

Abstract
Inflammatory bone resorption is a hallmark of periodontitis, and Tregs and Th2 cells are independently associated with disease progression attenuation. In this study, we employed an infection-triggered inflammatory osteolysis model to investigate the mechanisms underlying Treg and Th2 cell migration and the impact on disease outcome. Aggregatibacter actinomycetemcomitans–infected C57Bl/6 (wild-type [WT]) mice develop an intense inflammatory reaction and alveolar bone resorption, and Treg and Th2 cell migration is temporally associated with disease progression attenuation. Tregs extracted from the lesions preferentially express CCR4 and CCR8, whereas Th2 cells express CCR3, CCR4, and CCR8. The absence of CCR5 and CCR8 did not significantly impact the migration of Tregs and Th2 cells or affect the disease outcome. CCR4KO mice presented a minor reduction in Th2 cells in parallel with major impairment of Treg migration, which was associated with increased inflammatory bone loss and higher proinflammatory and osteoclastogenic cytokine levels. The blockade of the CCR4 ligand CCL22 in WT mice resulted in an increased inflammatory bone loss phenotype similar to that in the CCR4KO strain. Adoptive transfer of CCR4+ Tregs to the CCR4KO strain revert the increased disease phenotype to WT mice–like levels; also, the in situ production of CCL22 in the lesions is mandatory for Tregs migration and the consequent bone loss arrest. The local release of exogenous CCL22 provided by poly(lactic-co-glycolic acid) (PLGA) microparticles promotes migration of Tregs and disease arrest in the absence of endogenous CCL22 in the IL-4KO strain, characterized by the lack of endogenous CCL22 production, defective migration of Tregs, and exacerbated bone loss. In summary, our results show that the IL-4/CCL22/CCR4 axis is involved in the migration of Tregs to osteolytic lesion sites, and attenuates development of lesions by inhibiting inflammatory migration and the production of proinflammatory and osteoclastogenic mediators.
Keywords: OSTEOIMMUNOLOGY, CHEMOKINES, CYTOKINES, BONE RESORPTION, PERIODONTAL DISEASE
Introduction
Periodontal diseases (PD) are chronic inflammatory diseases characterized by inflammatory bone resorption of the supporting structures of the teeth; these diseases are the most prevalent form of bone pathology in humans and are a factor in modifying the systemic health of patients.(1,2) Although host inflammatory immune reactions in response to periodontopathogens are thought to protect the host against those infectious agents, advances in the field of osteoimmunology clearly show that cytokines produced as a result of the associated host response are ultimately responsible for disrupting local bone homeostasis (ie, increased bone resorption and decreased bone formation). This disruption results in the irreversible tissue destruction that characterizes PD.(3) In this context, proinflammatory and pro-osteoclastogenic leukocyte subsets, such as Th1 and Th17 cells, are thought to contribute to the progression of lesions by mediating the influx of leukocytes and osteoclast precursors into periodontal tissues and by increasing local RANKL levels.(4) Indeed, the involvement of Th1 and Th17 cell subsets and their prototypic cytokines in inflammatory osteolysis have been showed in a cause-and-effect manner in PD and rheumatoid arthritis (RA) models, which share several immunopathological phenotypical characteristics.(4) However, the isolated analysis of each factor does not allow the determination of the possible interplay between these Th-cell subsets in the inflammatory bone resorption process. In contrast to Th1 and Th17 cells, Th2 cell and Treg subsets are known to restrain the proinflammatory response and to restore the RANKL/osteoprotegerin (OPG) ratio to physiological levels, arresting PD development.(4,5) However, as with studies on proinflammatory and destructive mediators, the protective role of Th2 cell and Treg subsets against inflammatory osteolysis have been independently shown in a cause-and-effect manner, but the possible interplay between these T-cell subsets remains unexplored.(6,7)
Previous studies showed that after oral infection of C57Bl/6 mice, the occurrence of Th2 cells and Tregs in periodontal tissues is temporally associated with a decrease in proinflammatory, Th1 and Th17 cell responses, in parallel with a significant inflammatory attenuation of bone loss, suggesting a possible interplay (and even collaboration) between Th2 cells and Tregs.(8–10) A recent study reinforces this hypothesis, showing that the coexistence of high levels of IL-4 (the prototypical Th2 cytokine), FOXp3 (the Tregs master transcription factor), and IL-10 (a major product of Tregs) are associated with an inactive profile of osteolytic periapical lesions.(11) Such protective interplay does not seem to be limited to PD, because a positive correlation between increased levels of IL-4 and IL-10 was reported to be associated with the attenuation of RA severity.(12) It is also worth mentioning that Tregs and Th2 cells appear to share the expression of the chemokine receptors, such as CCR4 and CCR8, in theory making these cells similarly responsive for chemokines such as CCL17 and CCL22.(13) Indeed, these chemokines/chemokine receptors have been identified in inflammatory osteolytic lesions, reinforcing their potential role in the immunoregulation of bone pathologies.(4)
However, besides CCR4 and CCR8, Th2 cells and Tregs subsets can express distinct chemokine receptors, such as CX3CR1, CCR3, CCR5, CCR6, CCR7, and CRTH2, which also could each play important roles in migratory and nonmigratory and functions, such as retention, egress, and survival.(13) It is also important to consider that although the preferential expression of chemokine receptors by T-cell subsets is apparent in vitro, there is some overlapping expression in vivo, which is not surprising given the relative plasticity of the various T-cell subsets, adding significant complexity to the selective cell-recruitment process.(13) Indeed, the exact mechanisms underlying the migration of Th2 cells and Tregs in PD remain unclear and the mechanisms underlying the Tregs/Th2 protective interplay remains elusive.
We hypothesize that Th2 cells and Tregs may act in a cooperative manner to suppress inflammatory bone resorption, and that common cell migration properties may take place in this process. To investigate this hypothesis, we used a model of infection-triggered inflammatory osteolysis in an experimental setting involving WT and genetically-deficient C57Bl/6 mice, along with treatment targeting specific chemokines, to explore how Th2 cells and Tregs may impact inflammatory bone loss throughout experimental periodontitis in mice.
Materials and Methods
Experimental groups
Experimental groups comprised 8-week-old male WT C57BL/6 mice, and C57BL/6 mice with targeted disruption of the CCR4 (CCR4KO), CCR5 (CCR5KO), CCR8 (CCR8KO), and IL-4 (IL4KO), bred and maintained in the animal facilities of USP (breeding pairs obtained from Jackson Laboratory, Bar Harbor, ME, USA). Over the course of the study, the mice were fed with standard solid mice chow (Nuvital, Curitiba, PR, Brazil) and sterile water. The experimental protocol follows the principles of the Guide for the Care and Use of Laboratory Animals and was approved by the local Institutional Committee for Animal Care and Use.
Experimental periodontitis
Bacterial culture and the oral inoculation to achieve periodontal infection were performed as described.(14) In brief, the animals received an oral delivery of 1 × 109 colony-forming units (CFUs) of a diluted culture of Aggregatibacter actinomycetemcomitans JP2 (grown anaerobically in supplemented agar medium, tryptic soy-serum-bacitracin-vancomycin [TSBV]) in 100μL of phosphate-buffered saline (PBS) with 2% of carboxymethylcellulose, placed/inoculated (ie, not directly injected into the gingival tissue) in the oral cavity of mice with a micropipette. After 48 hours and 96 hours, this procedure was repeated. Negative controls comprised sham-infected mice, which received PBS with carboxymethylcellulose in solution without A. actinomycetemcomitans. The evaluation of the extent of alveolar bone loss was performed as described.(14) The maxillae were hemisected, exposed overnight in 3% hydrogen peroxide, and defleshed mechanically. The palatal faces of the molars were photographed at ×20 magnification using a dissecting microscope (Leica, Wetzlar, Germany), with the occlusal face of the molars positioned perpendicularly to the base. The images were digitized and analyzed using ImageTool 2.0 software (University of Texas Health Science Center, San Antonio, TX, USA). Quantitative analysis was used for the measurement of the area between the cement–enamel junction (CEJ) and the alveolar bone crest (ABC) in the three posterior teeth, in arbitrary units of area (AUA). At each time point 5 animals were analyzed, and for each animal the alveolar bone loss was defined as the increase in CEJ-ABC area in comparison with control mice at each time point.
Isolation of inflammatory cells from periodontal tissues and flow cytometric analysis
The isolation and characterization of leucocytes present in the lesion site were performed as described.(15) The entirety of buccal and palatal periodontal tissues of upper molars were collected, weighed, and incubated for 1 hour at 37°C, dermal side down (Sigma-Aldrich Co., St. Louis, MO, USA), in RPMI-1640, supplemented with NaHCO3, penicillin/streptomycin/gentamycin and 0.28 Wunsch units/mL of Liberase Blendzyme CI (Roche–F. Hoffmann-La Roche Ltd, Basel, Switzerland). The tissues of 5 mice, at each time point per group, were processed in the presence of 0.05% DNase (Sigma-Aldrich, Steinhein, Germany) using Medimachine (BD Biosciences PharMingen, San Diego, CA, USA), according to the manufacturer’s instructions. After processing, cell viability was assessed by Trypan blue exclusion, and the cell count was performed in a Neubauer chamber; the data depicted in the graphs refers to the total inflammatory cell count. For flow cytometry analysis, after cell counting the cells were stained for 20 min at 4 °C with the optimal dilution of each antibody; phycoerythrin (PE)-conjugated and fluorescein isothiocyanate (FITC)-conjugated antibodies against CD4, CD25, FOXP3, CCR4, CCR5, CCR7, CCR8, CXCR3, IL-4, IL-10, IL-17, IFN-γ, TGF-β, and CTLA-4 antibodies, as well as their respective isotype controls (BD Biosciences PharMingen). Cells were washed again and analyzed by FACScan and CellQuest software (BD Biosciences PharMingen). Results are presented as the number of cells (mean±SD) in the periodontal tissues of each mouse or the number of positive cells for each marker in the CD4+FOXp3+ and CD4+IL-4+ subpopulations; data were derived from independent experiments.
Isolation, adoptive transfer, and determination of suppressive activity of Tregs of CD4+CD25+ T cells
Tregs were isolated from PBMCs and gingival tissues using a CD4+CD25+ Regulatory T Cells Isolation Kit (Miltenyi Biotec, Bergisch Gladbac, Germany), as described.(16) Prior to the isolation procedures, gingival tissues were submitted to enzymatic digestion protocol as described in the Isolation of inflammatory cells from periodontal tissues and flow cytometric analysis section. Briefly, CD4+ T cells were pre-enriched by depletion of non-CD4+ T cells using a cocktail of biotin-labeled antibodies, anti-biotin magnetic beads, and an magnetic bead column (Miltenyi Biotec, Bergisch Gladbac, Germany); to isolate CD4+CD25+ T cells, enriched CD4+ T cells were incubated with PE-labeled anti-CD25 antibody and anti-PE magnetic beads. Then CD4+CD25+ T cells were positively selected using a MS magnetic bead column. The purity of CD4+CD25+ T cells was more than 95%, as confirmed by flow cytometry. To determine the isolated Treg cells’ suppressive activity, 5 × 104 CD4+CD25− cells were treated with 2μg/mL anti-CD3 and anti-CD28 (eBioscience) for 12 hours as effector cells, then incubated with or without isolated Tregs at 2:1 ratios of for 72 hours in complete medium containing RPMI 1640 (Sigma, St. Louis, MO, USA) supplemented with 5% FCS, as described.(17) Cell proliferation was assessed by [3H]thymidine incorporation as measured by scintillation counting.(18)
Tregs inhibition in vivo
Treg function was inhibited in vivo by treatment with anti–glucocorticoid-induced TNF-receptor-related protein (anti-GITR) antibodies, as described.(8) The anti-GITR (DTA-1) hybridomas were grown intraperitoneally in mineral oil–injected nude mice. The antibodies were purified from ascites by precipitation using ammonium sulfate (45%, wt/vol), and subsequently purified by G protein column (Amersham Biosciences USA, Piscataway, NJ, USA), as described.(8) Protein was quantified through the bicinchoninic method. The in vivo blockage of GITR molecules was performed by intraperitoneal injection of 500μg/mouse of purified mAb anti-GITR diluted in PBS. Control mice received 500 μg of normal rat IgG diluted in PBS at the same time points.
Chemokine-targeted treatments
The treatment with anti-CCL17 and anti-CCL22 neutralizing antibodies was performed as described,(19) consisting of an intraperitoneal injection of a 20-μg dose of either anti-CCL17 or anti-CCL22 antibodies (goat anti-mouse antibodies; R&D Systems, Minneapolis, MN, USA) dissolved in sterile normal saline, on alternate days, initiated at day 30 postinfection. Control groups received a 20-μg dose of normal goat IgG (R&D Systems) dissolved in 500μL of sterile normal saline via the intraperitoneal route over the same time course.(19) The treatment with met-RANTES (Serono Pharmaceutical Institute of Research, Geneva, Switzerland), a CCR5 inhibitor, was used as a control and performed as described.(20) Specifically 0.5 μg/kg of met-RANTES diluted in sterile normal saline was administered through intraperitoneal injection on alternate days, initiated at day 30 postinfection. The manufacture of poly(lactic-co-glycolic acid) (PLGA) microparticles containing recombinant mouse was performed as described,(21) and the treatment with CCL22-releasing PLGA microparticles was performed similarly to the method described.(5) Briefly, microparticles were administered to four sites via 2% carboxy methyl cellulose (CMC) in PBS suspension. Specifically, 2 to 5μL of solution containing 25 mg/mL of particles was administered to the proximal side of the first molar, each interdental site, and distal to the third molar of the right maxilla of the mice. Microparticles were administered to mice 7 days after bacterial inoculation. Control mice received blank particles, administered similarly to the CCL22-releasing particles (data not shown).
Protein extraction and ELISA
Measurements of cytokines and chemokines in periodontal tissue were performed by ELISA, as described.(14) For protein extraction, palatal periodontal tissue was homogenized in PBS, pH 7.4, centrifuged at 100g at 4°C, and the concentrations of cytokines/chemokines in periodontal extracts were determined by ELISA/ELISA multiplex using commercially available kits (R&D Systems) according to the manufacturer’s instructions. The results were expressed as picograms of cytokine (mean±SD) per milligram of periodontal tissue, for one experiment (representative of three experiments).
qPCR reactions
The extraction of total RNA from periodontal tissues was performed with the RNeasyFFPE kit (Qiagen Inc, Valencia, CA, USA) according to the manufacturer’s instructions. The integrity of RNA samples was checked by analyzing 1mg of total RNA on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions. Real-time quantitative PCR mRNA analyses were performed in Viia7 equipment (Applied Biosystems, Warrington, UK) by RealTimePCR using TaqMan chemistry (Invitrogen, Carlsbad, CA, USA) using inventoried optimized primers/probes sets (Invitrogen). The results, depicted as the relative level of gene expression, were calculated in reference to internal controls GAPDH and β-actin expression in the sample using the threshold cycle (Ct) method and the comparative Ct (2−ΔΔCt) calculation.
Statistical analysis
Data are presented as means±SD, and the statistical significance between the groups was analyzed by Kruskal-Wallis followed by Dunn’s posttest, or by Mann-Whitney test, both performed with GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA). Values of p<0.05 were considered statistically significant.
Results
Kinetics of T-helper subsets migration to periodontal tissues and profile of Tregs and Th2 cells chemokine receptor expression
To explore the possible involvement of T-helper subsets in inflammatory bone loss, and more specifically to investigate the involvement of Tregs and Th2 cells in the attenuation of inflammatory bone loss in experimental periodontitis, we first investigated the kinetics of T-helper subsets migration to periodontal tissues postinfection, as well as the expression of chemokine receptors on Tregs and Th2 cells (Fig. 1). The induction of experimental periodontitis resulted in a significant alveolar bone loss, represented by the increase in the in the area between the cement-enamel junction (CEJ) and the alveolar bone crest (ABC) in comparison with control mice (Fig. 1A). In the initial periods, until 30 days postinfection, a high rate of bone loss was observed, whereas in the later disease stage (ie, after 30 days) the rate of bone loss advance significantly decreased. A similar pattern was observed to the inflammatory cell influx into periodontal tissues, because the total leukocytes number as well as the number of CD4+CD25+ cells increases at a high rate in the initial disease period, and becomes relatively stable after 30 days postinfection (Fig. 1B). Th1 (CD4+IFN-γ+) cells were found in periodontal tissues in elevated numbers at the 15-day and 30-day time points, decreasing in number at the subsequent (45 and 60 days) time points (Fig. 1B). The Th17 (CD4+IL17+) cell count presented an initial peak at the 15-day time point, followed by a decrease at 30 days postinfection, and a later increase at the 45-day and 60-day time points (Fig. 1B). Tregs (CD4+FOXP3+) and Th2 (CD4+IL4+) cells were detected in the periodontal tissues in very low numbers until 15 days postinfection, being a significant increase in its counts observed at 30 days postinfection, and an additional increase in numbers was verified at the 45-day time point, and remained relatively stable until the 60-day time point (Fig. 1C). The phenotypic analysis (analyzed at 45 days postinfection) of Tregs from PBMCs revealed that CD4+FOXp3+ cells were also positive for CCR4, CCR5, and CCR8, and also for the Treg markers IL-10, TGF-β, and CTLA-4, whereas Tregs from periodontal tissues were positive for CCR4, CCR8, IL-10, TGF-β, and CTLA-4 (Fig. 1D, E). The analysis of CD4+IL4+ cells from PBMCs and periodontal tissues revealed that these cells presented a similar pattern of chemokine receptor expression, being positive for CCR3, CCR4, and CCR8 (Fig. 1F, G). For histological images of the inflammatory infiltrates and periodontal bone loss, please refer to other publications by our group.(10,22)
Fig. 1.
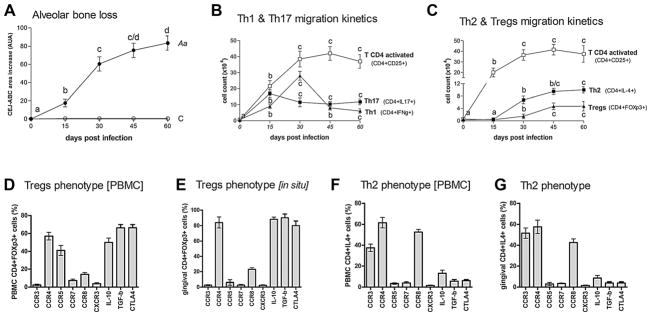
Migration kinetics and profile of chemokine receptors expression of CD4 T-cell subsets, and phenotypic analysis of Tregs and Th2 cells during the course of experimental periodontal disease. C57Bl/6 (WT) mice were infected orally with A. actinomycetemcomitans (Aa) and evaluated for: (A) alveolar bone loss, depicted as the increase in the area between the CEJ and the ABC in comparison with control mice in each time point, quantitatively measured in the palatal face of the three maxillary molars in AUA; (B) activated CD4 T cells (CD4+CD25+), Th17 cells (CD4+IL17+), and Th1 cells (CD4+IFNg+) cells count in periodontal tissues analyzed by flow cytometry, at 0, 15, 30, 45, and 60 days postinfection, depicted as the cell number ×104; (C) count of activated CD4 T cells (CD4+CD25+), Th2 cells (CD4+IL4+), and Tregs (CD4+FOXp3+) in periodontal tissues analyzed by flow cytometry, at 0, 15, 30, 45, and 60 days postinfection, depicted as the cell number ×104; (D–G) the phenotype of Th2 cells (CD4+IL4+) and Tregs (CD4+FOXp3+) from PBMC (D–F) and gingival tissues (E–G) at 45 days postinfection, evaluated by flow cytometry and depicted as the number of positive cells for each marker, all performed as described in the Materials and Methods. Values (mean±SD) obtained from 6 animals at each point, from one experiment representative of three. In A, B, and C, different letters (a, b, c, and d) represent statistically significant differences (p<0.05; Kruskal-Wallis and Dunn’s posttest) within the different time points for each individual parameter (bone loss in A, and for each CD4 T-cell subset count). CEJ=cement–enamel junction; ABC=alveolar bone crest; AUA=arbitrary units of area.
The role of chemokine receptors in the migration of Tregs and Th2 cells and the corresponding impact in the modulation of the inflammatory response and of alveolar bone loss
In view of the significant expression of CCR4 and CCR8 by Tregs and Th2 cells, we next investigated their possible role in the cell migration process and in the modulation of the inflammatory bone loss at the 45-day postinfection time point (Fig. 2). Our results show that the absence of CCR4 resulted in a modest, though significant, reduction in the influx of CD4+IL4+ cells into periodontal tissues, whereas the lack of CCR5 and CCR8 did not impact the migration of Th2-type cells (Fig. 2A). Our data also revealed that CCR4KO mice presented a marked impairment in the migration of CD4+FOXp3+ cells, whereas the lack of CCR5 and CCR8 did not appear to obstruct the migration of Tregs (Fig. 2B). Notably, the adoptive transfer (at the 30-day time-points) of Tregs (from WT, CCR5KO, and CCR8KO strains) to CCR4KO mice appears to completely revert the defective influx of CD4+FOXp3+ cells into periodontal tissues, showing the necessity of CCR4 for the process of migration of Tregs into periodontal tissues (Fig. 2C). Furthermore, our results show that the CCR4KO mice presented a significant increase in both alveolar bone loss (Fig. 2C) and in the inflammatory cell migration (Fig. 2D), whereas CCR5KO strains showed a low disease severity phenotype, and no significant changes were observed in the CCR8KO strain (Fig. 2C, D). Importantly, we also show that Tregs from WT, CCR4, CCR5, and CCR8 mice are equally immunosuppressive in vitro (Fig. 2E). In order to investigate the possible mechanisms underlying the exacerbated inflammatory bone loss observed in CCR4KO mice, we evaluated the degree of protein expression in these knockout mice. Specifically, ELISA analysis showed that CCR4 deficiency was associated with lower levels of IL-4, IL-10, TFG-β, and OPG in the tissues, whereas TNF-α, IFN-γ, IL-17, IL-6, and RANKL were significantly increased (Fig. 2F). Our results also show that adoptive transfer of CCR4+ Tregs to CCR4 mice reverse the alterations observed in this strain, indeed the overall cytokine levels were similar in WT and CCR4KO mice that received Tregs adoptive transfer, with the exception of the degree of expression of IL-4 (Fig. 2F). It is also worthwhile to mention that the evolution of bone loss in WT and CCR4KO mice strains is similar until the 30-day time point (Fig. 2G), when the influx of CCR4-mediated Tregs attenuates the disease development in the WT strain. Importantly, the adoptive transference of WT Tregs to the CCR4KO strain restores the kinetics of inflammatory bone loss to levels similar to those observed in the WT strain at the 45-day and 60-day time points (Fig. 2G).
Fig. 2.
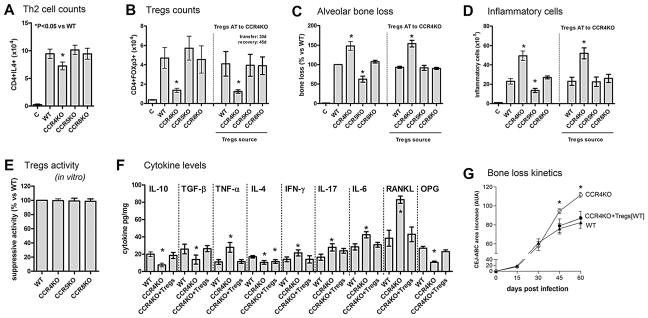
The role of chemokine receptors in the migration of Tregs and Th2 cells and the associated impact on the modulation of the inflammatory response and of alveolar bone loss during the course of experimental periodontal disease. C57Bl/6 (WT), CCR4KO, CCR5KO, and CCR8KO mice strains were infected orally with A. actinomycetemcomitans (Aa) and evaluated at the 45-day time point for: (A) the number of Th2 (CD4+IL4+) cells ×104 in periodontal tissues of WT and KO strains, as determined by flow cytometry; (B) the number of Treg (CD4+FOXp3+) cells ×104 in periodontal tissues of WT and KO strains, and in CCR4KO mice after AT of Tregs from WT and KO strains (Tregs AT transfer performed at the 30-day time point, analysis at the 45-day time point); (C) alveolar bone loss in WT and KO strains, and in CCR4KO mice after AT of Tregs from WT and KO strains (Tregs AT transfer performed at the 30-day time point, analysis at the 45-day time point); depicted as the % of bone loss (increase in CEJ-ABC area in the palatal face of maxillary molars) at the 45-day time point in comparison with infected WT mice; (D) total leukocyte count in periodontal tissues (performed in a Neubauer chamber) depicted as the number of leukocytes ×105, in WT and KO strains, and in CCR4KO mice after adoptive transfer of Tregs from WT and KO strains (Tregs AT transfer performed at the 30-day time point, analysis at the 45-day time point); (E) the suppressive capacity of Tregs from WT and KO strains, analyzed in vitro against CD3/CD28 activated T cells, depicted as the suppressive activity versus Tregs from WT mice; (F) the levels of cytokines in the periodontal tissues of WT, CCR4KO, and CCR4KO mice that received AT of Tregs from WT mice (+Tregs), as measured by ELISA; and (G) kinetics of alveolar bone loss in WT and CCR4KO strains, and CCR4KO mice that received AT of Tregs from WT mice (at Tregs AT transfer performed at the 30-day time point; analysis at the 45-day and 60-day time points); depicted as increase in CEJ-ABC area in the palatal face of maxillary molars (in arbitrary units of area). Values (mean±SD) obtained from 6 animals at each point, from one experiment representative of three; *p<0.05 (Mann-Whitney test) represent a statistically significant difference versus WT. AT=adoptive transfer; OPG=osteoprotegerin; CEJ-ABC=cement-enamel junction–alveolar bone crest.
Involvement of CCR4 ligands CCL22 and CCL17 in the migration of Tregs and Th2 cells and the associated impact on the modulation of the inflammatory response and alveolar bone loss
In view of the role of CCR4 in the migration of Tregs, we next investigated which ligands could effectively interact with CCR4 in this process (Fig. 3). Our results show that CCR4 ligands CCL17 and CCL22 (Fig. 3A, B) are produced in periodontal tissues in significant levels after 30 days postinfection, which correlates with the presence of Tregs and Th2 cells in the tissues (Fig. 1). When neutralizing antibodies were used along the periodontal infection, our results show that anti-CCL17 did not impact the migration of Tregs and Th2 cells into the periodontal tissues. In contrast, anti-CCL22 treatment resulted in a significant decrease in the number of CD4+FOXp3+ cells, whereas only a minor (statistically nonsignificant) reduction of CD4+IL4+ counts was observed (Fig. 3C, D). The blockade of CCL22 also resulted in an exacerbated disease phenotype, as showed by the increased bone loss (Fig. 3E) and increased inflammatory cell counts (Fig. 3F) when compared with untreated mice. Our results also show that the blockade of CCL22 resembles the response observed in the CCR4KO strain, characterized by lower levels of IL-10, TGF-β, and OPG, and increased TNF-α and RANKL levels (Fig. 3G). Interestingly, administration of CCL22 neutralizing antibody only resulted in a minor (statistically nonsignificant) reduction of IL-4 levels (Fig. 3G). In an attempt to confirm the role of CCL22 in Tregs migration, WT Tregs were adoptively transferred to CCR4KO mice at two different time points (7 days and 30 days postinfection), CCR4+ Tregs were observed to only effectively migrate to periodontal tissues and attenuate inflammatory bone loss when transferred at 30 days postinfection (Fig. 3F, G). Notably, this is the timeframe when CCL22 was found to be produced in the tissues (Fig. 2). The injection of CCL22-releasing PLGA particles in the periodontal tissues of mice in the early disease stage (7 days postinfection), when the endogenous CCL22 is not produced, allowed the early migration of Tregs transferred at 7 days postinfection (Fig. 4A), which was accompanied by a significant decrease in the inflammatory bone loss (Fig. 4B). Not surprisingly, this early protective migration was prevented by anti-CCL22 treatment (Fig. 4A, B), but not by anti-CCL17 or met-RANTES treatment (data not shown).
Fig. 3.

Involvement of CCL22 and CCL17 in the migration of Tregs and Th2 cells and the associated impact in inflammatory alveolar bone loss. C57Bl/6 (WT) and CCR4KO mice strains were infected orally with A. actinomycetemcomitans (Aa) and evaluated for: the levels of (A) CCL22 and (B) CCL17 in the periodontal tissues at 0, 7, 15, 21, 30, 45, and 60 days postinfection, as measured by ELISA; (C) the number of Th2 (CD4+IL4+) and (D) Treg (CD4+FOXp3+) cells in periodontal tissues of WT mice, at 45-days postinfection, upon the treatment with anti-CCL22, anti-CCL17, or met-RANTES, as determined by flow cytometry; (E) alveolar bone loss, depicted as the % of bone loss (increase in CEJ-ABC area in the palatal face of maxillary molars) at the 45-day time point in comparison with infected WT mice upon the treatment with anti-CCL22, anti-CCL17, or met-RANTES; (F) inflammatory cell count, depicted as the total leukocyte count ×105 in periodontal tissues (performed in a Neubauer chamber) in WT mice upon the treatment with anti-CCL22, anti-CCL17, or met-RANTES; (G) the cytokine levels in the periodontal tissues at the 45-day time point of WT upon the treatment with anti-CCL22, as measured by ELISA. Values (mean±SD) obtained from 6 animals at each point, from one experiment representative of three. In the graphs in A and B, different letters represent statistically significant differences (p<0.05; Kruskal-Wallis and Dunn’s posttest) within the different time points in each mice strains; in all the graphs *p<0.05 (unpaired t test) represent statistically significant differences versus WT. OPG=osteoprotegerin; CEJ-ABC=cement-enamel junction–alveolar bone crest.
Fig. 4.
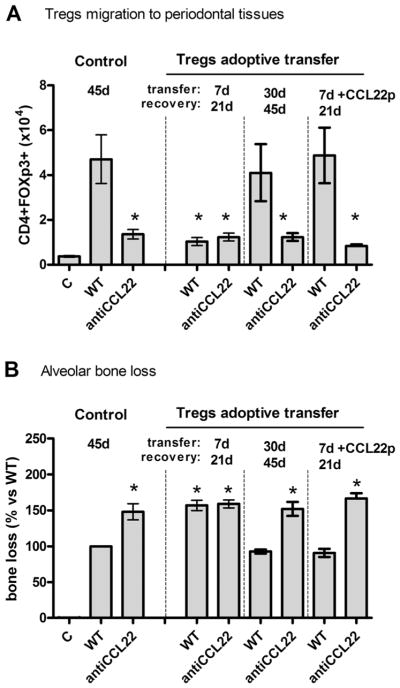
CCL22 is required for the migration and protective action of adoptively transferred Tregs. C57Bl/6 (WT) mice strain was infected orally with A. actinomycetemcomitans (Aa) and received the adoptive transference of Tregs (from WT mice). Tregs were transferred at 7 days and 30 days postinfection and the subsequent evaluations were performed at 21 days and 45 days postinfection, respectively, associated or not with antiCCL22 treatment and/or with injection of CCL22-releasing particles (CCL22p). After the treatments, mice were evaluated for (A) the number of Tregs in periodontal tissues, as determined by flow cytometry; and (B) alveolar bone loss quantification in these same experimental groups. Values (mean±SD) obtained from 6 animals at each point, from one experiment representative of three; *p<0.05 (unpaired t test) represent statistically significant differences versus WT.
The production of endogenous CCL22 and the consequent migration of Tregs is IL-4–dependent
In view of the involvement of CCL22-CCR4 axis in Tregs migration, we next investigated the possible involvement of IL-4 as the trigger of endogenous CCL22 expression. Our data show a dramatic decrease in CCL22 levels in the IL-4KO strain (Fig. 5A, B), which was associated with reduced Treg counts in the tissues (Fig. 5C), exacerbated bone loss (Fig. 5D), and increased total inflammatory cell counts (Fig. 5E). Interestingly, when the lack of endogenous CCL22 was overcome by the injection of CCL22-releasing particles in the periodontal tissues of IL-4KO mice, the migration of Tregs was fully restored (Fig. 5C), and the exacerbated inflammatory bone loss phenotype was completely reverted to levels similar to those observed in WT mice (Fig. 5D, E). When the cytokine levels in the lesions of IL4KO mice were evaluated (Fig. 5F), we observed that IL-4KO mice were characterized by a decreased production of IL-10, TGF-β, and OPG, and increased levels of TNF-α and RANKL when compared to WT mice. Notably, the restoration of Tregs migration by CCL22 particles resulted in the restoration of cytokine levels to those observed in WT mice (Fig. 5F). Interestingly, the injection of CCL22-releasing particles, and the consequent Tregs migration, do not result in the expression of CCL22mRNA in the tissues of WT and IL-4KO mice (Fig. 5B) or the production of IL-4 in WT mice (at 15 days postinfection) (Fig. 5G). Consistent with this result, the inhibition of Tregs (using anti-GITR) in WT mice does not compromise the production of IL-4 in periodontal lesions (Fig. 5G).
Fig. 5.

The production of CCL22 and the consequent migration of Tregs is IL-4–dependent. C57Bl/6 (WT) and IL4KO mice strains were infected orally with A. actinomycetemcomitans (Aa), treated or not with anti-GITR (to disable Tregs) or with CCL22-releasing particles (CCL22p), were evaluated at the 45-day time point for: the levels of CCL22 protein (A) and mRNA (B) in the periodontal tissues of WT and IL4KO strains, and WT mice treated with anti-GITR, as measured by ELISA; (B) the levels of CCL22 mRNA in the periodontal tissues, at the 45-day time point, of WT and IL4KO mice, treated or not with CCL22-releasing particles, as measured by qPCR; (C) the number of Treg (CD4+FOXp3+) cells in periodontal tissues of WT and IL4KO mice, treated or not with CCL22-releasing particles, as determined by flow cytometry; (D) alveolar bone loss, evaluated through measurement of the CEJ-ABC area in the palatal face of maxillary molars at the 45-day time point, depicted as the % bone loss in WT and IL4KO mice, treated or not with CCL22-releasing particles, in comparison with control (infected) mice; (E) total leukocyte count in periodontal tissues (performed in a Neubauer chamber) depicted as the number of leukocytes ×105, in WT and IL4KO mice, treated or not with CCL22-releasing particles, in comparison with control (infected) mice. Also, WT and IL4KO mice strains, and IL4KO mice that received the adoptive transfer of Tregs from WT mice (at the 30-day time point) were evaluated for (F) the levels of cytokines in the periodontal tissues, at the 45-day time point, as measured by ELISA; (G) the levels of IL-4 in periodontal tissues were also measured by ELISA in WT mice, treated or not with anti-GITR, and IL4KO mice that received the adoptive transfer of Tregs from WT mice. Values (mean±SD) obtained from6 animals at each point, from one experiment representative of three; *p<0.05 (Mann-Whitney test) represent statistically significant differences versus WT; # denotes “undetectable levels.” OPG=osteoprotegerin; GITR=glucocorticoid-induced TNF-receptor-related protein; CEJ-ABC=cement-enamel junction-alveolar bone crest.
Discussion
The chronic inflammatory immune response associated with PD pathogenesis mediates tissue destruction by means of the upregulation of chemoattraction of osteoclast precursors and the proinflammatory and osteoclastogenic mediators production, which collectively accounts for the irreversible bone resorption that typify PD.(4,23) However, some T-cell subsets, such as Th2 and Treg cells, have been associated the arrest of inflammatory osteolysis and the consequent attenuation of PD progression.(4) Although the role of these T-cell subsets have each been investigated individually, the possible interaction between these T helper subsets, as well as the exact mechanisms underlying its alleged protective role, remains unknown.
In this context, our data initially show that a significant influx of Th1 and Th17 cells is verified in the initial time points evaluated, being at this initial disease stage characterized by the development of inflammatory cellular infiltrate and by high rates of alveolar bone loss. Accordingly, Th1 and Th17 cells were previously described as mediators of inflammatory and osteoclastogenic processes.(4,11) Following the experimental disease development, both Th2 cells (CD4+IL4+) and Tregs (CD4+FOXp3+) migrate into periodontal tissues along experimental periodontitis.(8,10) In accordance with our data, the late disease period was previously described as a relatively stable lesion characterized by the steadiness of inflammatory cell migration and the reduction of alveolar bone loss evolution rate.(9) In order to dissect the mechanisms involved in Th2 and Tregs cells migration, we next showed that, although Tregs from PBMCs express high levels of CCR4 and CCR5, Tregs extracted from periodontal tissues express high levels of CCR4 and CCR8, suggesting that these receptors could be responsible for Treg influx into inflamed periodontium. Accordingly, previous studies report that Tregs can present a variable chemokine receptor repertoire including CCR4, CCR5, CCR7, and CCR8, and that such variability may confer the specificity of tissue/organ homing of Tregs.(13) Indeed, CCR4 and CCR8 have been implicated in the migration of Tregs to skin and mucosal tissues. CCR5 is described to mediate Treg migration to non-mucosal tissues. CCR7 is thought to mediate Treg migration to secondary lymphoid organs.(13) Indeed, current evidence shows that Tregs in vivo are comprised of heterogeneous subsets with chemokine receptor repertoires that mimic many/some effector and memory CD4+FOXp3− populations, which may account for the differential migration in health or disease conditions.(13) However, in view of the absence of definitive markers to unquestionably subset Tregs, the possible variation in chemokine receptor expression by these virtual Treg subpopulations awaits further confirmation.(24,25) Our results also show that Th2 cells (CD4+IL4+) from PBMCs and gingival tissues are similar with respect to chemokine receptor expression, being that CCR3, CCR4, and CCR8 are expressed by Th2 cells, suggesting that these three receptors contribute to Th2 cell migration to periodontal tissues along PD development. In accordance with our data, previous studies show a similar pattern of chemokine receptor expression by Th2 cells.(26)
In view of the chemokine receptor expression profile identified in Tregs and Th2 cells, we next evaluated the role of CCR4, CCR5, and CCR8 in their migration by means of genetically deficient mice strains. Our results showed that the lack of CCR4 critically impaired the migration of Tregs to periodontal tissues, whereas CCR5KO and CCR8KO mice presented a normal influx of Tregs to periodontium. Indeed, despite the variable chemokine receptor expression by Tregs, CCR4 has been described as the main receptor responsible for directing Tregs to peripheral tissues during inflammation.(27,28) Interestingly, the number of Tregs in the draining submandibular lymph nodes is not impacted by the lack of CCR4 (data not shown), suggesting that its deficiency specifically impacts its peripheral function at inflammatory sites. Interestingly, the migration of Tregs in the CCR8-deficient mice reinforces the preferential role of CCR4, as described.(13) The lack of impact of the absence of CCR5 on the influx of Tregs into the periodontal tissues seems to be consistent with the low expression of this receptor in Tregs from gingival tissue in contrast with the Tregs from PBMCs. Collectively, these data reinforce the existence of distinct Treg subsets with distinct migratory properties (as discussed in the beginning of this paragraph).(13,28) Also consistent with the role of CCR4 in directing migration of Tregs, in the late disease period (which can be characterized by influx of Tregs), there is a characteristic decline of CCR5 ligand expression and a significant increase in the expression of CCR4 ligands.(10,23) Interestingly, whereas the migration of Tregs was markedly compromised by the absence of CCR4, the reduction in the number of Th2 cells (CD4+IL4+), as well the local levels of IL-4, was smaller in percentage terms than the reduction in the number of Tregs and also the production of Treg-characteristic cytokines (IL-10 and TGF-β). However, the simultaneous expression of chemokine receptors CCR3, CCR4, and CCR8 by Th2 cells, suggests that these redundancies compensate for the lack of individual receptors, allowing the local development of Th2-type responses.(13,27,28)
Importantly, the CCR4 deficiency and the consequent defective migration of Tregs result in a clear disease exacerbation phenotype, characterized by increased bone loss and inflammatory cell infiltration, perceptible only in the late disease stage when CCR4-mediated Treg influx attenuates disease development in the WT strain. Interestingly, such a phenotype recapitulates the one presented by WT mice treated with anti-GITR in order to ablate the function of Tregs.(8) Indeed, the comparative analysis of the cytokine profile in WT and CCR4KO mice show that the lack of CCR4 results in lower levels of Tregs-associated cytokines IL-10 and TGF-β, and in higher levels of proinflammatory, Th1-type and Th17-type cytokines. Accordingly, Tregs were previously shown to attenuate such inflammatory and osteoclastogenic mediators/responses production in PD and RA models.(8,29) It is imperative to mention that in vitro CCR4KO Tregs display intact immunosuppressive function, suggesting that the compromised migratory properties of Treg, and not an intrinsic defective function, is responsible for the exacerbated PD phenotype. In fact, the adoptive transfer of WT CCR4+ Tregs to the CCR4KO strain completely reverts the exacerbated PD phenotype, resulting in bone loss and inflammation levels that were similar to those observed in WT mice. Indeed, the adoptive transfer of Tregs results in the restoration of the local “WT” profile of cytokines (including RANKL and OPG) emphasizing the central role of CCR4 in allowing Treg migration that attenuates inflammatory bone resorption. Similarly, blockade of Treg mobilization via CCR4 inhibition results in the exacerbation of inflammation in allergy models.(18,27)
In view of the critical role of CCR4 in the control of Treg migration, we performed a detailed investigation of the kinetic profile of CCR4 ligands. The two main CCR4 ligands, CCL17 and CCL22, were found to be produced in the periodontium after 30 days of infection, which corresponded to the timeframe of the influx of Tregs. Accordingly, both CCL17 and CCL22 were previously described to be expressed in human periodontal lesions.(30) In order to determine the importance of local chemokine expression to the migration of Tregs in a cause-and-effect manner, we showed that CCL22 inhibition significantly decreased the number of Tregs in the tissues, and resulted in increased bone loss and influx of inflammatory cells. Conversely, CCL17 blockade neither significantly impaired the migration of Tregs, nor resulted in changes in the disease severity parameters. Accordingly, despite the sharing of CCR4 specificity/binding by CCL17 and CCL22, CCL17 seems to be less efficient in CCR4+ cells chemoattraction in vivo.(19,26,31) Indeed, in vitro data shows that at similar concentrations CCL22 result in higher migration of CCR4+ cells than CCL17 (data not shown). To confirm the involvement of CCL22/CCR4 in the Treg migration process, we next performed the adoptive transfer of Tregs (CCR4+ WT Tregs to CCR4KO mice), in two different time intervals, where CCL22 is absent (7 to 21 days) or present (30 to 45 days) in periodontal tissues. Interestingly, transferred Tregs only can migrate to periodontal tissues and suppress experimental PD/inflammatory bone loss in the late period, when CCL17 and CCL22 are produced in the lesions. Interestingly, the CCL22 blockade along adoptive transfer of Tregs confirmed the major role for CCL22 in the migration of Tregs, with this blockade impairing the migration of Tregs and exacerbating the disease phenotype. In contrast, CCL17 blockade did not significantly impact the migration of Tregs or the disease outcome.
It is also important to consider that other factors, such as T cell receptor (TCR) activation and expansion status, may interfere not only in the CCR4 expression profile, but also in the responsiveness of CCR4+ cells to the ligands CCL17 and CCL22,(26) adding a new layers of complexity to the chemokine/chemokine receptors system. Certifying the role of CCL22 in Treg chemoattraction via CCR4, we show that when the early (ie, in the absence of endogenous CCL22 in gingival tissue) adoptive Tregs transfer is accompanied by the injection in the periodontal tissues of a CCL22-releasing formulation, Tregs regularly can infiltrate the inflamed periodontium and exert their suppressive function, attenuating PD-associated inflammatory bone loss. Accordingly, we previously showed that the therapeutic effect of CCL22-releasing systems was Treg-dependent.(5)
In addition, we investigated which factor would be responsible for promoting the increased expression of endogenous CCL22 in experimental PD, and the consequent impact on migration of Tregs. Considering that CCL22 was originally described as a Th2 chemokine, whose expression was induced on macrophages upon stimulation with Th2 cytokines,(32) we hypothesized that IL-4 could be a key factor in the Treg migration process. As expected, our data revealed an almost complete inhibition of CCL22 production/expression along experimental PD in IL-4KO mice, accompanied of a dramatically reduced migration of Tregs, exacerbated inflammation, and bone loss. Accordingly, IL-4 gene therapy was previously described to mediate Treg migration via the induction of CCL1, CCL17, and CCL22 in the central nervous system, mediating clinical recovery in mouse models of multiple sclerosis.(33) Interestingly, we show that the restoration of Treg migration in IL4KO mice by CCL22-releasing formulation completely reverts the exacerbated disease phenotype to levels similar to those observed in WT mice, suggesting that the previously alleged/described protective role of IL-4 in inflammatory osteolysis scenario(34) could actually rely on Treg chemoattraction. Also, it is worthwhile to mention that Treg inhibition (by anti-GITR) or recruitment (by CCL22p) does not significantly alter the endogenous expression/production of IL-4 and CCL22, showing that IL-4 is indeed upstream in the axis that governs Treg migration, and that IL-4 production is not under the control/influence of Tregs in our murine models of PD. Interestingly, whereas Tregs are described to effectively attenuate Th1 and Th17 responses, previous studies show that Tregs do not suppress Th2 cells’ ability to produce IL-4,(35) which fits with our data showing the central role of IL-4 in initiating the axis (ie, IL-4/CCL22/CCR4) that leads to migration of Tregs. Additional evidence supports the hypothesis that after the migration of Tregs, their immunoregulatory action toward attenuation of the inflammatory bone resorption is IL-4–independent. Previous studies show that gene therapy–induced expression of IL-4 in an RA model resulted in upregulation of IL-10, a known product of Tregs, whereas gene therapy–induced expression of IL-10 did not result in IL-4 production.(36) Interestingly, both situations resulted in reduction in bone erosion and cartilage degradation.(36) Also, the protective effects of IL-4 against exacerbated Th1-type responses appear to be mediated by IL-10.(37,38) However, because some studies report that IL-4 can directly stimulate the production of OPG and downregulate the levels of RANKL, proinflammatory, and osteoclastogenic cytokines,(7,39,40) it is possible that IL-4 exerts additional protective roles in the inflammatory osteolysis scenario other than mediating the migration of Tregs.
Finally, in view of the central role of IL-4 in triggering Treg migration via CCL22/CCR4, it is important to discuss the development of Th2 responses in the absence of CCR4. Accordingly, previous studies show a partial (~50%) decrease of Th2 response in the absence of CCR4.(41) However, genetic or pharmacological ablation of CCR4 function results in controversial data regarding its role in the development of Th2-type responses.(13,26,35) Indeed, whereas the majority of research has centered on the role of CCR4 as a key CCR4+ cell chemoattractant, Th2 cells express a variety of chemokine receptors, such as CCR3, CCR8, and the DP2 or CRTH2.(13,26) Accordingly, our data show only a modest (<15%) decrease in IL-4 levels in CCR8KO mice, suggesting that CCR3 and CCR4 receptors may compensate for the lack of CCR8.(42) Indeed, recent reports show that CCR3 inhibition impacts Th2 cell trafficking similarly to CCR4 blockade, reinforcing the involvement of multiple chemokine/chemokine receptors in Th2 response development.(26) Another possibility is that the initial expression of IL-4 is not dependent on the influx of polarized Th2 cells into periodontal tissues, and that the plasticity of T cells that reached the response site with a different phenotype (such as Th17 cells) could result in the initial IL-4 production.(43–45) Once produced in the tissue, IL-4 would induce CCL17 and CCL22 expression,(32) which in turn can support/increase Th2 response as well as support CCR4-dependent migration of Tregs. In accordance with this hypothesis, the lack of CCR4 does not eliminate the IL-4 expression, but only prevent its subsequent increase as observed in the WT strain. However, at this point our data do not support strong statements regarding the rise of IL-4 production along with periodontitis, and further studies are required to elucidate the mechanisms responsible for the generation/migration of Th2 cells to periodontal tissues. Also, it is important to consider that as in every experimental model, the model used in this study has some limitations, and ideally should be replicated with different bacterial challenges in mice strains with different genetic background to confirm the findings described here.
In summary, our results show the involvement of IL-4/CCL22/CCR4 axis in the migration of Tregs to osteolytic lesions sites, and that such a process attenuates lesion development by inhibiting inflammatory migration and the production of proinflammatory and osteoclastogenic mediators. These data suggest that chemokine receptors appear to be attractive potential therapeutic targets to limit the inflammatory and bone-resorptive process characteristic of PD.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) 1R01DE021058-01 A1 and 1R56DE021058-01 through the National Institute of Dental and Craniofacial Research (to SRL, CS, and GPG) and F31 DE021297-01 (to A.J. G.); Wallace H. Coulter Foundation (to SRL and CS); Camille and Henry Dreyfus Foundation (to SRL); Arnold and Mabel Beckman Foundation (to SRL); Commonwealth of Pennsylvania (to SRL); and Fundação de Amparo à Pesquisa do Estado de São Paulo (Foundation for Research Support of the State of São Paulo, or FAPESP (to GPG, AEV, CFF, and CCB).
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Authors’ roles: Study design: CSF, SRL, GPG Experimental conduct: ACAP, AEV, CFF, CCB, AG, SY Animal work: ACAP, AEV, CFF, CCB Data collection, analysis and interpretation: ACAP, AEV, CFF, CCB, APC, APFT, CSF, SRL, GPG Manuscript preparation: CSF, SRL, GPG Approving final version of the manuscript: all authors.
References
- 1.Tonetti MS, Claffey N European Workshop in Periodontology Group C. Advances in the progression of periodontitis and proposal of definitions of a periodontitis case and disease progression for use in risk factor research. Group C consensus report of the 5th European Workshop in Periodontology. J Clin Periodontol. 2005;32 (Suppl 6):210–3. doi: 10.1111/j.1600-051X.2005.00822.x. [DOI] [PubMed] [Google Scholar]
- 2.Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J Intern Med. 2007;261(6):511–28. doi: 10.1111/j.1365-2796.2007.01821.x. [DOI] [PubMed] [Google Scholar]
- 3.Kornman KS, Crane A, Wang HY, et al. The interleukin-1 genotype as a severity factor in adult periodontal disease. J Clin Periodontol. 1997;24(1):72–7. doi: 10.1111/j.1600-051x.1997.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 4.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89(12):1349–63. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 5.Glowacki AJ, Yoshizawa S, Jhunjhunwala S, et al. Prevention of inflammation-mediated bone loss in murine, canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci U S A. 2013;110(46):18525–30. doi: 10.1073/pnas.1302829110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochetkova I, Thornburg T, Callis G, Pascual DW. Segregated regulatory CD39 + CD4+ T cell function: TGF-beta-producing Foxp3- and IL-10-producing Foxp3+ cells are interdependent for protection against collagen-induced arthritis. J Immunol. 2011;187(9):4654–66. doi: 10.4049/jimmunol.1100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lubberts E, Joosten LA, Chabaud M, et al. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105(12):1697–710. doi: 10.1172/JCI7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garlet GP, Cardoso CR, Mariano FS, et al. Regulatory T cells attenuate experimental periodontitis progression in mice. J Clin Periodontol. 2010;37(7):591–600. doi: 10.1111/j.1600-051X.2010.01586.x. [DOI] [PubMed] [Google Scholar]
- 9.Garlet GP, Cardoso CR, Silva TA, et al. Cytokine pattern determines the progression of experimental periodontal disease induced by Actinobacillus actinomycetemcomitans through the modulation of MMPs, RANKL, and their physiological inhibitors. Oral Microbiol Immunol. 2006;21(1):12–20. doi: 10.1111/j.1399-302X.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 10.Garlet GP, Avila-Campos MJ, Milanezi CM, Ferreira BR, Silva JS. Actinobacillus actinomycetemcomitans-induced periodontal disease in mice: patterns of cytokine, chemokine, and chemokine receptor expression and leukocyte migration. Microbes Infect. 2005;7(4):738–47. doi: 10.1016/j.micinf.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Araujo-Pires AC, Francisconi CF, Biguetti CC, et al. Simultaneous analysis of T helper subsets (Th1, Th2, Th9, Th17, Th22, Tfh, Tr1 and Tregs) markers expression in periapical lesions reveals multiple cytokine clusters accountable for lesions activity and inactivity status. J Appl Oral Sci. 2014;22(4):336–46. doi: 10.1590/1678-775720140140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brownlie RJ, Myers LK, Wooley PH, et al. Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4-producing regulatory T cells: a novel function for an ancient protein. Arthritis Rheum. 2006;54(3):854–63. doi: 10.1002/art.21654. [DOI] [PubMed] [Google Scholar]
- 13.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 14.Repeke CE, Ferreira SB, Jr, Claudino M, et al. Evidences of the cooperative role of the chemokines CCL3, CCL4 and CCL5 and its receptors CCR1+ and CCR5+ in RANKL+ cell migration throughout experimental periodontitis in mice. Bone. 2010;46(4):1122–30. doi: 10.1016/j.bone.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Garlet GP, Cardoso CR, Campanelli AP, et al. The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: host protection and tissue destruction. Clin Exp Immunol. 2007;147(1):128–38. doi: 10.1111/j.1365-2249.2006.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against Coxsackievirus B3-induced cardiac fibrosis. PLoS One. 2013;8 (9):e74955. doi: 10.1371/journal.pone.0074955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L, Xu W, Wen Z, Xiong S. In situ prior proliferation of CD4+ CCR6+ regulatory T cells facilitated by TGF-beta secreting DCs is crucial for their enrichment and suppression in tumor immunity. PLoS One. 2011;6(5):e20282. doi: 10.1371/journal.pone.0020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iellem A, Mariani M, Lang R, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194 (6):847–53. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakubzick C, Wen H, Matsukawa A, Keller M, Kunkel SL, Hogaboam CM. Role of CCR4 ligands, CCL17 and CCL22, during Schistosoma mansoni egg-induced pulmonary granuloma formation in mice. Am J Pathol. 2004;165(4):1211–21. doi: 10.1016/S0002-9440(10)63381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Repeke CE, Ferreira SB, Jr, Vieira AE, et al. Dose-response met-RANTES treatment of experimental periodontitis: a narrow edge between the disease severity attenuation and infection control. PLoS One. 2011;6 (7):e22526. doi: 10.1371/journal.pone.0022526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jhunjhunwala S, Raimondi G, Glowacki AJ, et al. Bioinspired controlled release of CCL22 recruits regulatory T cells in vivo. Adv Mater. 2012;24(35):4735–8. doi: 10.1002/adma.201202513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trombone AP, Ferreira SB, Jr, Raimundo FM, et al. Experimental periodontitis in mice selected for maximal or minimal inflammatory reactions: increased inflammatory immune responsiveness drives increased alveolar bone loss without enhancing the control of periodontal infection. J Periodontal Res. 2009;44(4):443–51. doi: 10.1111/j.1600-0765.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira SB, Jr, Repeke CE, Raimundo FM, et al. CCR5 mediates pro-osteoclastic and osteoclastogenic leukocyte chemoattraction. J Dent Res. 2011;90(5):632–7. doi: 10.1177/0022034510395021. [DOI] [PubMed] [Google Scholar]
- 24.Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev. 2014;259(1):159–72. doi: 10.1111/imr.12175. [DOI] [PubMed] [Google Scholar]
- 25.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14(10):1007–13. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komiya T, Sugiyama T, Takeda K, et al. Suppressive effects of a novel CC chemokine receptor 4 antagonist on Th2 cell trafficking in ligand- and antigen-induced mouse models. Eur J Pharmacol. 2013;720(1–3):335–43. doi: 10.1016/j.ejphar.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 27.Faustino L, da Fonseca DM, Takenaka MC, et al. Regulatory T cells migrate to airways via CCR4 and attenuate the severity of airway allergic inflammation. J Immunol. 2013;190(6):2614–21. doi: 10.4049/jimmunol.1202354. [DOI] [PubMed] [Google Scholar]
- 28.Afshar R, Strassner JP, Seung E, et al. Compartmentalized chemokine-dependent regulatory T-cell inhibition of allergic pulmonary inflammation. J Allergy Clin Immunol. 2013;131(6):1644–52. doi: 10.1016/j.jaci.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan ME, Flierman R, van Duivenvoorde LM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 2005;52(7):2212–21. doi: 10.1002/art.21195. [DOI] [PubMed] [Google Scholar]
- 30.Cardoso CR, Garlet GP, Moreira AP, Junior WM, Rossi MA, Silva JS. Characterization of CD4 + CD25+ natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J Leukoc Biol. 2008;84(1):311–8. doi: 10.1189/jlb.0108014. [DOI] [PubMed] [Google Scholar]
- 31.Wu C, Zhou Q, Qin XJ, Qin SM, Shi HZ. CCL22 is involved in the recruitment of CD4 + CD25 high T cells into tuberculous pleural effusions. Respirology. 2010;15(3):522–9. doi: 10.1111/j.1440-1843.2010.01719.x. [DOI] [PubMed] [Google Scholar]
- 32.Andrew DP, Chang MS, McNinch J, et al. STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytokines IL-4 and IL-13. J Immunol. 1998;161(9):5027–38. [PubMed] [Google Scholar]
- 33.Butti E, Bergami A, Recchia A, et al. IL4 gene delivery to the CNS recruits regulatory T cells and induces clinical recovery in mouse models of multiple sclerosis. Gene Ther. 2008;15(7):504–15. doi: 10.1038/gt.2008.10. [DOI] [PubMed] [Google Scholar]
- 34.Myers LK, Tang B, Stuart JM, Kang AH. The role of IL-4 in regulation of murine collagen-induced arthritis. Clin Immunol. 2002;102(2):185–91. doi: 10.1006/clim.2001.5162. [DOI] [PubMed] [Google Scholar]
- 35.Faustino L, Mucida D, Keller AC, et al. Regulatory T cells accumulate in the lung allergic inflammation and efficiently suppress T-cell proliferation but not Th2 cytokine production. Clin Dev Immunol. 2012;2012:721817. doi: 10.1155/2012/721817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim SH, Evans CH, Kim S, Oligino T, Ghivizzani SC, Robbins PD. Gene therapy for established murine collagen-induced arthritis by local and systemic adenovirus-mediated delivery of interleukin-4. Arthritis Res. 2000;2(4):293–302. doi: 10.1186/ar104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han WG, van der Voort EI, el Bannoudi H, Louis-Plence P, Huizinga TW, Toes RE. DX5(+)CD4(+) T cells modulate cytokine production by CD4 (+) T cells towards IL-10 via the production of IL-4. Eur J Immunol. 2010;40(10):2731–40. doi: 10.1002/eji.201040574. [DOI] [PubMed] [Google Scholar]
- 38.Charbonnier LM, Han WG, Quentin J, et al. Adoptive transfer of IL-10-secreting CD4 + CD49b+ regulatory T cells suppresses ongoing arthritis. J Autoimmun. 2010;34(4):390–9. doi: 10.1016/j.jaut.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Stein NC, Kreutzmann C, Zimmermann SP, et al. Interleukin-4 and interleukin-13 stimulate the osteoclast inhibitor osteoprotegerin by human endothelial cells through the STAT6 pathway. J Bone Miner Res. 2008;23(5):750–8. doi: 10.1359/jbmr.080203. [DOI] [PubMed] [Google Scholar]
- 40.Yamada A, Takami M, Kawawa T, et al. Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology. 2007;120(4):573–9. doi: 10.1111/j.1365-2567.2006.02538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mikhak Z, Fukui M, Farsidjani A, Medoff BD, Tager AM, Luster AD. Contribution of CCR4 and CCR8 to antigen-specific T(H)2 cell trafficking in allergic pulmonary inflammation. J Allergy Clin Immunol. 2009;123(1):67–73.e3. doi: 10.1016/j.jaci.2008.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277(5334):2005–7. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 43.Cosmi L, Maggi L, Santarlasci V, Liotta F, Annunziato F. T helper cells plasticity in inflammation. Cytometry A. 2014;85(1):36–42. doi: 10.1002/cyto.a.22348. [DOI] [PubMed] [Google Scholar]
- 44.Hirahara K, Poholek A, Vahedi G, et al. Mechanisms underlying helper T-cell plasticity: implications for immune-mediated disease. J Allergy Clin Immunol. 2013;131(5):1276–87. doi: 10.1016/j.jaci.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20(1):4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]