

Abstract
Background
Childhood separation anxiety disorder (SAD) is hypothesized to share etiologic roots with panic disorder. The aim of this study was to estimate the genetic and environmental sources of covariance between childhood SAD and adult onset panic attacks (AOPA), with the primary goal to determine whether these two phenotypes share a common genetic diathesis.
Methods
Participants included parents and their monozygotic or dizygotic twins (n = 1,437 twin pairs) participating in the Virginia Twin Study of Adolescent Behavioral Development and those twins who later completed the Young Adult Follow-Up (YAFU). The Child and Adolescent Psychiatric Assessment was completed at three waves during childhood/adolescence followed by the Structured Clinical Interview for DSM-III-R at the YAFU. Two separate, bivariate Cholesky models were fit to childhood diagnoses of SAD and overanxious disorder (OAD), respectively, and their relation with AOPA; a trivariate Cholesky model also examined the collective influence of childhood SAD and OAD on AOPA.
Results
In the best-fitting bivariate model, the covariation between SAD and AOPA was accounted for by genetic and unique environmental factors only, with the genetic factor associated with childhood SAD explaining significant variance in AOPA. Environmental risk factors were not significantly shared between SAD and AOPA. By contrast, the genetic factor associated with childhood OAD did not contribute significantly to AOPA. Results of the trivariate Cholesky reaffirmed outcomes of bivariate models.
Conclusions
These data indicate that childhood SAD and AOPA share a common genetic diathesis that is not observed for childhood OAD, strongly supporting the hypothesis of a specific genetic etiologic link between the two phenotypes.
Keywords: separation anxiety disorder, panic attacks, twins, genetic, environment
INTRODUCTION
Klein[1] first proposed that childhood separation anxiety disorder (SAD) was a precursor of panic disorder (PD), a condition that generally emerges in early adulthood.[2] This “separation anxiety hypothesis” was founded on retrospective reports of an association between childhood separation anxiety and adult PD, relative to other psychiatric groups. During the past three decades, mixed evidence has emerged from three types of studies that either support or challenge the possibility of an etiologic link between SAD and PD; these studies either examine retrospective report, the prospective course of childhood SAD into adulthood, or parent–child aggregation of PD–SAD.
The strongest support for the developmental link between childhood SAD and adult onset PD comes from prospective studies. In a follow-up study of 54 children referred for treatment of SAD, 7% of individuals with a history of SAD developed PD whereas no participant in the comparison group developed PD.[3] Moreover, no other anxiety disorder was more prevalent in adults with a history of SAD relative to comparison participants, although the overwhelming majority of SAD children did not develop PD. A second longitudinal study reinterviewed children (ages 9–13) approximately 7.5 years after receiving treatment and observed a higher rate (27.8%) of PD among children receiving a principal diagnosis of childhood SAD compared to children with a primary diagnosis of social phobia (SP; 12.5%) or overanxious disorder (OAD; 14.3%).[4] Although these studies support the SAD–PD connection, most of the participants had not yet passed through the age of risk for PD (i.e. mid-late 20s;[5]), which likely diminished the strength of association between the two disorders. The most striking support for a developmental link between SAD and PD comes from a follow-up study of children at high and low risk for PD and major depressive disorder (MDD) ascertained through parent probands treated for PD, MDD, or control parents (total n = 233;[6]). Results indicated that offspring with a history of childhood SAD were at greatest risk for developing PD and agoraphobia, with an OR of approximately 9.0. By comparison, odds of developing MDD were 3.2 for children with a history of SAD.
Family studies examining parent–child transmission provide only modest support for the SAD–PD hypothesis. Several family aggregation studies document greater frequency of SAD diagnoses among offspring of parents with PD, relative to offspring of healthy parents,[7–10] These results appear relatively consistent, suggesting that PD in a parent confers risk for development of SAD. On the other hand, because SAD also has been linked to parental MDD with some consistency, it may represent a nonspecific correlate of parental mood or anxiety disorders.[9] Moreover, some studies link development of PD to OAD;[11]) and some retrospective studies link childhood SAD to adult onset disorders besides PD (e.g. SP;[12]).
The high-risk family design also has been used to study carbon dioxide (CO2) sensitivity in offspring. Although sensitivity to breathing CO2-enriched air has emerged as a robust biological marker of PD,[13,14] this sensitivity is not evident among offspring of parents with PD. Rather, presence of a childhood anxiety disorder (including SP, generalized anxiety disorder [GAD], and SAD) is related to Acute Panic Inventory scores assessed in response to CO2 breathing, with anxious children reporting a significantly greater number of symptoms.[13] In a second study, the interaction between offspring SAD and parental PD was examined. Results indicated that children with both risk factors manifest a ventiliatory response similar to that observed among adult panic patients including heightened respiratory rate, decreased tidal volume, and increased number of panic-like symptoms.[14] Thus, children with SAD, particularly those of parents with PD, manifest CO2 sensitivity, which suggests this population is at increased risk for development of PD.
Although family studies suggest some level of coaggregation of offspring SAD among parental PD, family studies cannot distinguish between genetic and shared environmental causes. Genetic and shared environmental factors can be disaggregated using twin methods, however. Of the twin studies available, they suggest a heritable component to SAD, with the genetic contribution being higher for females (0.46–0.59) compared with males (0.21–0.31) and unique environmental factors being more significant for males (0.69–0.92) relative to females (0.41–0.55).[15] These estimates are consistent with other studies observing robust heritability estimates for anxiety symptoms in females compared with males.[15,16] The adult PD literature also suggests that only genetic (0.47) and unique environmental factors (0.57) emerge as significant influences on PD.[17] Collectively, the extant literature largely supports the hypothesized link between SAD–PD and, while SAD confers risk for PD within individuals, risk appears to be higher for offspring of PD probands.
The current study utilizes the power of the twin design to determine whether genetic risk factors associated with childhood SAD also relate specifically to development of panic attacks in young adulthood. We define our phenotype here as a lower bound of lifetime panic attacks versus PD for several reasons: (1) most of the adult twins in our sample had not yet passed through the age of risk for PD; (2) individuals may carry a genetic risk for a particular psychiatric syndrome that may not be expressed at a diagnostic level, but may be detectable at subthreshold levels; and (3) PD has a low lifetime prevalence rate (approximately 1.5–3.5%; APA, 2000) thereby decreasing the amount of information available for complex twin modeling. Studies of nonclinical panickers also suggest several similarities with PD patients, with nonclinical panickers reporting many of the same symptoms as persons with PD.[18] Prevalence estimates indicate that about 9–14% of the general population report at least one DSM-defined panic attack in their lifetime.[19,20] In our sample, approximately 10.6% of respondents met DSM-IV criteria for panic attack(s). Of this 10.6%, approximately 6.75% met lifetime criteria for panic attacks and 3.9% met the more stringent PD criteria. Thus, our adult onset panic attack (AOPA) phenotype includes a mix of persons experiencing panic attacks and those meeting criteria for PD.
We predict that genetic factors associated with childhood SAD will influence AOPA. As a second step, we will examine the relation between childhood OAD and AOPA to determine whether SAD has a unique relationship with PD relative to childhood OAD. We predict that genetic factors associated with childhood OAD will not contribute as strongly to AOPA as compared to SAD. Finally, we will conduct a third analysis where we model childhood SAD, childhood OAD, and AOPA in one model. We again predict a unique genetic relationship between childhood SAD and AOPA in the context of childhood OAD. Moreover, because childhood OAD shares diagnostic overlap with GAD, the results observed for OAD serve as a proxy for the relation between childhood GAD and AOPA.
METHODS
PARTICIPANTS
Participants in this study included child respondents and their parents who participated in the Virginia Twin Study of Adolescent Behavioral Development (VTSABD). The VTSABD was designed as an epidemiological study of children and as an epidemiological study of twins for genetically informative analysis. Childhood psychiatric data comprise intensive, juvenile, and young adult longitudinal, multirater (child, parent) clinical interview data in a large sample of male and female Caucasian twin pairs. Psychiatric outcomes assessed in twins and their parents included symptoms; severity; impairment; and treatment relating to a range of anxiety disorders, depression, conduct disorder, oppositional-defiant disorder, attention-deficit hyperactivity disorder, and substance use disorders. Parents also were queried about their twins’ zygosity and the similarity of the twins’ early environment. All interviewers received 3 weeks of intensive training, and all interviews were audiotaped and reviewed by senior monitors. Regular meetings were held to prevent interviewer drift and review other aspects of the research protocol. A residential meeting also was held annually to ensure that the research protocol was being practiced in a standardized format.
The total sample included 1,437 twin pairs (27.1% monozygotic [MZ] female, 16.1% dizygotic [DZ] female, 20.7% MZ male, 15.0% DZ male, 21.0% opposite-sex DZ pairs). Three longitudinal waves of the VTSABD were conducted during childhood and adolescence. The mean age was 12.35 years (±2.59; range 8.2–18.2) at the wave one assessment, 13.58 years (±2.4; range 9.3–18.3) at the wave two assessment, and 15.5 years (±1.6; range 12.0–18.0) at the wave three assessment. About 6.45% (n = 176) subjects met lifetime criteria for SAD based on a positive diagnosis from at least one of the three assessment waves. Most youth were diagnosed with SAD at the wave one assessment (80.1%), however, with few cases being diagnosed at waves two (18.2%) and three (1.7%). About half (50.6%) of all children receiving a diagnosis of SAD also met criteria for OAD at some point in their lifetime. Approximately 19.6% (n = 563) of the sample met criteria for OAD at either wave one, two, or three. Of those youth meeting criteria for OAD, 58.7% were diagnosed at wave one, 31.6% at wave two, and 9.7% at wave three. Participants with a positive history for SAD (29.5%) were more likely not to participate in the Young Adult Follow-up (YAFU) compared to persons without a history of SAD (22.4%). We used the maximum likelihood estimator in our analyses, which is robust to this type of missingness. Moreover, we used all available twin data (i.e. data of twins who did not participate in the YAFU and data of twins whose cotwin did not participate).
Clinical information was assessed using the Child and Adolescent Psychiatric Assessment (CAPA), a semistructured, investigator-based psychiatric interview administered by separate interviewers to both twins and at least one of the twins’ biological parents. The CAPA evaluates a total of 15 areas of functional impairment associated with specific symptom clusters. Items are rated on a 3-point scale (i.e. no, partial, or severe impairment). Ratings are then aggregated within and across symptom domains to arrive at an estimate overall level of impairment associated with specific symptom clusters (e.g. depression) and across all psychiatric symptoms.
The YAFU study represents an outcome study of all the twins who participated in the first wave of the VTSABD. These twins were contacted by phone and evaluated as young adults (i.e. age 18 or older) using the Structured Clinical Interview for DSM-III-R (SCID).[21] The mean age at the single YAFU assessment was 21.4 years (±2.0; range 18–27).
STATISTICAL ANALYSES
Within all twin models, SAD, OAD, and AOPA were binary variables (0 = no, 1 = yes). For childhood SAD and OAD, participants were coded a “1” if they ever meet criteria for either respective disorder at the wave one, two, or three assessments. Participants were coded as a “1” for AOPA if they endorsed the panic attack probe in the PD section of the SCID, reported experiencing at least 4 of the 13 DSM-III-R panic attack symptoms, and reported their panic attack as reaching peak intensity within 10 min of first noticing symptoms. Of the twins who participated in the YAFU, approximately 265 of 2,275 (11.65%) individuals met our criteria for a panic attack.
We used bivariate and trivariate Cholesky decomposition to address questions regarding the magnitude of genetic and environmental influences between childhood SAD and AOPA. Two or more traits, such as childhood SAD and AOPA, can be correlated because they share common genes and/or common environmental influences. Twin data on multiple traits allows for the partitioning of the covariation between traits to be disaggregated into its genetic and environmental components. The Cholesky decomposition is a statistical procedure that allows for the disaggregation of covariance into three possible sources including additive genetic (A), common (shared) environmental (C), and unique or nonshared environmental (E) components. We also conducted a trivariate Cholesky, with childhood SAD, childhood OAD, and AOPA serving as outcome variables in one comprehensive model that allowed for the examination of the genetic and environmental influences associated with childhood SAD on AOPA in the context of childhood OAD.
Figure 1 provides an illustration of the relations between the additive genetic component of twin 1 and twin 2 for phenotype 1 (SAD) and phenotype 2 (AOPA). Correlations between twins for additive genetic factors were fixed at 1 for MZ twin pairs, as they share 100% of their genes, and 0.5 for DZ pairs as they share an average of 50% of their genes identical by descent. Common environment correlations between cotwins were fixed at 1 for both MZ and DZ pairs based on the rigorous and frequent testing that has supported the assumption that environments for MZ and DZ twins are comparable. By definition, nonshared environment is uncorrelated in twins. Genetic modeling was performed using the statistical package Mx[22] using maximum likelihood estimation.
Figure 1.

Diagram depicting a bivariate Cholesky decomposition of the additive genetic component. A1 represents the additive genetic component for phenotype 1 and A2 represents the additive genetic component for phenotype 2 measured for twin 1 and twin 2. X1 represents phenotype 1 and Y1 represents pheno-type 2. A1 for Twin 1 and Twin 2 correlate 1 for MZ twins and 0.5 for DZ twins. Path estimate a21 reflects the additive genetic contribution of phenotype 1 on phenotype 2.
Full ACE (genetic, common, and unique environment) models, which tested for quantitative and qualitative sex effects, were first examined, with the Akaike Information Criterion (AIC[23]) used to determine the best fitting model. A lower AIC (i.e. more negative) value represents a better balance between goodness of fit and parsimony. Models with fewer parameters also are preferred if they do not result in a significant deterioration of fit. The best fitting ACE model was then simplified by successively eliminating parameters [i.e. submodels: AE (genetic and unique environment), CE (common and unique environment)] to determine whether these factors contribute to phenotypic variance/covariance, resulting in improved model fit using the AIC statistic.
Inclusion of both male and female same sex twins allows a test of quantitative sex differences, i.e. whether the magnitudes of genetic and environmental effects differ in males and females for a specific trait or disorder. This hypothesis is tested by constraining these effects to be equal in males and females and examining degradation in model fit. In addition, by including opposite sex twins, it is possible test for qualitative sex differences, which test whether different genetic and shared environmental effects are important for males and females. If different genetic influences are important for males and females, then opposite sex twins will be less genetically similar for the trait relative to DZ same-sex twins and their genetic correlation will be less than 0.5. This can be tested by allowing the genetic correlation for opposite sex twin pairs (rG) to be freely estimated in the model, rather than being fixed.[24]
RESULTS
Results of all Cholesky decompositions are presented in Tables 1 (bivariate) and 2 (trivariate). For all models, Model I always included specified pathways from all latent variables (A1, C1, E1 and A2, C2, and E2), allowed path coefficients to differ between the sexes (i.e. quantitative sex effects), and freely estimated the genetic correlation (rG; i.e. qualitative sex effects). Model II was the same as model I except rG was fixed to 1 (i.e. no qualitative sex effects). Model III was the same as Model I except that male and female parameters were equated (i.e. no quantitative sex effects). While Model IV also included all ACE parameters, male and female parameters were constrained to equality, and the rG was fixed at 1.
TABLE 1.
Results of the bivariate Cholesky decomposition for childhood SAD (top) and childhood OAD (bottom) and AOPAs
| Model parameters | Sex effects quan/qual | –2LL | AIC | df | Δ–2LL | ΔAIC | |
|---|---|---|---|---|---|---|---|
| SAD and AOPAs | I. ACE | +/+ | 2,730.77 | –7,221.23 | 4,976 | — | — |
| II. ACE | +/– | 2,730.29 | –7,223.71 | 4,977 | — | — | |
| III. ACE | –/+ | 2,739.68 | –7,226.32 | 4,983 | — | — | |
| IV. ACE | –/– | 2,741.61 | –7,226.39 | 4,984 | — | — | |
| Submodels | |||||||
| V. AE* | –/– | 2,741.67 | –7,232.33 | 4,987 | –0.06 | –5.94 | |
| VI. CE | –/– | 2,746.75 | –7,227.25 | 4,987 | +5.14 | –0.86 | |
| OAD and AOPAs | I. ACE | +/+ | 4,223.81 | –5,836.19 | 5,030 | — | — |
| II. ACE | +/– | 4,221.89 | –5,840.11 | 5,031 | — | — | |
| III. ACE | –/+ | 4,230.73 | –5,843.27 | 5,037 | — | — | |
| IV. ACE | –/– | 4,236.05 | –5,839.96 | 5,038 | — | — | |
| Submodels | |||||||
| V. AE* | –/– | 4,236.42 | –5,845.58 | 5,041 | –0.37 | –5.62 | |
| VI. CE | –/– | 4,241.47 | 5,840.53 | 5,041 | +5.42 | –0.57 |
Note: Qual, qualitative sex effects; Quan, quantitative sex effects; “+” the presence of either qualitative or quantitative sex effects; or “–” their absence from the model. The AE and CE models are nested submodels of the ACE model.
The best fitting model.
TABLE 2.
Results of the trivariate Cholesky decomposition for childhood SAD, childhood OAD, and AOPAs
| Model parameters | Sex effects quan/qual | –2LL | AIC | df | Δ–2LL | ΔAIC |
|---|---|---|---|---|---|---|
| I. ACE | +/+ | 5,399.15 | –10,078.85 | 7,739 | — | — |
| II. ACE | +/– | 5,397.99 | –10,082.01 | 7,740 | — | — |
| III. ACE | –/+ | 5,412.94 | –10,095.06 | 7,754 | — | — |
| IV. ACE | –/– | 5,416.31 | –10,093.69 | 7,755 | — | — |
| Submodels | ||||||
| VII. AE* | –/– | 5,416.63 | –10,099.37 | 7,758 | 0.32 | –5.68 |
| VIII. CE | –/– | 5,423.55 | –10,092.44 | 7,758 | 7.24 | +1.25 |
Note: Qual, qualitative sex effects; Quan, quantitative sex effects; “+” the presence of either qualitative or quantitative sex effects; or “–” their absence from the model. The AE and CE models are nested submodels of the ACE model.
The best fitting model.
CHILDHOOD SAD AND AOPAs
The best fitting and most parsimonious of the four models was Model IV, suggesting neither qualitative nor quantitative sex effects. Next, two submodels (Models V and VI) associated with Model IV were examined. Model V includes the AE parameters only and determines whether dropping the C parameter (i.e. shared environment) results in an improvement or worsening of fit relative to the full ACE model (Model IV). Results indicated that Model V, the AE model, resulted in improved fit based on the AIC (ΔAIC = −5.94), whereas the model that included no genetic effects (CE; Model VI) resulted in no significant change in model fit (ΔAIC = −0.86).
Figure 2 provides the path coefficients for the best-fitting AE model. Path estimates indicate that approximately 35% (0.592) of the genetic variance in AOPA is accounted for by the genetic factors for childhood SAD, with no additional genetic contribution unique to AOPA (0.0012 = 0%) and little influence associated with unique childhood environment (112 = 1.2%). Unique adult environment also was strongly predictive of adult onset panic, accounting for a somewhat larger proportion of variance (64%).
Figure 2.
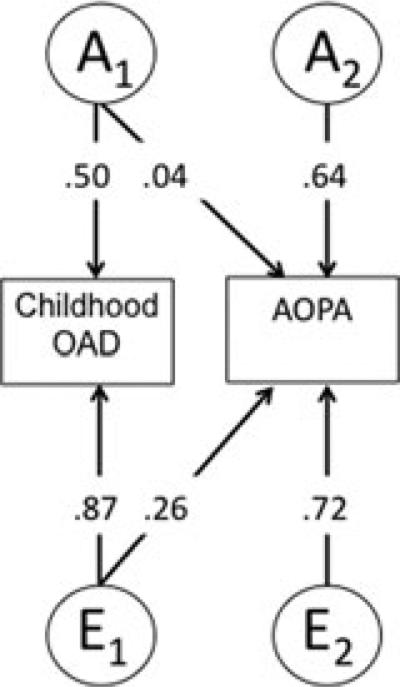
Path estimates from the best-fitting bivariate Cholesky model for childhood SAD and AOPAs. “A,” additive genetic factors; “E,” nonshared environmental factors.
CHILDHOOD OAD AND AOPAs
Examination of AIC values for models examining the relation of childhood OAD and AOPA indicate that dropping qualitative sex effects results in improved fit (i.e. Model II). Dropping quantitative sex effects (Model III) also improves model fit. Dropping both the qualitative and qualitative sex effects, however, does not result in even greater improvement of fit, suggesting that there is likely something different about males and females in the context of childhood OAD and AOPA, but our sample is underpowered to detect this difference. Thus, the most parsimonious model (Model IV), which does not include qualitative or quantitative sex effects was interpreted. Two submodels (Models V and VI) associated with Model IV were examined. Model V again includes the AE parameters only and determines whether dropping the C parameter worsens the fit relative to the full ACE model (Model IV). Results indicated that the AE model resulted in improved fit based (ΔAIC = −5.94) whereas the CE model did not (ΔAIC = − 0.57).
Figure 3 provides the path coefficients for the best-fitting AE model. Unlike SAD, path estimates for childhood OAD indicate that this childhood disorder did not predict genetic variance in AOPA but, rather, there was evidence of genetic factors unique to AOPA emerging, explaining approximately 41% (0.642) of the variance in AOPA. Unique environment also strongly predicted adult panic attacks, accounting for the largest portion of variance (52%).
Figure 3.
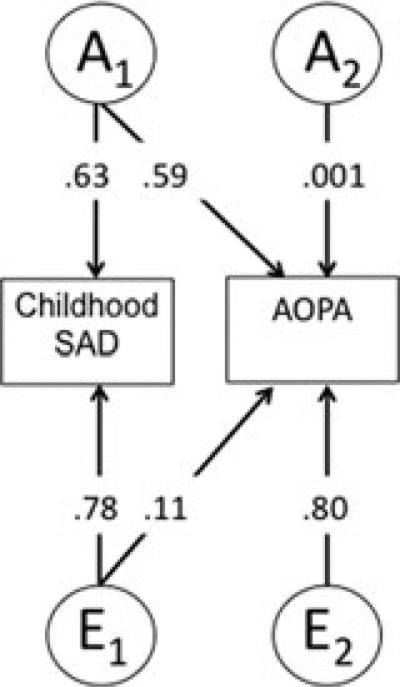
Path estimates from the best-fitting bivariate Cholesky model for childhood OAD and AOPAs. “A,” additive genetic factors; “E,” nonshared environmental factors.
CHILDHOOD SAD, CHILDHOOD OAD, AND AOPAs
For the trivariate Cholesky, which included childhood SAD, OAD, and AOPA, the AIC again indicates that dropping the qualitative sex effect (Model II) and quantitative sex effects (Model III), respectively, improve model fit. Simultaneously dropping the qualitative and qualitative sex effects, however, does not result in even greater improvement of fit. Thus, the most parsimonious model (Model IV), which does not include qualitative or quantitative sex effects, was interpreted. Submodels of Model IV suggested that the AE Model resulted in improved fit (ΔAIC = −5.68) whereas the CE model resulted in a slight worsening of model fit (ΔAIC = +1.25). Figure 4 presents path estimates that largely con firm previous findings of the bivariate models: childhood SAD predicting significant genetic variance in AOPA (36%) with unique environment being the other major contributor, explaining 53% of the variance in AOPA. Variables were ordered in the analysis to map onto their typical developmental/temporal order (i.e. SAD generally precedes OAD). Model results and path estimates did not fluctuate significantly when the order of SAD and OAD was reversed.
Figure 4.

Path estimates for the best-fitting trivariate Cholesky model for childhood SAD, childhood OAD, and AOPAs. “A,” additive genetic factors; “E,” nonshared environmental factors.
DISCUSSION
We found evidence of a shared genetic diathesis between childhood SAD (versus childhood OAD) and AOPA in a large general population twin sample. Specifically, childhood SAD was uniquely, genetically associated with development of AOPA whereas childhood OAD was not. Collectively, our results implicate two primary factors as being critical to the development of panic attacks and, in part PD: genetic factors manifest during childhood that are phenotypically expressed as separation anxiety coupled with unique environmental influences. Thus, our findings suggest that a genetic liability is set “in motion” during childhood, and this liability contributes to development of panic in the presence of environmental events experienced into adulthood.
Although results of the trivariate Cholesky decomposition largely confirmed outcomes of the two separate bivariate models, it also shed light on the genetic and environmental relation between the two childhood disorders. Results suggested moderate (21%) shared environmental risk between childhood SAD and OAD, with no significant, shared genetic pathway. In a related manner, while OAD was not genetically associated with AOPA, this childhood syndrome did share about 10% unique environmental risk with AOPA.
Longitudinal studies provide valuable information regarding the developmental unfolding and fluctuating nature of psychiatric dysfunction. Current data suggests that some disorders exhibit homotypic continuity, onsetting in childhood and continuing into adulthood with the same symptom profile and clinical manifestation.[25] For example, symptoms of SP in adolescence are highly predictive of similar symptoms in adulthood.[26] The prediction of a disorder by a different disorder is referred to as heterotypic continuity.[25] Our results suggest heterotypic continuity between childhood SAD and adult panic, and the mechanism that accounts for this continuity is that of a common genetic vulnerability.
The lack of different “genes” coming online in adulthood for AOPA was rather unexpected. For this reason, a second set of analyses (supplemental Table 1) using a threshold model was undertaken wherein participants were coded as either having (1) no history of panic attack, PD, (2) history of panic attack, or (3) history of DSM-IV PD. We tested whether modeling the pheno-type on a panic continuum would result in instability of model outcomes. Regardless of the model (i.e. bivariate, threshold), childhood SAD continued to contribute significantly to the genetic factor associated with AOPA, accounting for about 36% of the variance, and no different genetic factors emerging into adulthood. Moreover, we collectively examined childhood SAD, childhood OAD, and AOPA in the context of a trivariate Cholesky and again observed similar results to that of the original bivariate model, suggesting relative stability of the model findings and bolstering our confidence in model results.
Little is known about the pathogenesis, course, and nature of SAD. For this reason, theoretical models of SAD are virtually nonexistent. A developmental psychopathology perspective that focuses on the ethologic roots of SAD and its developmental trajectory is clearly needed to guide clinical research questions. Because children with SAD, such as PD patients, exhibit hypersensitivity to cues of impending suffocation during exposure to rising levels of CO2; Klein's suffocation false-alarm theory[27] may have important implications in the understanding of this childhood disorder.
Limitations of the current study include a somewhat low prevalence of AOPAs (for the purpose of twin modeling) coupled with categorical outcomes. Future studies could investigate continuous measures of SAD and PD to estimate genetic and environmental sources of covariation, which would overcome this limitation. Moreover, the prevalence of OAD was higher compared to epidemiological estimates of GAD, with 6% of children and adolescents meeting lifetime criteria for GAD in the National Comorbidity Survey Replication-Adolescent.[28]. In treatment seeking samples, most youth who qualify for a diagnosis of OAD also meet criteria for GAD,[29] but this high rate of agreement is likely due to increased severity levels among juveniles referred for treatment. The agreement rate for OAD and GAD in an epidemiological sample is not known, but is likely lower than that observed in clinical samples. Moreover, the threshold to qualify for a diagnosis of OAD is probably somewhat lower than that of GAD, as the criteria for OAD were less well-defined and did not include some of the hallmark criteria now associated with GAD (e.g. inability to control worry).
Strengths of this study include its prospective, genetically informed design to determine whether childhood SAD and AOPAs/PD share a common genetic and environmental diathesis. Although our results require replication, they provoke interesting thought regarding the time course of SAD and PD and provide a level of insight into the factors that play substantive roles in their covariation. Like most other anxiety disorders, multiple etiological pathways are causal (i.e. equifinality) in the development of PD and we only investigated two possible developmental courses. Thus, additional childhood anxiety disorders (e.g. SP) and their relation to PD onset should be considered as well as the relation between childhood SAD and development of other adult anxiety syndromes. Finally, our study focused on latent genetic and environmental factors. Future studies should include measured genes (e.g., Catechol-O-methyltransferase[30]), environmental risk factors (childhood parental loss[31]), and their interactive effects to verify their role in SAD and PD phenotypes.
REFERENCES
- 1.Klein DF. Delineation of two drug-responsive anxiety syndromes. Psychopharmacology. 1964;3:397–408. doi: 10.1007/BF02193476. [DOI] [PubMed] [Google Scholar]
- 2.Weissman MM, Bland RC, Canino GJ, et al. The cross-national epidemiology of panic disorder. Arch Gen Psychiatry. 1997;54:305–309. doi: 10.1001/archpsyc.1997.01830160021003. [DOI] [PubMed] [Google Scholar]
- 3.Gittelman-Klein R, Klein DF. Controlled imipramine treatment of school phobia. Arch Gen Psychiatry. 1971;25:204–207. [PMC free article] [PubMed] [Google Scholar]
- 4.Aschenbrand SG, Kendall PC, Webb A, Safford SM, Flannery-Schroeder E. Is childhood separation anxiety disorder a predictor of adult panic disorder and agoraphobia? A seven-year longitudinal study. J Am Acad Child Adolesc Psychiatry. 2003;42:1478–1485. doi: 10.1097/00004583-200312000-00015. [DOI] [PubMed] [Google Scholar]
- 5.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 4th ed., revised American Psychiatric Association; Washington, DC: 2000. [Google Scholar]
- 6.Biederman J, Petty CR, Hirshfeld-Becker DR, et al. Developmental trajectories of anxiety disorders in offspring at high risk for panic disorder and major depression. Psychiatry Res. 2007;153:245–252. doi: 10.1016/j.psychres.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weismann MM, Leckman JF, Merikangas KR, Gammon GD, Prusoff BA. Depression and anxiety disorders in parents and children: Results from the Yale family study. Arch Gen Psychiatry. 1984;41:845–852. doi: 10.1001/archpsyc.1984.01790200027004. [DOI] [PubMed] [Google Scholar]
- 8.Capps L, Sigman M, Sena R, Henker B, et al. Fear, anxiety and perceived control in children of agoraphobic parents. J Child Psychol Psychiatr. 1996;37:445–452. doi: 10.1111/j.1469-7610.1996.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 9.Biederman J, Faraone SV, Hirshfeld-Becker DR, Friedman D, Robin JA, Rosenbaum JF. Patterns of psychopathology and dys-function in high-risk children of parents with panic disorder and major depression. Am J Psychiatry. 2001;158:49–57. doi: 10.1176/appi.ajp.158.1.49. [DOI] [PubMed] [Google Scholar]
- 10.Biederman J, Monuteaux MC, Faraone SV, et al. Does referral bias impact findings in high-risk offspring for anxiety disorders? A controlled study of high-risk children of non-referred parents with panic disorder/agoraphobia and major depression. J Aff Disorders. 2004;82:209–216. doi: 10.1016/j.jad.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 11.Last CG, Hersen M, Kazdin A, Orvaschel H, Perrin S. Anxiety disorders in children and their families. Arch Gen Psychiatry. 1991;48:928–934. doi: 10.1001/archpsyc.1991.01810340060008. [DOI] [PubMed] [Google Scholar]
- 12.Otto MW, Pollack MH, Maki KM, et al. Childhood history of anxiety disorders among adults with social phobia: rates, correlates, and comparisons with patients with panic disorder. Depress Anxiety. 2001;14:209–213. doi: 10.1002/da.1068. [DOI] [PubMed] [Google Scholar]
- 13.Pine DS, Klein RG, Roberson-Nay R, et al. Response to 5% carbon dioxide in children and adolescents: relationship to panic disorder in parents and anxiety disorders in subjects. Arch Gen Psychiatry. 2005;62:73–80. doi: 10.1001/archpsyc.62.1.73. [DOI] [PubMed] [Google Scholar]
- 14.Roberson-Nay R, Klein DF, Klein RG, et al. Carbon dioxide hypersensitivity in separation-anxious offspring of parents with panic disorder. Biol Psychiatry. 2010;67:1171–1177. doi: 10.1016/j.biopsych.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eaves LJ, Silberg JL, Maes HH, et al. Genetics and developmental psychopathology: 2. The main effects of genes and environment on behavioral problems in the Virginia Twin Study of Adolescent Behavioral Development. J Child Psychol Psychiatr. 1997;38:965–980. doi: 10.1111/j.1469-7610.1997.tb01614.x. [DOI] [PubMed] [Google Scholar]
- 16.Feigon SA, Waldman ID, Levy F, Hay DA. Genetic and environmental influences on separation anxiety disorder symptoms and their moderation by age and sex. Behav Genet. 2001;31:403–411. doi: 10.1023/a:1012738304233. [DOI] [PubMed] [Google Scholar]
- 17.Hettema JM, Neale MC, Kendler KS. A review of meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry. 2001;158:1568–1578. doi: 10.1176/appi.ajp.158.10.1568. [DOI] [PubMed] [Google Scholar]
- 18.Norton GR, Dorward J, Cox BJ. Factors associated with panic attacks in nonclinical subjects. Behav Ther. 1986;17:239–252. [Google Scholar]
- 19.Salge RA, Beck JG, Logan AC. A community survey of panic. J Anxiety Disord. 1988;2:157–167. [Google Scholar]
- 20.Wittchen HU, Essau CA. Epidemiology of panic disorder: Progress and unresolved issues. J Psychiatric Res. 1993;27:47–68. doi: 10.1016/0022-3956(93)90017-v. [DOI] [PubMed] [Google Scholar]
- 21.Spitzer RL, Williams JB, Gibbon M, First MB. The structured clinical interview for the DSM-III-R(SCID): I. History, rationale, and description. Arch Gen Psychiatry. 1992;49:624–629. doi: 10.1001/archpsyc.1992.01820080032005. [DOI] [PubMed] [Google Scholar]
- 22.Neale MC, Boker SM, Xie G, Maes HH. Mx: Statistical Modeling. 6th ed. Commonwealth University, Medical College of Virginia; Richmond, VA: 2003. [Google Scholar]
- 23.Akaike H. Factor-analysis and AIC. Psychometrica. 1987;52:317–332. [Google Scholar]
- 24.Blokland GAM, McMahon KL, Zhu G, et al. Quantifying the heritability of task-related brain activation and performance during the N-back working memory task: a twin fMRI study. Genet Imaging Neurosci. 2008;79:70–79. doi: 10.1016/j.biopsycho.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costello JE, Mustillo S, Erkanli A, Keeler G, Angold A. Prevalence and development of psychiatric disorders in childhood and adolescence. Arch Gen Psychiatry. 2003;60:837–844. doi: 10.1001/archpsyc.60.8.837. [DOI] [PubMed] [Google Scholar]
- 26.Pine DS, Coplan JD, Papp LA, et al. Ventilatory physiology of children and adolescents with anxiety disorders. Arch Gen Psychiatry. 1998;55:123–129. doi: 10.1001/archpsyc.55.2.123. [DOI] [PubMed] [Google Scholar]
- 27.Klein DF, Mannuzza S, Chapman T, Fyer AJ. Child panic revisited. J Am Acad Child Adolesc Psychiatry. 1992;31:112–114. doi: 10.1097/00004583-199201000-00017. [DOI] [PubMed] [Google Scholar]
- 28.Merikangas KR, He J, Burstein M, et al. Lifetime prevalence of mental disorders in U.S. adolescents: results from the National Comorbidity Survery Replication-Adolescent Supplement (NCS-A). J Am Acad Child Adolsc Psychiatry. 2010;49:980–989. doi: 10.1016/j.jaac.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kendall PC, Warman MJ. Anxiety disorders in youth: diagnostic consistency across DSM-III-R and DSM-IV. J Anxiety Disord. 1996;10:452–463. [Google Scholar]
- 30.Maron E, Hettema JM, Shlik J. Advances in molecular genetics of panic disorder. Mol Psychiatry. 2010;15:681–701. doi: 10.1038/mp.2009.145. [DOI] [PubMed] [Google Scholar]
- 31.Battaglia M, Pesenti-Gritti P, Medland SE, Ogliari A, Tambs K, Spatola CAM. A genetically informed study of the association between childhood separation anxiety, sensitivity to CO2, panic disorder, and the effect of childhood parental loss. Arch Gen Psychiatry. 2009;66:64–71. doi: 10.1001/archgenpsychiatry.2008.513. [DOI] [PubMed] [Google Scholar]