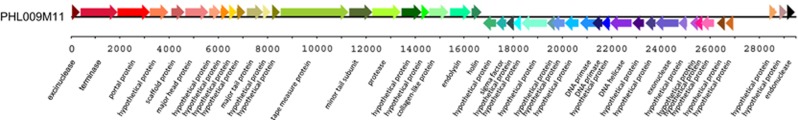
A representative genome of the newly sequenced P. acnes phages. The annotated genome of PHL009M11 is shown as a representative of the 48 newly sequenced P. acnes phage genomes. On average 45 open reading frames are encoded in each phage genome.
The P. acnes phage genomes are highly similar to each other (Supplementary Figure S2). The 48 phage genomes have comparable sizes (29.0–29.8 Kb) and GC contents (53.7–54.5%) (Supplementary Table S1). The sequence identity between any pair of genomes ranges from 85.2 to 100%. On average 45 open reading frames were predicted in each genome. Consistent with previous reports (Farrar et al., 2007; Lood and Collin, 2011; Marinelli et al., 2012), these open reading frames were arranged compactly within the left and right arm regions of the genome (Figure 1, Supplementary Figure S2). Our analysis of the 48 new phage genomes supports the annotation of the gp22/gp23 locus as a single open reading frame (495 to 522 bp) on the minus strand (Supplementary Figure S2). This is different from previous annotations based on a small number of genomes (Farrar et al., 2007; Lood and Collin, 2011).
Phylogenetic relationships among the phage genomes
To determine the genome diversity of P. acnes phages, we compared 62 sequenced phage genomes, including our 48 phage genomes and 14 previously published genomes (Farrar et al., 2007; Lood and Collin, 2011; Marinelli et al., 2012). Similar to their bacterial host, P. acnes phages have limited genomic diversity. All 62 phages are highly similar in genome sequence. The core regions, which are shared by all sequenced genomes, consist of 22 348 bp (76% of the average genome length) and contain 7232 SNPs. The average distance among the phages was 0.257 (substitution rate at the SNP sites). A phylogenetic tree constructed based on these SNPs (Figure 2), or only the non-synonymous SNPs (Supplementary Figure S3), shows that no particular phylogenetic clades were found among the phages.
Figure 2.

P. acnes phages are highly similar to each other with no significant phylogenetic lineages observed. A phylogenetic tree of the 62 currently sequenced phage genomes was constructed based on the 7232 SNPs in the core regions. No significant lineages were observed. Nine groups of nearly identical phages are indicated, highlighting that the phages within each group belong to the same strain. Branches with bootstrap values <80 (based on 1000 resamplings) were collapsed.
Despite the lack of phylogenetic lineages among the P. acnes phage genomes, we observed several groups (I–IX), each of which consists of nearly identical phages (Figure 2). The phages within each group (I–IX) differ by no greater than 14 bp within the entire 29 kb genome (Supplementary Table S2). As a comparison, the average pairwise difference among all 62 phages is 3176 bp. To determine whether the nearly identical phages within each group represent clones of the same phage strain, we estimated the frequency of genetic polymorphism in each of our phage isolates. We mapped all available sequence reads of each phage to its assembled consensus genome, including the sequence reads not used in the genome assembly process. We identified the nucleotide positions with a minor allele frequency ⩾10% and covered by at least 30 reads with Phred quality ⩾30. We found that each phage genome contains 0–11 polymorphic sites (Supplementary Table S1). The number of polymorphisms in each assembled genome did not increase beyond 11 sites despite the large numbers of sequencing reads obtained (up to 9120 × genome coverage). From this analysis, we conclude that the background level of genetic polymorphism within a clonal P. acnes phage isolate is ~11 bp. Thus, phage isolates with a similar or smaller number of nucleotide differences throughout the entire genome can be considered as belonging to the same phage strain. Since the phages within each group (I–IX) differ by at most 14 bp (Supplementary Table S2), they likely represent clones of the same phage strain.
Diversity of P. acnes phages in the human skin
The relationships among the phages within each group (I–IX) provide insights on the diversity of P. acnes phages in the skin microbiota. We observed three types of relationships among the highly similar phages within the groups (Figure 2 and Supplementary Table S2). First, phages within the same group were isolated from the same sample of the same individual. These include PHL067M01, PHL067M09 and PHL067M10 in Group I; PHL082M00, PHL082M02, PHL082M03 and PHL082M04 in Group II; PHL064M01 and PHL064M02 in Group V and PHL116M00 and PHL116M10 in Group IX. Only one other pair of phages, PHL117M00 and PHL117M01, which were isolated from the same sample, was not the same strain. Our data suggest that while an individual microbiota can harbor multiple strains of phages, it is likely more common that one strain of P. acnes phage dominates the phage population. Second, phages within the same group were isolated from different samples of the same individuals over a period of 14 to 21 weeks. These include PHL085M01 and PHL085N00 in Group VI; PHL114L00 and PHL114N00 in Group VII and PHL151M00 and PHL151N00 in Group VIII. All paired phages from our longitudinal samples were nearly identical to each other. This suggests that the same phage strain can persist in an individual skin microbiota. Third, some phages within the same group were isolated from different individuals, such as the phages in Groups III, IV, V, VI and VIII. Of the 43 unique phage strains represented by all 62 isolates sequenced to date, 5 strains from our study currently show evidence of inhabiting more than one individual. This suggests that a pool of common P. acnes phage strains exists in the human population. Interestingly, among the five shared strains, two inhabited related individuals. In Group IV, the two nearly identical phages, PHL150M00 and PHL308M00, were isolated from two brothers. Two of the four phages in Group VIII, PHL010M04 and PHL066M04, and their closely related phage strain PHL073M02 were isolated from three siblings (Figure 2 and Supplementary Table S2). This suggests that transmissions of skin bacteriophages, either directly or via the transmission of phage-carrying bacterial hosts, are likely to occur between related individuals.
P. acnes phage populations in the skin microbiome
To validate that our above findings on the phage diversity in the human skin microbiota were not biased due to isolated phages, we analyzed the skin metagenomic shotgun sequencing data collected from healthy individuals in the HMP (The Human Microbiome Project Consortium, 2012). Although P. acnes phages were previously found in metagenomic sequencing data, their diversity was not analyzed. This is the first time that the population diversity of P. acnes phages in the skin microbiome is characterized. Among the available 27 HMP skin samples, 9 were collected from the left retroauricular crease and 18 were collected from the right retroauricular crease (Supplementary Table S3). Some samples were collected from the same individuals. We first extracted the P. acnes phage reads from each sample. The number of phage reads was from 24–612 512 (0–2060 × coverage on PA6 genome, Supplementary Table S3), independent of the sequencing depth of each sample, suggesting that the relative abundance of P. acnes phages in the skin microbiota can vary in a large range among individuals.
To determine P. acnes phage populations in individual skin microbiota, we next assembled P. acnes phage genomes in each metagenomic data set. Seven samples (25.9% of total samples) had a modest to high sequencing coverage of P. acnes phages (17 × –2060 × ) (Supplementary Table S3), which allowed metagenomic assembly. We performed the SNP analysis on the assembled phage genomes using the same criteria as in the analysis of complete phage genomes described above. To evaluate the effect of sequencing depth on the detection rate of SNPs, for samples with >30 × phage coverage, we repeated the phage genome assembly and SNP analysis using only portions of phage reads at various coverages (20 × , 100 × , 250 × , 500 × , 1000 × and 1500 × ), as applicable. One of the samples, HMP20, had >1000 SNPs in the core regions of the assembled phage genome, which leveled off to 1353 sites when all the sequence reads (538 × ) were used in the assembly (Figure 3, Supplementary Table S4). This suggests that the P. acnes phage pool in HMP20 was adequately sampled and that it likely consists of two dominant phage strains based on phage genome comparison (Supplementary Materials).
Figure 3.

P. acnes phage population in the skin microbiota is often dominated by a single strain. The numbers of SNPs identified in the core regions of P. acnes phage genomes are shown at different sequencing coverages. The phage genomes were assembled from the HMP metagenomic shotgun sequencing data.
On the other hand, three other samples, HMP04, HMP09 and HMP15, each had six or fewer SNPs in the core genome regions, despite having substantially higher sequencing coverages than HMP20. This suggests that these samples each harbor only one dominant P. acnes phage strain. This result supports our earlier conclusion that an individual skin microbiota is often dominated by one P. acnes phage strain. We were able to assemble high-quality draft genomes of P. acnes phages from these three metagenomic data sets, which cover 96.4–96.7% of the core genome regions. They are typical of the P. acnes phage strains, with high similarities to the 62 sequenced genomes. A phylogenetic tree including these three new genomes with the 62 isolated phage genomes is shown in Supplementary Figure S4. Samples HMP04 and HMP09 were collected from the left and right retroauricular crease of the same individual, and the phage genomes assembled from these two samples are highly similar, potentially originating from the same strain.
The assembled phage genomes in the remaining three samples, HMP03, HMP08 and HMP24, contained <20 SNPs, which are in the range of the SNPs found in a single phage strain. Due to their lower phage coverages, it is yet inconclusive whether single or multiple dominant phage strains were present in these communities.
Our analysis of the phage diversity from the HMP metagenomic data showed that among the skin communities that had detectable P. acnes phages, three harbored only one dominant strain while one harbored two different strains. These findings are consistent with our conclusions based on the isolated phages from our study cohort.
Phage genes under diversification
Phylogenetically related strains can reflect phage diversification under selection. Two phages, PHL037M02 and PHL073M02, are highly related to the members of Groups VI and VIII, respectively (Figure 2). However, they contain many more nucleotide variations than 11 bp, and thus are considered separate strains based on our criterion. Nonetheless, their high degree of similarity to the members of these groups may reflect recent selective pressures driving phage diversification. We identified 160 sites of nucleotide variations between PHL037M02 and the Group VI members, 50 of which are non-synonymous. All but one of these genetic differences is located in a region encoding Gp16, Gp17 and Gp18, as annotated in the genome of phage PA6 (Supplementary Figure S5A). The exact functions of these genes are unknown, but their location near the 3' end of the left arm between structural protein genes and lysis protein genes suggests that they could encode late-acting proteins. Based on the McDonald–Kreitman test (Egea et al., 2008), gene gp17 is under selection (P=0.011).
We identified 81 sites of nucleotide variations between PHL073M02 and the Group VIII members. The sequence differences lie primarily within the region encoding an endolysin and a putative type II holin (Gp20 and Gp21, Supplementary Figure S5B). These lytic cycle proteins permeabilize the cell membrane and degrade the extracellular peptidoglycan layer to release new phage particles from the bacterial host. As previously mentioned, PHL073M02 and two Group VIII members, PHL010M04 and PHL066M04, were isolated from three siblings living in the same household. Thus, they likely originated from the same ancestral phage strain. This suggests that the endolysin and holin genes, which are essential for phage multiplication, are under rapid evolution. Consistent with this finding, in all sequenced phages, we found frequent amino acid variations in these two gene products. This suggests that mechanisms determining host bacterium lysis specificity and kinetics may be under selection in these phages. The sequence variation sites in these two genes among the phages may be potential targets for phage engineering to manipulate their lytic activities against bacterial hosts.
The two highly similar phage genomes assembled from the HMP samples, HMP04 and HMP09, which were collected from the same individual, differed by 222 nucleotides. Most of these variations are centered at the 5′ end of the right arm of the genome, which encodes putative regulatory or DNA-binding proteins of largely unknown functions.
Range and specificity of Propionibacteria–phage interactions
To determine whether P. acnes phages modulate the relative abundances of different P. acnes strains in the skin microbiota by selective killing, we characterized the host range and specificity of P. acnes phages. We tested 15 of the 48 sequenced phages against a panel of 74 Propionibacterium strains, including 67 P. acnes strains, 3 P. humerusii strains, 1 P. granulosum strain and 3 P. avidum strains. Except for the P. acnes strains KPA171202, ATCC11828, HL201PA1 and HL202PA1, all of these Propionibacterium strains were isolated from the same cohort of subjects sampled for phages. The genomes of all 67 P. acnes strains and 3 P. humerusii strains have been sequenced (Fitz-Gibbon et al., 2013; Tomida et al., 2013). Our bacterial collection included all major lineages of P. acnes found on the human skin, with multiple strains representing each of the major clades, IA-1, IA-2, IB-1, IB-2, IB-3 and II, as well as one type III strain. We constructed a phylogenetic tree of the 67 P. acnes strains based on the SNPs in their core genomic regions (Figure 4a) (Tomida et al., 2013). Using a modified cross-streak method, we determined the resistance/susceptibility of each of the 74 bacterial strains against the 15 phages. In total, 1110 bacterium–phage interactions were measured. Each experiment was repeated a minimum of five times. For the bacterial strains that showed resistance to phages, we determined the fold increase in resistance by measuring efficiency of plaquing (EOP) relative to the P. acnes strain ATCC6919, which is known to be susceptible to all tested phages.
Figure 4.

Host range and specificity of P. acnes phages. Resistance/susceptibility of Propionibacterium strains (in rows) against 15 of the 48 sequenced phages (in columns) is shown. (a) Phage infection outcomes of 67 P. acnes strains. All P. acnes strains in clades IA-1, IA-2, IB-1 and IB-2 were susceptible to the tested phages, while phage resistance was found in strains of clades IB-3, II and III, colored in pink. The dendrogram to the left shows the phylogenetic clades of P. acnes strains (Fitz-Gibbon et al., 2013). Only topology is shown. (b) Phage infection outcomes of three P. humerusii strains, one P. granulosum strain and three P. avidum strains show that P. acnes phages could infect and lyse P. humerusii strains, while P. granulosum and P. avidum were resistant to all tested phages.
We found that the outcome of the bacterium–phage interactions is P. acnes lineage dependent. All type I P. acnes strains (clades IA-1, IA-2, IB-1 and IB-2) except clade IB-3 were susceptible to all tested phages (Figure 4a). Among them, the phages often formed turbid plaques on P. acnes strains of clade IA-1, but clear plaques on strains of clades IB-1 and IB-2, as summarized in Figure 5. This suggests that these phages engage in two different states depending on the host strains: a pseudolysogenic response in clade IA-1 strains, and a lytic cycle in clade IB-1 and IB-2 strains.
Figure 5.

The frequency of phage pseudolysogeny varies among different P. acnes strains. The turbidity of the phage plaques formed on P. acnes culture plates was examined. Data from 60 P. acnes strains challenged by 13 phages were recorded and summarized. The 60 P. acnes strains belong to five clades: IA-1 (n=16), IA-2 (n=14), IB-1 (n=6), IB-2 (n=17) and II (n=7). The average turbidity score of the bacterial strains in each clade challenged by each phage (in colors) is shown. Independent of the phages tested, the plaque turbidity score varied among different P. acnes strains: it is high in clade IA-1 and low in clades IB-1 and IB-2. This suggests that the phages tend to enter a pseudolysogenic state in clade IA-1 strains and a lytic cycle in clades IB-1 and IB-2 strains.
Certain P. acnes strains of clades IB-3, II and III are highly resistant to multiple phages. Two strains of clade IB-3 (KPA171202 and HL030PA1) were highly resistant to most of the tested phages with a ⩾100-fold increase in resistance. The genomes of clade IB-3 show that these strains encode components of a restriction modification system (genes PPA1611 and PPA1612 in KPA171202). This may explain their resistance to phages. Among the nine type II strains, two strains, HL001PA1 and HL042PA3, were highly resistant to some of the phages. This is consistent with previous observations that strains of this type were more frequently resistant to phages (Webster and Cummins, 1978). The resistance to phages observed in type II strains could be partially attributed to the CRISPR mechanism encoded in their genomes, which is addressed below. The only type III strain tested, HL201PA1, was resistant to all 15 phages. Since type III strains are not commonly found on the skin of the face, it is possible that these P. acnes phages isolated from the face have not yet evolved a mechanism to infect type III strains.
To determine whether P. acnes phages modulate the abundance and diversity of other species in addition to P. acnes in the skin microbiota, we investigated the host range of P. acnes phages in related species. Strains of other human skin-associated Propionibacteria, including 3 strains of P. humerusii, 1 strain of P. granulosum and 3 strains of P. avidum, were tested against the 15 phages (Figure 4b). P. humerusii is a newly defined species (Butler-Wu et al., 2011). In our previous study, P. humerusii was one of the major species found on the skin with a relative abundance of 1.9% in the pilosebaceous unit based on 16S ribosomal RNA analysis (Fitz-Gibbon et al., 2013). It is closely related to P. acnes with >98% identity in the 16S ribosomal RNA gene sequence. P. granulosum and P. avidum are common skin commensals (Cummins, 1976; Ördögh and Hunyadkürti, 2013). While all tested P. granulosum and P. avidum strains showed strong resistance to all the phages, two P. humerusii strains, HL037PA2 and HL037PA3, were susceptible to all the phages tested. The third P. humerusii strain, HL044PA1, was susceptible to 10 of the 15 phages tested. Our results show that the host range of P. acnes phages is not limited to P. acnes but also includes a closely related Propionibacterium species, suggesting that P. acnes phages may also be able to modulate P. humerusii populations in the skin microbiota.
P. acnes phages can adopt a pseudolysogenic state depending on the host P. acnes strains
It has been suggested that P. acnes phages may enter pseudolysogeny as an alternative to the lytic cycle (Farrar et al., 2007; Lood and Collin, 2011). As described above, we discovered that P. acnes phages often adopt a pseudolysogenic state in clade IA-1 strains, but rarely in clade IB-1 and IB-2 strains, suggesting that their pseudolysogeny is dependent on the host P. acnes strains (Figure 5). In support of the existence of pseudolysogeny in P. acnes phages and consistent with the result by Marinelli et al., (2012), our genome sequencing data revealed the ends of the phage genomes to be flanked by 11-nucleotide single-stranded overhangs. Previous reports suggested that these overhangs may be involved in the circularization of the phage genomes (Farrar et al., 2007; Lood and Collin, 2011). To test this hypothesis, we designed PCR primers annealing to the ends of the phage genomes. A PCR product spanning the two ends with the predicted size (~735 bp) and sequence was amplified from all the phages tested, suggesting that the phage DNA can exist in a circular form mediated by the overhangs at the ends (Supplementary Figure S6A).
To demonstrate the pseudolysogenic properties of P. acnes phages, we infected P. acnes strain ATCC6919, a clade IA-1 strain (Liu et al., 2014), with five different phages (PHL060L00, PHL112N00, PHL037M02, PHL073M02 and PHL114L00). Following the formation of plaques by each phage, re-growth of the bacteria was observed starting in the center of the plaque regions. The re-grown bacteria showed no evident lysis when challenged by 13 phages that previously could lyse this P. acnes strain, including the phages of the initial exposure (Supplementary Figure S6B). This suggests that the bacterial host gained superinfection immunity after infection by these phages.
To further determine whether the phage DNA exists as an episome in the bacterial host after infection, we tested the presence of phage DNA in four distinct colonies of the re-grown ATCC6919 culture that was initially infected with PHL060L00 and subsequently passaged. Two of the four colonies produced an expected 437 bp amplicon in a PCR targeting the gp11 gene (Supplementary Figure S6C), supporting the presence of phage in a subpopulation of the re-grown P. acnes as an episome. In addition, we sequenced the genomic DNA extracted from two ATCC6919 cultures, each of which was passaged from a re-grown culture after phage infection. In both cases, the phage reads obtained from whole genome sequencing were assembled into complete genomes that are separate from host P. acnes contigs, supporting earlier evidence that P. acnes phage DNA does not integrate into the host genome (Farrar et al., 2007; Lood and Collin, 2011), but exists as an extrachromosomal element.
In summary, all above results suggest that P. acnes phages can adopt a pseudolysogenic state in clade IA-1 strains.
Resistance to bacteriophages does not correlate with the presence of matching CRISPR spacers in type II P. acnes strains
Since certain type II P. acnes strains are resistant to phages, we next investigated whether the CRISPR/Cas mechanism could explain the resistance of the type II strains against phages. Among the 67 P. acnes strains, 9 belong to type II and encode CRISPR/Cas elements. Each of the type II strains has two to nine 33-bp spacers in their CRISPR arrays (Tomida et al., 2013). In total, they encode 44 spacers, 28 of which are unique. We identified 42 unique protospacers in the 15 tested phage genomes that match any of the 28 unique spacer sequences in the 9 type II P. acnes strains. Because the CRISPR/Cas system has been shown to tolerate a limited number of mutations in protospacer targets (Semenova et al., 2011; Manica et al., 2013), in our analysis we allowed up to two mismatches for a sequence to be considered a recognizable protospacer. Similar results were obtained when only perfectly matching protospacers were considered. We found that all identified protospacers are located primarily on the left arm of the phage genomes, which is more conserved than the right arm (Supplementary Figure S7). In addition, the locations of the protospacers are generally conserved among all phage genomes that harbor the same protospacer sequences. These suggest that the CRISPR/Cas system tends to target the more conserved regions of the phage genomes.
In contrast to a prior report (Marinelli et al., 2012), we found that the resistance/susceptibility of the nine type II P. acnes strains against phages did not correlate with the presence/absence of at least one phage-matching CRISPR spacer (r=0.234, Figure 6). There were multiple observations that even though the strain encodes a matching CRISPR spacer, it was susceptible to the corresponding phage. To determine whether more than one matching spacer is needed to confer phage resistance, we analyzed the correlation between the resistance/susceptibility and the number of matching spacers encoded in the CRISPR array. No strong correlation (r=0.26) was found, suggesting that encoding multiple matching spacers does not necessarily protect the strain from phage infection. In addition, no presence of particular spacer sequences can explain the resistance/susceptibility to phages. When we allowed no mismatches or up to five mismatches for a sequence to be considered a recognizable protospacer, the correlations were not much improved. This suggests that having matching CRISPR spacers is insufficient for these strains to be resistant to phages and that other regulation mechanisms are likely to be involved.
Figure 6.

Phage resistance is not correlated with the presence of matching CRISPR spacers in type II P. acnes strains. The pattern of resistance/susceptibility of the 9 type II P. acnes strains against 15 phages did not correlate with the presence/absence of matching CRISPR spacers, that is, the pink cells do not perfectly overlap with the cells containing red or orange pixels. P. acnes type II strains are shown in rows and the phages are shown in columns. Pink cells indicate that the bacterial strains are resistant to the corresponding phages. The colored pixels in each cell represent the CRISPR spacers encoded in each P. acnes strain. Each red pixel indicates that this spacer has an exact protospacer match in the corresponding phage. Each orange pixel indicates that this spacer has a partially matched protospacer (with one to two mismatches) in the corresponding phage. Gray pixels indicate no matched protospacers found in the corresponding phage.
Phages can escape the CRISPR/Cas mechanism through mutations at sites involved in protospacer recognition. The short nucleotide motif adjacent to the protospacer, known as the protospacer-adjacent motif (PAM), is highly conserved among targets of CRISPR/Cas systems (Mojica et al., 2009). Mutations in these nucleotides have been found to disrupt CRISPR-mediated resistance despite complete identity in the protospacer sequence (Semenova et al., 2011, 2009; Westra et al., 2012).
To determine whether the lack of correlation between bacterial resistance and the presence of matching spacer sequences is due to mutations in the PAM sequence, we examined the PAM regions of the 137 protospacers identified in the phages from this study and from the study by Marinelli et al., (2012). The protospacers can be separated into two groups depending on the outcomes of the bacterium–phage interaction—resistant or susceptible to the phages (Figure 7). Fifty-three protospacers belong to the first group, where the bacterial hosts with matching spacer sequences were resistant to the phages. The other 84 protospacers belong to the second group, where lysis of the bacterial hosts occurred despite the presence of a matching spacer. Among the first group of protospacers, we observed a conserved ‘AA' motif immediately upstream of the protospacer sequence (Figure 7a). The AA motif is also conserved in the PAM sequences of the 84 protospacers from the second group where the bacterial hosts were susceptible to the phages, except in 3 cases, where an A-to-G mutation occurred. These three mutated positions were found in phages PHL060L00 and PHL112N00, which are targeted by spacers encoded in P. acnes strain HL042PA3, and in phage PHL113M00, which is targeted by a spacer encoded in strain HL110PA3 (Figure 7b). However, these mutations in the PAM sequence cannot explain the lack of resistance against these phages, as all three phages contain one to three additional protospacers that are recognizable by the spacers encoded in the corresponding bacterial strains. All of these additional protospacers have intact PAMs. Our findings are corroborated by the previous observation of unexplained susceptibility to P. acnes phage despite the presence of matching spacers (Marinelli et al., 2012). In summary, our data demonstrate that encoding CRISPR spacers that match against the genome of an invading P. acnes phage is often insufficient for an effective defense in P. acnes and that other regulation mechanisms are likely to be involved.
Figure 7.

Sequence conservation in protospacers and PAMs. A logo representation of the protospacer sequences that match P. acnes CRISPR spacers (within two mismatches) is shown. The PAM sequence of the protospacers is also shown. (a) Sequence conservation among the protospacer motifs from the phages unable to lyse the corresponding bacterial strains. (b) Sequence conservation among the protospacer motifs from the phages able to lyse the corresponding bacterial strains. The PAM sequences are conserved in both cases.
Discussion
The skin microbial community has an essential role in defending against the invasion and colonization of pathogens. Although the bacterial and fungal compositions of human skin have been characterized at multiple sites in healthy individuals (The Human Microbiome Project Consortium, 2012; Gao et al., 2007; Paulino et al., 2008; Grice et al., 2009; Gao et al., 2010; Ursell et al., 2012; Blaser et al., 2013; Findley et al., 2013; Fitz-Gibbon et al., 2013; Nakatsuji et al., 2013), we have limited understanding of the skin virome (Foulongne et al., 2012; Ma et al., 2014). Whether bacteriophages have a role in skin health and disease by modulating the diversity and dynamics of the bacterial community on the skin is poorly understood.
The lack of knowledge of the skin virome is partly due to the limited microbial biomass of skin samples, which poses challenges in determining the composition and diversity of skin viruses. As methods are being developed in recent years for high-throughput sequencing from a limited amount of DNA (Duhaime and Sullivan, 2012; Duhaime et al., 2012; Solonenko et al., 2013) and for capturing metagenomic viral sequences in metagenomic sequencing data (DeLong et al., 2006; Mizuno et al., 2013), it is becoming feasible to study lowly abundant viral communities directly from metagenomic samples without enriching the viral portion. In this study, using genome sequencing and metagenomic analysis of the skin microbiome, we characterized a large number of P. acnes bacteriophages and their genome diversity in the skin of healthy individuals and acne patients. This effort significantly increased our knowledge about this viral population and allowed us to investigate its population diversity in the human skin microbiota.
Unlike phages of other species (Nolan et al., 2006; Dorscht et al., 2009; Sepúlveda-Robles et al., 2012), P. acnes phages show a high genetic similarity. The phages may have evolved recently under limited selection, as no clear lineages can be found among the phages studied, suggesting that these phages may be relatively new to their hosts (Marinelli et al., 2012). Alternatively, the high similarity among the phages may be attributed to the low genetic diversity of their host strains, as P. acnes has limited genome expansion among different lineages (Tomida et al., 2013).
By comparing the phages isolated from different individuals (Figure 2), we found that in individuals harboring P. acnes phages, the skin community is often dominated by one P. acnes phage strain. Our metagenomic data analysis of the skin samples from the HMP cohort confirmed this finding (Figure 3), despite the differences between the two analyses in methodology, study cohorts and skin sites that were sampled. We also observed the same phages in the skin microbiota when the individuals were sampled 14–21 weeks later (Supplementary Table S2). Our findings are consistent with the intra-individual temporal stability found in the human oral and gut viromes (Pride et al., 2012; Minot et al., 2013). Further studies at multiple time points are needed to determine the dynamics of P. acnes phage populations in the skin microbiota. In addition, we found that while many individuals harbor unique phage strains (Figure 2), some individuals share the same phage strains in the skin microbiota, suggesting that a pool of common phages exists among the human population. Lastly, we isolated identical phage strains from siblings (Supplementary Table S2), suggesting possible transmission of skin viruses between related individuals.
Pseudolysogeny occurs abundantly in natural environments and is an important part of bacteria–phage interactions (Ripp and Miller, 1997). It has been suggested to influence phage survival (Ripp and Miller, 1997), phage-dependent bacterial mortality (Ripp and Miller, 1998) and the virulence of certain bacterial strains (Sakaguchi et al., 2005). Although it has been observed that P. acnes phages can adopt pseudolysogeny (Lood and Collin, 2011), for the first time we revealed that this behavior is largely dependent on the lineages of the host strains and is not a property of the phages. All tested P. acnes phages more frequently become pseudolysogenic when the host P. acnes strains are from clade IA-1, compared with other lineages (Figure 5). We further determined several pseudolysogenic characteristics of P. acnes phage infection (Supplementary Figure S6). These include the ability of the phage genome to exist as a circular episome in the host cells, superinfection immunity of the host bacteria gained from prior phage infection and phage DNA persistence in a subpopulation of the host cells following infection. While the term ‘pseudolysogeny' is used here and in the previous studies (Lood and Collin, 2011; Marinelli et al., 2012) to describe this behavior of P. acnes phages, further mechanistic definitions are needed to determine whether this infection state represents phages undergoing extrachromosomal lysogeny, inefficient lysis or other potential mechanisms.
We found that although P. acnes phages can lyse P. acnes strains from most lineages, certain strains from clades IB-3, II and III are resistant (Figure 4a). This bacterial lineage-dependent resistance to phages suggests that antiviral mechanisms may have a role in defining the P. acnes strain populations in the presence of phages. Based on the genomes of Propionibacterium strains, we identified two antiviral mechanisms that may explain phage resistance in different P. acnes lineages, restriction modification and CRISPRs. Both of these systems act against phage DNA that has successfully entered the host. It is possible that these P. acnes lineages employ additional mechanisms that act at other stages of infection, such as receptor binding or DNA injection, which are commonly responsible for phage resistance in mycobacteria (Jacobs-Sera et al., 2012).
We observed that phage DNA recognition by P. acnes CRISPRs often did not protect the tested bacterial strains from phage lysis (Figure 6). This result contrasts with a previous study reporting a correlation between matching spacers and phage resistance (Marinelli et al., 2012). While this earlier study similarly reported 1 unexplained lytic outcome out of 20 tested interactions, where 2 matched spacers failed to confer phage resistance, in our study we observed a much higher proportion of lytic outcomes, 27 out of 37 non-redundant interactions. The contrasting conclusions from the two studies may be attributed to the differences in the bacterial host strains and phage strains tested. To demonstrate the robustness of our results, we included redundant bacterial host strains and phage strains in our experiments. Both RT2 and RT6 strains, which belong to two different lineages of P. acnes that encode CRISPR/Cas systems, and several members of the same phage strains were tested in the analysis and yielded consistent results.
Several possible reasons may explain the low correlation between spacer matching and phage resistance. The CRISPR/Cas system in P. acnes may be used for purposes other than defense against foreign DNA, as suggested for the Type I-E CRISPR/Cas system of Escherichia coli (Touchon and Rocha, 2010; Touchon et al., 2011). Alternatively, other molecular regulations employed by bacterial host or phage may be important for CRISPR-mediated resistance and may contribute to the infection outcome. Such regulation mechanisms potentially include transcriptional and/or translational regulation of CRISPR RNA and Cas genes and anti-CRISPR genes encoded by phages (Bondy-Denomy et al., 2013; Pawluk et al., 2014). Interactions between P. acnes and phages may also depend on additional mechanisms involved in phage binding, entry, replication or release.
To our knowledge, this study is the first large-scale investigation of bacterium–phage interaction in the human skin microbiota using both genetically defined bacterial strains and phage strains. Our study suggests that the prey–predator relationship between P. acnes and its phages may have a role in modulating the composition of the skin microbiota. The finding that the infection outcome of P. acnes phages varies depending on P. acnes lineages suggests that the presence of phages may alter the P. acnes population structure by targeting particular subsets of strains. Based on evidence of different propensities among P. acnes lineages to be lysed, to resist or to sustain a pseudolysogenic infection, it is plausible that in the presence of phages various P. acnes strains may populate at different rates in the community. On the other hand, the Propionibacterium strain composition may determine the abundance and dynamics of the phages in the community. Phage bursts may occur in specific skin community structures. Future studies utilizing recently developed techniques, such as microfluidic digital PCR (Tadmor et al., 2011), phageFISH (Allers et al., 2013) and viral tagging (Deng et al., 2012), will provide further insight on the diversity and dynamics of the phage populations in the human microbiome and the prey–predator relationship between the bacterial hosts and phages.
P. acnes phages may hold therapeutic potential for modifying the skin microbiota based on the individual bacterial community structure to restore a healthy microbial composition. We previously demonstrated that P. acnes strain population structure differed significantly between acne patients and healthy individuals (Fitz-Gibbon et al., 2013). Certain strains were highly associated with the disease, while some strains were highly associated with healthy skin. The interactions between genetically defined P. acnes strains and phages that we characterized in this study may be able to guide the selection of phage strains to inhibit specific P. acnes population types associated with acne or other skin diseases. For example, phage therapy may not be effective if the host microbiota contains phage-resistant pathogenic strains. Potential phage-based therapy should consider the microbiome structure of the individual to be effective and personalized. The host strain specificity of P. acnes phages may prove advantageous for the selective suppression of pathogenic P. acnes strains and preservation of commensal or beneficial strains in the skin microbial community.
Acknowledgments
This research was funded by NIH grants R01GM099530 and UH2AR057503 from NIGMS and NIAMS. JL was supported by the Microbial Pathogenesis Training Grant T32AI07323. Phage genomes were sequenced at the UCLA Genotyping and Sequencing Core. We thank Dr Emma Barnard and Emily Curd for providing assistance in preparation of MiSeq sequencing libraries, and Dr Baochen Shi for help with metagenomic data analysis.
Author contributions
JL and ST analyzed the data. RY, QZ, SN, NJB, TL and LN performed the experiments. ML helped perform the experiments. MCE collected the clinical samples. NC maintained the IRB protocol and collected the clinical samples. HL conceived and directed the project and analyzed the data. JL and HL wrote the paper.
The Regents of the University of California is the owner of a patent application based on some of the results presented, which names ST and HL as inventors.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Allers E, Moraru C, Duhaime MB, Beneze E, Solonenko N, Barrero-Canosa J, et al. Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environ Microbiol. 2013;15:2306–2318. doi: 10.1111/1462-2920.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergler-Czop B, Brzezińska-Wcisło L. Dermatological problems of the puberty. Postepy Dermatol Alergol. 2013;30:178–187. doi: 10.5114/pdia.2013.35621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Estrada I, et al. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 2013;7:85–95. doi: 10.1038/ismej.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojar Ra, Holland KT. Acne and Propionibacterium acnes. Clin Dermatol. 2004;22:375–379. doi: 10.1016/j.clindermatol.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzin B. Studies on the Corynebacterium acnes. Acta Pathol Microbiol Scand. 1964;60:599–608. doi: 10.1111/apm.1964.60.4.599. [DOI] [PubMed] [Google Scholar]
- Butler-Wu SM, Sengupta DJ, Kittichotirat W, Matsen FA, Bumgarner RE. Genome sequence of a novel species, Propionibacterium humerusii. J Bacteriol. 2011;193:3678. doi: 10.1128/JB.05036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S.1999Genome sequence assembly using trace signals and additional sequence informationIn: Wingender E (ed)Computer science and biology. Proceedings of the German Conference on Bioinformatics GCB, Hannover, Germany; 45–56. [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins CS. Identification of Propionibacterium acnes and related organisms by precipitin tests with trichloroacetic acid extracts. J Clin Microbiol. 1976;2:104–110. [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, Frigaard N-U, et al. Community genomics among stratified microbial assemblages in the ocean's interior. Science. 2006;311:496–503. doi: 10.1126/science.1120250. [DOI] [PubMed] [Google Scholar]
- Deng L, Gregory A, Yilmaz S, Poulos BT, Hugenholtz P, Sullivan MB. Contrasting life strategies of viruses that infect photo- and heterotrophic bacteria, as revealed by viral tagging. MBio. 2012;3:e00516–12. doi: 10.1128/mBio.00373-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorscht J, Klumpp J, Bielmann R, Schmelcher M, Born Y, Zimmer M, et al. Comparative genome analysis of Listeria bacteriophages reveals extensive mosaicism, programmed translational frameshifting, and a novel prophage insertion site. J Bacteriol. 2009;191:7206–7215. doi: 10.1128/JB.01041-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime MB, Deng L, Poulos BT, Sullivan MB. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: a rigorous assessment and optimization of the linker amplification method. Environ Microbiol. 2012;14:2526–2537. doi: 10.1111/j.1462-2920.2012.02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime MB, Sullivan MB. Ocean viruses: rigorously evaluating the metagenomic sample-to-sequence pipeline. Virology. 2012;434:181–186. doi: 10.1016/j.virol.2012.09.036. [DOI] [PubMed] [Google Scholar]
- Egea R, Casillas S, Barbadilla A. Standard and generalized McDonald-Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 2008;36:W157–W162. doi: 10.1093/nar/gkn337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MD, Howson KM, Bojar RA, West D, Towler JC, Parry J, et al. Genome sequence and analysis of a Propionibacterium acnes bacteriophage. J Bacteriol. 2007;189:4161–4167. doi: 10.1128/JB.00106-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz-Gibbon S, Tomida S, Chiu B-H, Nguyen L, Du C, Liu M, et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol. 2013;133:2152–2160. doi: 10.1038/jid.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulongne V, Sauvage V, Hebert C, Dereure O, Cheval J, Gouilh MA, et al. Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One. 2012;7:e38499. doi: 10.1371/journal.pone.0038499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Perez-Perez GI, Chen Y, Blaser MJ. Quantitation of major human cutaneous bacterial and fungal populations. J Clin Microbiol. 2010;48:3575–3581. doi: 10.1128/JCM.00597-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Tseng C, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci USA. 2007;104:2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, et al. On the nature of mycobacteriophage diversity and host preference. Virology. 2012;434:187–201. doi: 10.1016/j.virol.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong EC, Ko HL, Pulverer G. Studies on bacteriophages of Propionibacterium acnes. Med Microbiol Immunol. 1975;161:263–271. doi: 10.1007/BF02122714. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden JJ. The evolving role of Propionibacterium acnes in acne. Semin Cutan Med Surg. 2001;20:139–143. doi: 10.1053/sder.2001.28207. [DOI] [PubMed] [Google Scholar]
- Li H. Metagenomic study of the human skin microbiome associated with acne. J Invest Dermatol. 2010;133:2152–2160. doi: 10.1038/jid.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cheng A, Bangayan NJ, Barnard E, Curd E, Craft N, et al. Draft genome sequences of Propionibacterium acnes type strain ATCC6919 and antibiotic-resistant strain HL411PA1. Genome Announc. 2014;2:e00740–14. doi: 10.1128/genomeA.00740-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomholt HB, Kilian M. Population genetic analysis of Propionibacterium acnes identifies a subpopulation and epidemic clones associated with acne. PLoS One. 2010;5:e12277. doi: 10.1371/journal.pone.0012277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lood R, Collin M. Characterization and genome sequencing of two Propionibacterium acnes phages displaying pseudolysogeny. BMC Genomics. 2011;12:198. doi: 10.1186/1471-2164-12-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Madupu R, Karaoz U, Nossa CW, Yang L, Yooseph S, et al. Human papillomavirus community in healthy persons, defined by metagenomics analysis of human microbiome project shotgun sequencing data sets. J Virol. 2014;88:4786–4797. doi: 10.1128/JVI.00093-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manica A, Zebec Z, Steinkellner J, Schleper C. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2013;41:10509–10517. doi: 10.1093/nar/gkt767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli LJ, Fitz-gibbon S, Hayes C, Bowman C, Inkeles M, Loncaric A, et al. Propionibacterium acnes bacteriophages display limited genetic diversity and broad killing activity against bacterial skin isolates. MBio. 2012;3:e00279–12. doi: 10.1128/mBio.00279-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marples RR, McGinley KJ, Mills OH. Microbiology of comedones in acne vulgaris. J Invest Dermatol. 1973;60:80–83. doi: 10.1111/1523-1747.ep12724149. [DOI] [PubMed] [Google Scholar]
- McDowell A, Barnard E, Nagy I, Gao A, Tomida S, Li H, et al. An expanded multilocus sequence typing scheme for propionibacterium acnes: investigation of ‘pathogenic', ‘commensal' and antibiotic resistant strains. PLoS One. 2012;7:e41480. doi: 10.1371/journal.pone.0041480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Rapid evolution of the human gut virome. Proc Natl Acad Sci USA. 2013;110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno CM, Rodriguez-Valera F, Kimes NE, Ghai R. Expanding the marine virosphere using metagenomics. PLoS Genet. 2013;9:e1003987. doi: 10.1371/journal.pgen.1003987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- Nakatsuji T, Chiang H-I, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. doi: 10.1038/ncomms2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan JM, Petrov V, Bertrand C, Krisch HM, Karam JD. Genetic diversity among five T4-like bacteriophages. Virol J. 2006;3:30. doi: 10.1186/1743-422X-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ördögh L, Hunyadkürti J. Complete genome sequence of Propionibacterium avidum strain 44067, isolated from a human skin abscess. Genome Announc. 2013;1:5–6. doi: 10.1128/genomeA.00337-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino LC, Tseng C-H, Blaser MJ. Analysis of Malassezia microbiota in healthy superficial human skin and in psoriatic lesions by multiplex real-time PCR. FEMS Yeast Res. 2008;8:460–471. doi: 10.1111/j.1567-1364.2008.00359.x. [DOI] [PubMed] [Google Scholar]
- Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL, Davidson AR. A new group of phage Anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio. 2014;5:e00896–14. doi: 10.1128/mBio.00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pride DT, Salzman J, Haynes M, Rohwer F, Davis-Long C, White RA, et al. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012;6:915–926. doi: 10.1038/ismej.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhvel SM, Amirian DA. Bacterial flora of comedones. Br J Dermatol. 1979;101:543–548. doi: 10.1111/j.1365-2133.1979.tb11883.x. [DOI] [PubMed] [Google Scholar]
- Ripp S, Miller RV. Dynamics of the pseudolysogenic response in slowly growing cells of Pseudomonas aeruginosa. Microbiology. 1998;144:2225–2232. doi: 10.1099/00221287-144-8-2225. [DOI] [PubMed] [Google Scholar]
- Ripp S, Miller RV. The role of pseudolysogeny in bacteriophage-host interactions in a natural freshwater environment. Microbiology. 1997;143:2065–2070. doi: 10.1099/00221287-143-6-2065. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Valera F, Martin-Cuadrado A-B, Rodriguez-Brito B, Pasić L, Thingstad TF, Rohwer F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- Rohwer F. Global phage diversity. Cell. 2003;113:141. doi: 10.1016/s0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- Rohwer F, Thurber RV. Viruses manipulate the marine environment. Nature. 2009;459:207–212. doi: 10.1038/nature08060. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Sakaguchi Y, Hayashi T, Kurokawa K, Nakayama K, Oshima K, Fujinaga Y, et al. The genome sequence of Clostridium botulinum type C neurotoxin-converting phage and the molecular mechanisms of unstable lysogeny. Proc Natl Acad Sci USA. 2005;102:17472–17477. doi: 10.1073/pnas.0505503102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko Ka, Semenova A, Westra ER, Wanner B, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Nagornykh M, Pyatnitskiy M, Artamonova II, Severinov K. Analysis of CRISPR system function in plant pathogen Xanthomonas oryzae. FEMS Microbiol Lett. 2009;296:110–116. doi: 10.1111/j.1574-6968.2009.01626.x. [DOI] [PubMed] [Google Scholar]
- Sepúlveda-Robles O, Kameyama L, Guarneros G. High diversity and novel species of Pseudomonas aeruginosa bacteriophages. Appl Environ Microbiol. 2012;78:4510–4515. doi: 10.1128/AEM.00065-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solonenko SA, Ignacio-Espinoza JC, Alberti A, Cruaud C, Hallam S, Konstantinidis K, et al. Sequencing platform and library preparation choices impact viral metagenomes. BMC Genomics. 2013;14:320. doi: 10.1186/1471-2164-14-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA, Chan AM, Cottrell MT. Infection of phytoplankton by viruses and reduction of primary productivity. Nature. 1990;347:467–469. [Google Scholar]
- Tadmor AD, Ottesen EA, Leadbetter JR, Phillips R. Probing individual environmental bacteria for viruses by using microfluidic digital PCR. Science. 2011;333:58–62. doi: 10.1126/science.1200758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida S, Nguyen L, Chiu B-H, Liu J, Sodergren E, Weinstock GM, et al. Pan-genome and comparative genome analyses of propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. MBio. 2013;4:e00003–e00013. doi: 10.1128/mBio.00003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, Charpentier S, Clermont O, Rocha EPC, Denamur E, Branger C. CRISPR distribution within the Escherichia coli species is not suggestive of immunity-associated diversifying selection. J Bacteriol. 2011;193:2460–2467. doi: 10.1128/JB.01307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, Rocha EPC. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLoS One. 2010;5:e11126. doi: 10.1371/journal.pone.0011126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursell LK, Clemente JC, Rideout JR, Gevers D, Caporaso JG, Knight R. The interpersonal and intrapersonal diversity of human-associated microbiota in key body sites. J Allergy Clin Immunol. 2012;129:1204–1208. doi: 10.1016/j.jaci.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterbury JB, Valois FW. Resistance to co-occurring phages enables marine synechococcus communities to coexist with cyanophages abundant in seawater. Appl Envir Microbiol. 1993;59:3393–3399. doi: 10.1128/aem.59.10.3393-3399.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster GF, Cummins CS. Use of bacteriophage typing to distinguish propionibacterium acnes types I and II. J Clin Microbiol. 1978;7:84–90. doi: 10.1128/jcm.7.1.84-90.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PBG, Künne T, Wong SP, Staals RHJ, Seegers CLC, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and Cas3. Mol Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39:S34–S37. doi: 10.1016/s0190-9622(98)70442-6. [DOI] [PubMed] [Google Scholar]
- Wylie KM, Mihindukulasuriya KA, Zhou Y, Sodergren E, Storch GA, Weinstock GM. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014;12:71. doi: 10.1186/s12915-014-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierdt CH, Webster C, Rude WS. Study of the anaerobic corynebacteria. Int J Syst Bacteriol. 1968;18:33–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.