

Abstract
The mechanism of amiloride inhibition of Coxsackievirus B3 (CVB3) and poliovirus type 1 (PV1) RNA replication was investigated using membrane-associated RNA replication complexes. Amiloride was shown to inhibit viral RNA replication and VPgpUpU synthesis. However, the drug had no effect on polymerase elongation activity during either (−) strand or (+) strand synthesis. These findings indicated that amiloride inhibited the initiation of RNA synthesis by inhibiting VPg uridylylation. In addition, in silico binding studies showed that amiloride docks in the VPg binding site on the back of the viral RNA polymerase, 3Dpol. Since VPg binding at this site on PV1 3Dpol was previously shown to be required for VPg uridylylation, our results suggest that amiloride inhibits VPg binding to 3Dpol. In summary, our findings are consistent with a model in which amiloride inhibits VPgpUpU synthesis and viral RNA replication by competing with VPg for binding to 3Dpol.
Keywords: Coxsackievirus B3 (CVB3), poliovirus (PV), amiloride, VPg, VPg uridylylation, RNA replication complex, (−) strand RNA, (+) strand RNA
Introduction
Coxsackievirus B3 (CVB3) and poliovirus type 1 (PV1) are human enteroviruses that belong to the Picornaviridae family of (+) strand RNA viruses. The 5′ end of the single-stranded RNA genome is covalently linked to a virus-encoded protein, VPg, and the 3′ end is polyadenylated. The genomic RNA contains a large open reading frame flanked by 5′ and 3′ nontranslated regions (NTRs). Translation of the viral genome results in the synthesis of a polyprotein, which is processed by viral proteases 2Apro and 3Cpro/3CDpro into the mature viral proteins. The P1 region of the genome encodes the capsid proteins, and the P2 and P3 regions encode the non-structural proteins that are required for RNA replication including 3Dpol, the viral RNA-dependent RNA polymerase.
The 5′ terminal cloverleaf (5′CL), the 3′NTR including the poly(A) tail and the internal cre(2C) hairpin are cis-acting elements that are needed for viral RNA replication (Liu et al., 2009;Steil and Barton, 2009a;Ogram and Flanegan, 2011). The 5′CL is a multifunctional element that is required for translation, (−) and (+) strand synthesis and VPg uridylylation (Barton et al., 2001;Gamarnik and Andino, 1998;Gamarnik and Andino, 2000;Murray et al., 2001;Ogram et al., 2010;Sharma et al., 2005;Spear et al., 2008;Sharma et al., 2009;Teterina et al., 2001;Vogt and Andino, 2010). The conserved cre hairpin structure in the 2C coding region of the RNA genome serves as the primary template for VPg uridylylation by 3Dpol to form VPgpUpU (McKnight and Lemon, 1998;Goodfellow et al., 2000;Paul et al., 2000;Gerber et al., 2001). Results of many studies show that VPgpUpU serves as the primer for 3Dpol to initiate both (−) and (+) strand synthesis (Fogg et al., 2003;Morasco et al., 2003;Murray and Barton, 2003;Takegami et al., 1983a;Takegami et al., 1983b;Toyoda et al., 1987;Steil and Barton, 2009b;Morasco et al., 2003;Murray and Barton, 2003;van Ooij et al., 2006).
The drug, amiloride, was previously shown to inhibit CVB3 replication in infected HeLa cells by inhibiting viral RNA replication without affecting host or viral protein synthesis (Harrison et al., 2008). Amiloride was also shown to increase the mutation frequencies of both CVB3 and PV1 in infected cells. This was shown to be an indirect mutagenic effect that was mediated by an increase in the intracellular concentration of Mg+2 and Mn+2, which affected the fidelity of the viral polymerase (Levi et al., 2010). The effect of amiloride on single-nucleotide (AMP) incorporation was investigated in assays containing purified CVB3 3Dpol and a 10-nucleotide, self-annealing, RNA primer-template (SSU) (Gazina et al., 2011). A small inhibitory effect on nucleotide incorporation (<9%) was observed in these reactions when amiloride was added with ATP in the presence of Mg+2. Effective inhibition was only observed when 3Dpol was preincubated with amiloride for several minutes in the absence of ATP or Mg+2. Taken together, these results indicate that the kinetics of amiloride inhibition of 3Dpol catalytic activity is very slow compared to the rapid rate of ATP incorporation in the presence of Mg+2. Finally, amiloride was shown to inhibit VPgpUpU synthesis in reconstituted reactions containing purified CVB3 3Dpol, VPg and cre hairpin RNA (Gazina et al., 2011). Interestingly, the inhibition of VPgpUpU synthesis by amiloride was not dependent on preincubating 3Dpol with the drug prior to adding UTP to start the reaction.
In the current study, we examined the effect of amiloride on CVB3 and PV1 RNA replication in preinitiation replication complexes (PIRCs) isolated from HeLa cell-free reactions (Barton and Flanegan, 1997;Lyons et al., 2001;Morasco et al., 2003;Sharma et al., 2009). Our findings showed that amiloride had no measurable effect on the elongation activity of the viral polymerase, but specifically inhibited the initiation of RNA replication and VPg uridylylation. Furthermore, in silico binding studies showed that amiloride docks at the VPg binding site previously identified at the back of 3Dpol. Since previous genetic studies indicate that VPg binding to this site is required for efficient VPgpUpU synthesis (Lyle et al., 2002), our results suggest that amiloride inhibits VPg binding to 3Dpol. Based on these findings, we propose a model in which amiloride competes with VPg for binding to the site on the back of 3Dpol, which in turn inhibits VPgpUpU synthesis and viral RNA replication.
Results
Effect of amiloride on viral RNA replication
To investigate the underlying mechanism of amiloride inhibition of CVB3 RNA replication, we used membrane-associated preinitiation replication complexes (PIRCs) isolated from HeLa S10 reactions. In previous studies, we showed that PIRCs support efficient VPgpUpU synthesis and (−) and (+) strand synthesis (Sharma et al., 2009). CVB3 P23 RNA is a subgenomic RNA that contains sequences for the 5′NTR, the P23 coding region, which encodes the replication proteins, the 3′NTR and associated poly(A) tail. P23 RNA contains two non-viral G residues at the 5′ end which supports only (−) strand RNA synthesis. This RNA was added to HeLa S10 reactions, and PIRCs were isolated from the reactions at 3 h. The PIRCs were resuspended in replication assay buffer containing all four NTPs, [α-32P] CTP and Mg+2. Labeled (−) strand synthesis was measured in the presence or absence of amiloride as described in Materials and methods. The amount of labeled (−) strand RNA synthesized in each reaction was determined by electrophoresis in a denaturing agarose gel. In the presence of 0.4, 0.8 and 1.6 mM amiloride, (−) strand synthesis was 49%, 25% and 2%, respectively of the level observed in the absence of the drug (Fig. 1A). The concentration of amiloride required to observe 50% inhibition of (−) strand synthesis (IC50) was calculated and shown to be 0.41 mM in these reactions (Fig. 1B). These results demonstrated that amiloride inhibited (−) strand synthesis in a concentration dependent manner in PIRCs.
Fig. 1.
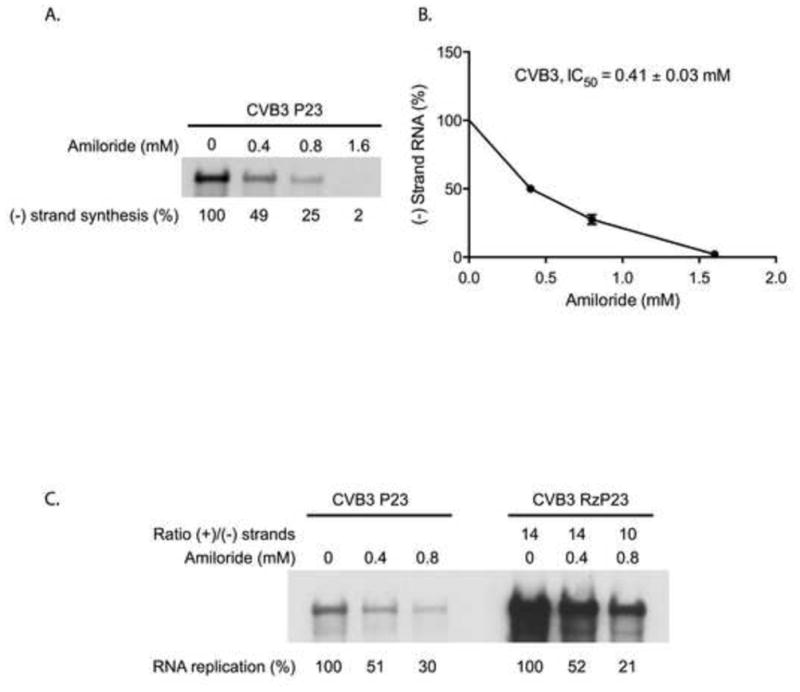
Effect of amiloride on CVB3 RNA replication and the ratio of (+)/(−) strand synthesis. (A) (−) Strand synthesis was measured in PIRCs containing CVB3 P23 RNA, which supports only (−) strand synthesis. PIRCs were resuspended in replication assay buffer containing [α-32P]CTP in the presence of increasing concentrations of amiloride and the reactions were incubated at 37°C for 1 h. The labeled product RNAs were analyzed by electrophoresis in a denaturing CH3HgOH-agarose gel visualized by autoradiography and quantitated using a PhosphorImager. (B) The effect of amiloride on (−) strand synthesis was plotted as a function of amiloride concentration. These results were used to calculate the 50% inhibitory concentration (IC50) of amiloride as described in Materials and methods. (C) The replication of CVB3 P23 RNA or CVB3 RzP23 RNA was measured in reactions containing increasing concentrations of amiloride. (−) Strand synthesis was measured in reactions containing CVB3 P23 RNA, and (−) and (+) strand synthesis was measured in reactions containing CVB3 RzP23 RNA. The labeled product RNAs were analyzed by denaturing agarose gel electrophoresis during which all RNA structure is completely disrupted, including RI and RF RNAs. The ratio of (+)/(−) strand synthesis was calculated as described in Materials and methods.
Since amiloride inhibited CVB3 (−) strand synthesis, a corresponding decrease in (+) strand synthesis should also be observed in the presence of the drug. To determine if amiloride differentially inhibited CVB3 (+) strand synthesis, we measured the effect of amiloride on the ratio of (+)/(−) strand synthesis (Sharma et al., 2009). A decrease in the (+)/(−) strand ratio should be observed if amiloride differentially inhibits (+) strand synthesis. The (+)/(−) strand ratio was calculated by measuring labeled RNA synthesis in reactions containing CVB3 P23 RNA or CVB3 RzP23 RNA as previously described (Sharma et al., 2009). (−) strand RNA is synthesized in reactions containing P23 RNA. In contrast, both (−) strand and (+) strand RNAs are synthesized in reactions containing RzP23 RNA. This RNA contains a 5′ hammerhead ribozyme (Rz) which upon cleavage generates an authentic 5′ terminus which supports both (−) and (+) strand synthesis. The effect of amiloride on labeled RNA synthesis was determined in separate reactions containing either P23 or RzP23 RNA. The results showed that amiloride inhibited (−) strand synthesis (Fig. 1C left) and overall synthesis (both (+) and (−) strand) (Fig. 1B right) by similar amounts. The amount of labeled RNA synthesized in each reaction was then used to calculate the ratio of (+)/(−) strand synthesis as described in Materials and methods. In the absence of the drug, the (+)/(−) strand ratio was 14 consistent with previous studies (Fig. 1C) (Sharma et al., 2009). In reactions containing 0.4 mM and 0.8 mM amiloride, the (+)/(−) strand ratio was 14 and 10, respectively (Fig. 1C). These results indicated that amiloride had no significant effect on the ratio of (+)/(−) strand synthesis and therefore, amiloride did not differentially inhibit CVB3 (+) strand synthesis.
We next determined if amiloride also inhibited PV1 RNA replication in PIRCs. The synthesis of (−) strand RNA was measured in the presence or absence of amiloride as described above. In the presence of 0.4, 0.8 and 1.6 mM amiloride, (−) strand synthesis was 58%, 28% and 2% of the level observed in the absence of the drug (IC50=0.48 mM )(Figs.2A and 2B). These results showed that amiloride inhibited PV1 (−) strand synthesis at levels similar to that observed with CVB3 P23 RNA (Fig. 1A).
Fig. 2.
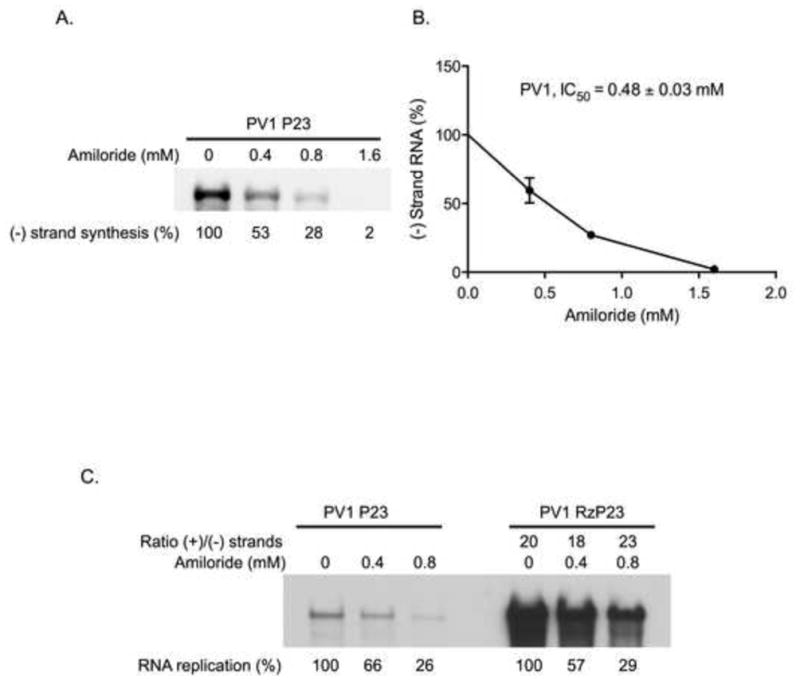
Effect of amiloride on PV1 RNA replication and the ratio of (+)/(−) strand synthesis. (A) The effect of amiloride on PV1 (−) strand synthesis was measured in PIRCs containing PV1 P23 RNA as described in Fig. 1A. (B) The IC50 for amiloride inhibition of PV1 (−) strand synthesis was determined as described in Fig. 1B. (C) The replication of PV1 P23 RNA or PV1 RzP23 RNA was measured in reactions containing increasing concentrations of amiloride. RNA replication and the ratio of (+)/(−) strand RNA synthesis were determined as described in Fig. 1C.
To determine the effect of amiloride on (+) strand synthesis in reactions containing PV1 RNA, we calculated the (+)/(−) strand ratio by measuring labeled RNA synthesis in reactions containing PV1 P23 RNA or PV1 RzP23 RNA. Amiloride inhibition of (−) strand synthesis (Fig. 2B left) and overall synthesis (Fig. 2B right) were similar in these reactions. The ratio of (+)/(−) strand synthesis was 20 in the absence of the drug (Fig. 2B). In reactions containing 0.4 mM or 0.8 mM amiloride, the (+)/(−) strand ratio was 18 and 23, respectively (Fig. 2B). As previously observed with CVB3, amiloride had no significant effect on the ratio of (+)/(−) strand synthesis during PV1 RNA replication and did not differentially inhibit (+) strand synthesis.
Effect of amiloride on (−) strand elongation
We next investigated if amiloride inhibited the elongation of CVB3 (−) strand RNA in replication complexes. To do this, we measured the effect of amiloride on the time required to synthesize full-length (−) strand RNA in PIRCs. In these reactions, removal of guanidine-HCl allows for the synchronous initiation of (−) strand synthesis. If amiloride inhibits the elongation rate of 3Dpol, then the time required to synthesize full-length (−) strand RNA will increase in the presence of the drug. In contrast, if amiloride inhibits the initiation of RNA synthesis, the total amount of RNA synthesized will decrease but the time required to synthesize full-length RNA will be the same. PIRCs were isolated from reactions containing CVB3 P23 RNA and resuspended in replication buffer in the presence or absence of amiloride. Aliquots were removed at the indicated times and labeled product RNA was analyzed by denaturing agarose gel electrophoresis. In the untreated reactions, the growing nascent RNA chains were observed at 5, 6 and 7 min and full-length (−) strand RNA was observed by 8 min (Fig. 3A). In the presence of 0.8 mM amiloride, full-length (−) strand RNA was detected by 8–9 min although the overall intensity of the product RNA was significantly reduced (Fig. 3A). These results indicated that amiloride had no significant effect on the time required to synthesize full-length product RNA even though the overall level of (−) strand synthesis was inhibited by fourfold (Fig. 1A). Taken together, these results indicated that amiloride inhibited CVB3 (−) strand initiation but had little effect on polymerase elongation activity.
Fig. 3.
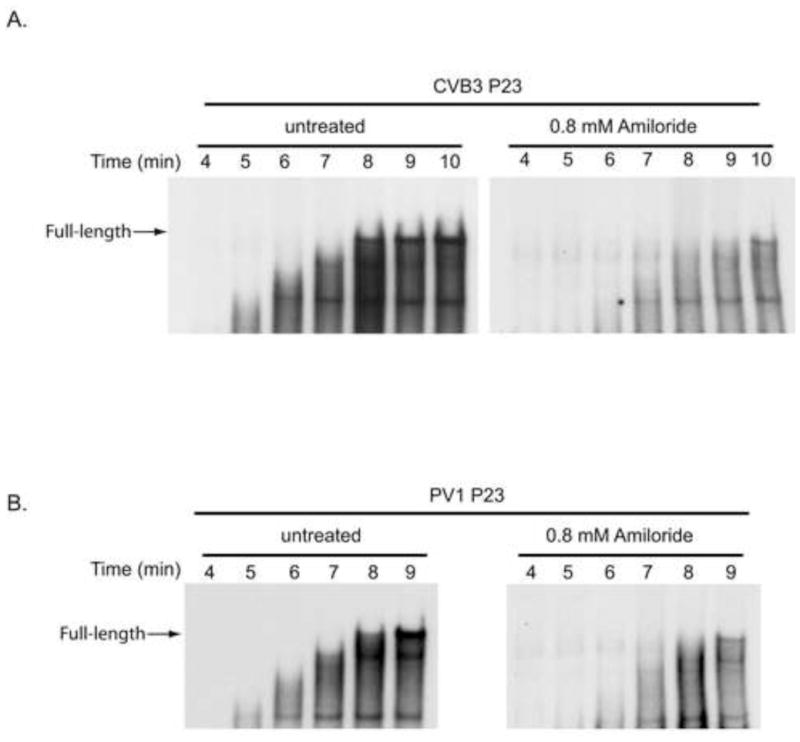
Effect of amiloride on (−) strand elongation in CVB3 and PV1 RNA replication complexes. (A) To measure CVB3 (−) strand elongation, PIRCs containing CVB3 P23 RNA were resuspended in replication assay buffer in the presence of 0.8 mM amiloride. The reactions were incubated at 37°C and aliquots were removed at the indicated time points. The labeled product RNAs were analyzed by denaturing agarose gel electrophoresis as described in Materials and methods. The gel panel on the right which contained labeled RNA synthesized in the presence of amiloride was exposed about 5-times longer time than the gel panel on the left. (B) PV1 (−) strand RNA elongation was measured as described in (A).
To examine if amiloride inhibited RNA elongation during PV1 (−) strand RNA synthesis a similar time-course experiment was performed using PIRCs containing PV1 P23 RNA. The growing nascent RNAs were observed between 5–8 min and full-length (−) strand RNA was observed at 8 min in the untreated reactions (Fig. 3B). In the presence of 0.8 mM amiloride, full-length (−) strand RNA was again detected at 8 min, although the overall level of (−) strand synthesis was significantly reduced by the drug (Fig. 3B). Since amiloride had no significant effect on the time required to synthesize full-length (−) strand RNA, we concluded that amiloride inhibited PV1 (−) strand initiation but had no significant effect on nascent chain elongation.
Effect of amiloride on (+) strand elongation
The effect of amiloride on the elongation of nascent (+) strand RNA in replication complexes was also investigated. To do this, preformed RNA replication complexes were isolated from reactions containing RzP23 RNA. The preformed replication complexes contain replication intermediate RNA, which consists of a (−) strand template and multiple nascent (+) strands (Fig. 4A). RNA replication assays were then performed in the presence or absence of amiloride. In these assays, the preformed nascent (+) strands were allowed to elongate into full-length (+) strand RNA in the presence of [α-32P] CTP (Fig. 4B). The reactions were incubated for a total of 5 min and aliquots were removed at the indicated time points (Fig. 4B). The 5 min incubation period was long enough to allow most of the nascent (+) strands to elongate into full-length RNA but was too short to allow for the re-initiation and synthesis of full-length RNA. Therefore, only elongation of preformed nascent (+) strands into full-length (+) strand RNA was measured in these reactions. The amount of full-length labeled RNA synthesized at each time point was quantitated and expressed as PI units in the bar graph shown below the gel panel. As expected, the amount of full-length RNA increased as a function of incubation time (Fig. 4B). If amiloride inhibited the elongation of nascent chains, the amount of labeled full-length RNA synthesized at each time point would be lower in the presence of the drug. However, there was no significant difference in the amount of labeled RNA synthesized at each time point in the absence or presence of the drug (Fig. 4B). The average incorporation rate of [32P] CMP into (+) strand RNA was 1.9±0.5 and 2.1±0.7 PI units/min, in the absence or presence of amiloride, respectively. These results clearly demonstrated that amiloride did not inhibit the elongation of CVB3 (+) strand RNA in replication complexes.
Fig. 4.
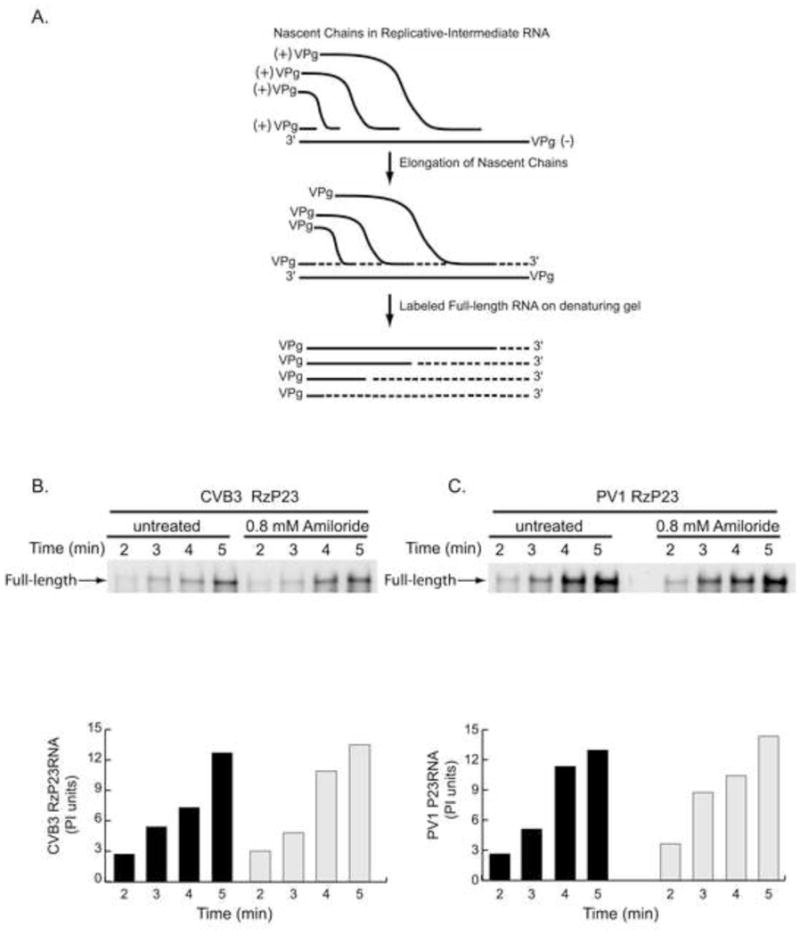
Effect of amiloride on elongation of (+) strand RNA in preformed replication complexes containing CVB3 or PV1 RNA. (A) Schematic representation of the elongation of nascent (+) strand RNA in replicative-intermediate (RI) RNA that is present in preformed replication complexes. (B) To measure (+) strand elongation, preformed replication complexes containing CVB3 RzP23 RNA were resuspended in replication assay buffer in the presence of 0.8 mM amiloride as described in Materials and methods. The reactions were incubated at 37°C and aliquots were removed at the indicated time points. The labeled product RNAs were analyzed by denaturing agarose gel electrophoresis. The amount of labeled product RNA synthesized at each time point was quantitated as PhosphorImager units (PI units) and is shown as a bar graph below the gel panel. (C) PV1 (+) strand RNA elongation was measured as described in (B).
Using the same experimental approach, the effect of amiloride on the elongation of nascent PV1 (+) strand RNA was also investigated. Once again, the amount of labeled full-length RNA increased as a function of incubation time, and there was no significant difference in the amount of labeled RNA synthesized at each time point in the absence or presence of the amiloride (Fig. 4C). The average incorporation rate of [32P] CMP into (+) strand RNA was 2.1±0.7 and 2.3±0.5 PI units/min in the absence or presence of amiloride, respectively. Based on these results, we concluded that amiloride did not inhibit the elongation of PV1 (+) strand RNA. Taken together, the above results confirmed that amiloride did not significantly inhibit the elongation activity of either CVB3 or PV1 3Dpol in replication complexes.
Effect of amiloride on cre-dependent VPgpUpU synthesis
The above results demonstrated that amiloride inhibited RNA replication but did not inhibit the elongation activity of 3Dpol. Therefore, these findings suggested that amiloride inhibited the initiation of (−) and (+) strand synthesis during RNA replication. Since uridylylation of VPg is required for the initiation of (−) and (+) strand synthesis, we investigated the effect of amiloride on cre-dependent VPgpUpU synthesis in PIRCs. To measure cre-dependent VPgpUpU synthesis, PIRCs were isolated from reactions containing CVB3 P23 RNA. The indicated concentrations of amiloride were added to the reactions and VPg uridylylation assays were performed as described in Materials and methods. VPgpUpU synthesis was inhibited by amiloride in a concentration dependent manner with an IC50 of 0.37 mM (Fig. 5A). We also examined if amiloride inhibited cre-dependent VPgpUpU synthesis in reactions containing PV1 P23 RNA. Once again, VPgpUpU synthesis was inhibited by amiloride in a concentration dependent manner with an IC50 of 0.67 mM (Fig. 5B). Taken together these findings demonstrated that amiloride inhibited cre-dependent VPgpUpU synthesis in replication complexes containing CVB3 or PV1 RNA.
Fig. 5.
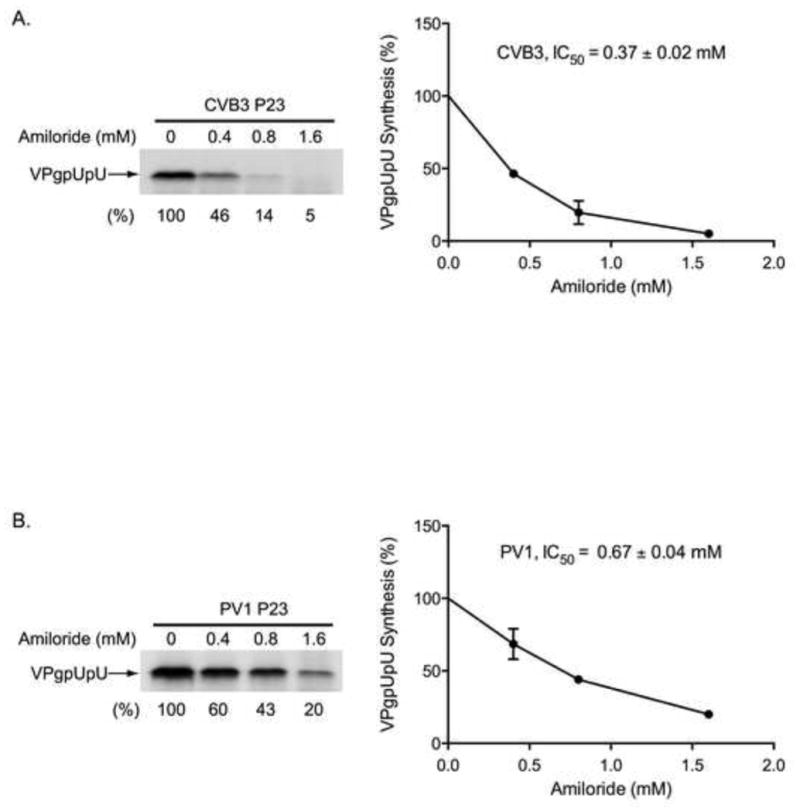
Effect of amiloride on CVB3 and PV1 cre-dependent VPgpUpU synthesis. (A) To measure VPgpUpU synthesis, PIRCs containing CVB3 P23 RNA were resuspended in reaction mixtures containing [α-32P]UTP in the presence of increasing concentrations of amiloride. The reactions were incubated at 37°C for 1 h. The labeled VPgpUpU synthesized was analyzed by SDS-PAGE (9–18% polyacrylamide), detected by autoradiography and quantitated using a PhosphorImager. The effect of amiloride on VPgpUpU synthesis was plotted as a function of amiloride concentration in the graph on the right. These results were used to calculate the IC50 for amiloride inhibition of CVB3 VPgpUpU synthesis (B) The inhibition of PV1 VPgpUpU synthesis by amiloride and the calculation of the IC50 was determined as described in (A).
Molecular Docking analysis of VPg-3Dpol and amiloride-3Dpol interactions
A previous study identified a VPg binding site in the back of the thumb region of 3Dpol in the CVB3 VPg-3Dpol crystal structure (Gruez et al., 2008). In this co-crystal structure, residues 7–15 of VPg were observed to bind along the back of the thumb region of CVB3 3Dpol (PDB: 3CDW) (Gruez et al., 2008). To generate a full-length model of VPg binding to this site in CVB3 and PV1 3Dpol, we performed molecular docking studies based on the published CVB3 VPg-3Dpol co-crystal structure. Using the known sequences for both CVB3 and PV1 VPg, the fragmented crystallographic model was extended bilaterally with subsequent rounds of simulated annealing for energy minimization and potential electrostatic interaction developments in DeepView (Guex and Peitsch, 1997). The models showing the interaction of the full-length VPg with the VPg binding site on the back of CVB3 and PV1 3Dpol are shown in Figs.6A and 7A, respectively.
Fig. 6.
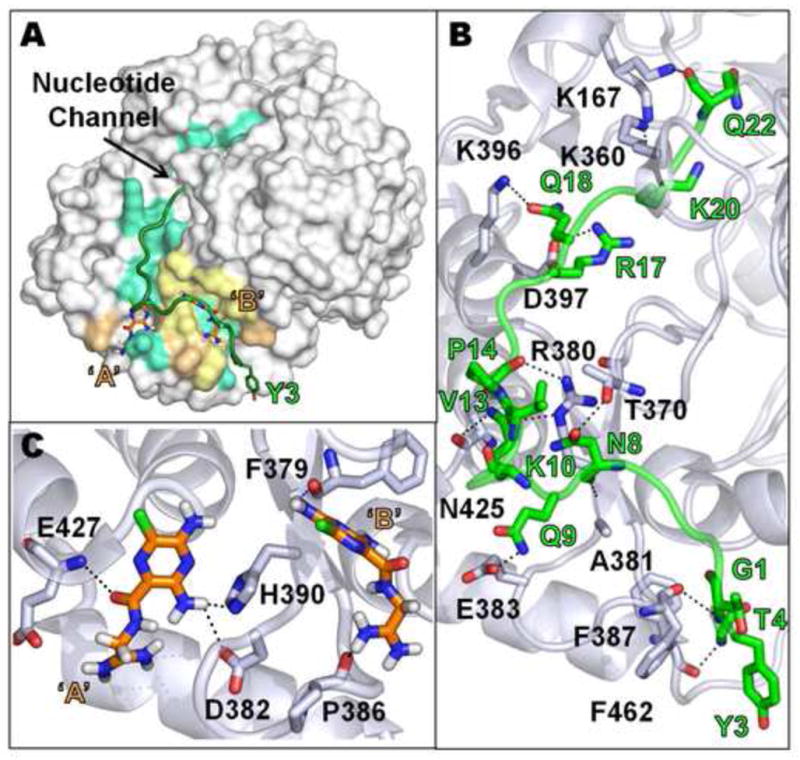
Docking Interactions of CVB3 3Dpol with full-length VPg and amiloride. (A) A surface representation of 3Dpol is shown in light gray. VPg and amiloride are shown in green cartoon view and orange stick view, respectively. The sites in 3Dpol that correspond to the VPg interacting residues are colored cyan and the amiloride interacting sites are colored light orange. The sites that are common for both ligands are colored pale yellow. The two amiloride binding sites in 3Dpol are labeled ‘A’ and ‘B’. The UMP-linkage site, Y3, in the N-terminus of VPg is also shown in stick view. (B) Model showing hydrogen bond interactions (dashed black lines) between VPg (green Cα) and 3Dpol (light gray Cα). The UMP-linkage site, Y3, and amino acid side chains involved in hydrogen bond interactions are also shown in the model. (C) Model showing the hydrogen bond interactions between amiloride (orange) and 3Dpol (light gray Cα). ‘A’ and ‘B’ are the two amiloride binding sites in 3Dpol. Nitrogen atoms are colored in blue, oxygen in red, chlorine in green and hydrogen in white. The specific hydrogen bond interactions shown in panels B and C are listed in Table 1. See Supplementary materials for PDB files of these figures.
Fig. 7.
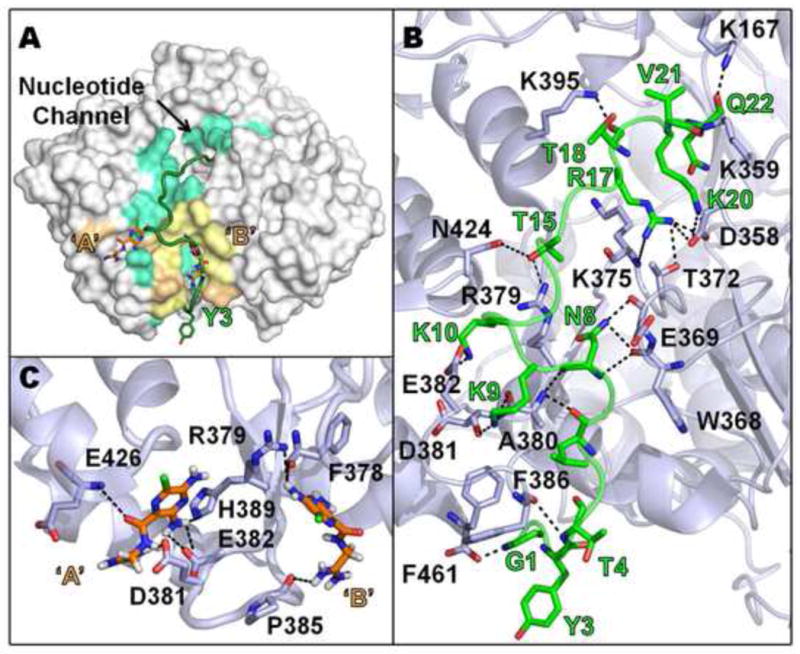
Docking Interactions of PV1 3Dpol with full-length VPg and amiloride. (A) A surface representation of 3Dpol is shown in light gray. VPg and amiloride are shown in green cartoon view and orange stick view, respectively. The sites in 3Dpol that correspond to the VPg interacting residues are colored cyan and the amiloride interacting sites are colored light orange. The sites that are common for both ligands are colored pale yellow. The two amiloride binding sites in 3Dpol are labeled ‘A’ and ‘B’. The UMP-linkage site, Y3, in the N-terminus of VPg is also shown in stick view. (B) Model showing hydrogen bond interactions (dashed black lines) between VPg (green Cα) and 3Dpol (light gray Cα). The UMP-linkage site, Y3, and amino acid side chains involved in hydrogen bond interactions are also shown in the model. (C) Model showing the hydrogen bond interactions between amiloride (orange) and 3Dpol (light gray Cα). ‘A’ and ‘B’ are the two amiloride binding sites in 3Dpol. Nitrogen atoms are colored in blue, oxygen in red, chlorine in green and hydrogen in white. The specific hydrogen bond interactions shown in panels B and C are listed in Table 2. See Supplementary materials for PDB files of these figures.
In both models, we observed that the tyrosine (Y3), which is the UMP-linkage site in the N-terminus of VPg, was distal from the nucleotide entry channel (Figs.6A and 7A). Furthermore, residues R380 and E383 of CVB3 3Dpol and R379 and E382 of PV1 3Dpol were seen to make hydrogen bond connections to several side-and main-chain atoms in VPg (Figs.6B and 7B). The guanidine group of R380 hydrogen bonds to the carbonyl groups of V13 and P14 of VPg; the carboxylate of E383 hydrogen bonds with Q9 of VPg (Fig. 6B). In the full-length model of CVB3 and PV1 VPg, there are other interactions sites between 3Dpol and VPg that could offer further binding stability (Tables 1 and 2). There are some minor local differences in VPg binding when comparing CVB3 to PV1 (Tables 1 and 2) such as the binding motif for K10 and R17 on the two polymerases but the overall interaction domains are similar (compare cyan colors in Figs.6A and 7A).
Table 1.
Comparative Binding Interactions of CVB3 VPg and Amiloride to 3Dpol.
| Ligand | 3Dpol H-Bond | 3Dpol van der Waals | |
|---|---|---|---|
| VPg | Q22 | K167 | K61, R163, S219, P222, Y234, S235, W369, Y378, F379, L388, V389, V392, P394, H424 |
| K20 | K360 | ||
| Q18 | K396 | ||
| R17 | D397 | ||
| V13, P14 | R380 | ||
| K10 | N425 | ||
| Q9 | E383 | ||
| N8 | T370, A381 | ||
| T4 | F387 | ||
| G1 | F462 | ||
| Amiloride | ‘A’ | D382, H390, E427 | A381, F387 |
| ‘B’ | F379, P386 | P222, V223, W369, T370, R380, A381 | |
Bold residues indicate a common binding motif between VPg and amiloride on 3Dpol.
Table 2.
Comparative Binding Interactions of PV1 VPg and Amiloride to 3Dpol.
| Ligand | 3Dpol H-Bond | 3Dpol van der Waals | |
|---|---|---|---|
| VPg | Q22 | K167 | K38, R163, S164, S219, P222, V223, F377, F378, P385, L387, I388, H389, V391, P393, E396, H423 |
| K20 | D358 | ||
| T18 | K395 | ||
| R17 | T372, K375, D358 | ||
| T15 | R379, N424 | ||
| K10 | D381, E382 | ||
| K9 | A380, D381 | ||
| N8 | A380, W368, E369 | ||
| T4 | F386 | ||
| G1 | F461 | ||
| Amiloride | ‘A’ | D381, E382, H389, E426 | A380, F386 |
| ‘B’ | F378, R379, P385 | P222, W368, E369, R379, A380 | |
Bold residues indicate a common binding motif between VPg and amiloride on 3Dpol.
The interaction energy of VPg binding to the back of 3Dpol was calculated via PDBePISA (Table 3) (Krissinel and Henrick, 2007). The higher binding energy observed with PV1 is in accordance with the increased number of hydrogen bonds and van der Waals interactions seen to be occurring between PV1 3Dpol and VPg compared to CVB3 3Dpol and VPg (Tables 1 and 2). For example, it can be seen that PV1 3Dpol presents several more hydrogen bonding partners to PV1 VPg residues N8, K9, K10 and R17 than the comparable residues in CVB3 VPg that interacted with 3Dpol. Based on the calculated binding energies for VPg and 3Dpol, it appears that PV1 VPg binds with a 2.3 fold higher affinity to PV1 3Dpol compared to the binding of CVB3 VPg to CVB3 3Dpol.
Table 3.
Binding energies of full-length VPg and amiloride to CVB3 and PV 3Dpol
| Ligand | CVB3 ΔG (kcal mol−1) | PV1 ΔG (kcal mol−1) | Fold Difference of Binding* |
|---|---|---|---|
| VPg 1 | −7.3 | −7.8 | 2.3 |
| Amiloride ‘A’ 2 | −4.7 | −4.7 | 1.0 |
| Amiloride ‘B’ 2 | −5.6 | −5.5 | 0.8 |
Binding energies calculated from the webserver PDBePISA (Krissinel and Henrick, 2007).
Binding energies calculated from the webserver PEARLS (Lang et al., 2009).
The fold difference of binding was calculated using the equation k=(kT/h)exp(−ΔG/RT) where k is the binding constant (assumed to be equal to the binding energy), k is the Boltzmann constant, h is Planck’s constant, ΔG is the binding energy (J mol−1), R is the gas constant and T is the temperature in Kelvin. The fold difference is the ratio of k for PV 3Dpol to that of CVB3 3Dpol.
Molecular docking studies were also performed to determine if amiloride docked at the VPg binding site on the back of 3Dpol as well as in the catalytic site of 3Dpol. Our results showed that amiloride did not dock in the catalytic site of 3Dpol in the presence of Mg2+, which was in agreement with the molecular docking results of a previous study, (Gazina et al., 2011). This finding was consistent with our results which showed that amiloride did not inhibit the catalytic activity of 3Dpol in RNA elongation assays. Importantly, our docking results revealed two potential binding pockets for amiloride (‘A’ and ‘B’) within the VPg binding site (Figs.6A and 6C; 7A and 7C). These two sites gave the tightest binding energies (i.e., more negative solvation energy) along the entire surface of the VPg binding channel and resulted in the docking of amiloride in these two sites in every docking attempt. Therefore, no other binding sites were identified in the search area. The docking energies were calculated using the webserver PEARLS (Han et al., 2006) and were ~ 5 kcal mol−1 at the two sites (Table 3). The calculated binding affinities for amiloride binding to both CVB3 and PV1 3Dpol were essentially the same. Our docking study showed that amiloride made several H-bond and van der Waals interactions with 3Dpol in sites ‘A’ and ‘B’ (Tables 1 and 2). Interestingly, there are several conserved residues in 3Dpol that interact with both amiloride and VPg (Tables 1 and 2; yellow residues in Figs.6A and 7A). The presence of two potential amiloride binding sites on 3Dpol may increase the ability of amiloride to inhibit VPg binding to 3Dpol. Taken together, these results suggest that amiloride competes with VPg for binding at the same site in the polymerase.
Discussion
In this study, we used membrane-associated replication complexes containing viral RNA to investigate the underlying mechanism of amiloride inhibition of CVB3 RNA replication. These complexes allowed us to independently analyze the effect of the drug on (−) and (+) strand synthesis as well as VPgpUpU synthesis. Furthermore, we were able to separately investigate the effect of amiloride on the initiation and elongation of RNA synthesis. Our results showed that amiloride inhibited overall CVB3 RNA replication without inhibiting the elongation activity of the viral polymerase. These results suggested that amiloride inhibited RNA replication at the initiation step. Consistent with this conclusion was our finding that amiloride inhibited the synthesis of VPgpUpU, the primer used by the viral polymerase to initiate RNA synthesis. In this study, we also investigated the effect of amiloride on the replication of poliovirus (PV1). Amiloride inhibited the overall replication of PV1 RNA but did not inhibit the polymerase elongation activity. In addition, amiloride inhibited VPgpUpU synthesis in reactions containing PV1 RNA. Taken together, these findings are consistent with a model in which amiloride inhibited the initiation of both CVB3 and PV1 RNA replication by inhibiting VPgpUpU synthesis.
We showed that amiloride inhibited (−) strand RNA synthesis in reactions containing CVB3 and PV1 P23 RNA transcripts, which supported only one round of (−) strand synthesis. Furthermore, overall RNA replication, which includes both (−) and (+) strand synthesis was also inhibited by amiloride. However, amiloride did not affect the ratio of (+)/(−) strand RNA synthesis, which indicated that amiloride did not differentially inhibit (+) strand synthesis. Therefore, amiloride inhibited RNA replication for both viruses and there was no significant difference in the observed IC50 values determined in our assays. In a previous study by Harrison et al.,2008, lower IC50 values for amiloride inhibition of CVB3 virus production were observed in cells infected at a low multiplicity of infection. In this case, however, the effect of the drug on virus production was measured during multiple rounds of replication over a period of 48 h. Therefore, the experimental conditions were very different in the two studies which would have affected the IC50 values determined.
We next asked the question at what step does amiloride inhibit RNA synthesis. Is it at the level of RNA elongation or RNA initiation? Given that PIRCs can be used to follow initiation and elongation of (−) strand RNA, we used this approach and showed that the time taken to make full-length (−) strand RNA was about the same in the presence or absence of amiloride. On the other hand, the overall amount of product RNA was reduced in the reactions containing amiloride. Based on these results, we concluded that amiloride did not significantly inhibit the elongation of (−) strand RNA during CVB3 or PV1 RNA replication even though the total amount of RNA synthesized was reduced. We also measured the effect of amiloride on the elongation of nascent (+) strand RNA in normal replication complexes containing either CVB3 or PV1 RNA. If the elongation of nascent (+) strand RNA was inhibited by amiloride then the amount of labeled full-length product RNA synthesized at each time point would be lower in the presence of the drug. Interestingly, the rate of elongation of labeled (+) strand RNA synthesized was equivalent in the presence and absence of the drug. Taken together, our findings demonstrated that amiloride did not significantly inhibit the elongation of either (−) or (+) strand RNA in replication complexes containing viral RNA and replication proteins.
Our finding that amiloride inhibited RNA synthesis but did not inhibit the elongation activity of the viral polymerase raised the possibility that amiloride inhibited RNA replication by inhibiting the initiation step of RNA replication. The first step in enteroviral RNA replication is the synthesis of VPgpUpU, which is the primer used to initiate viral RNA synthesis. In the absence of VPgpUpU, initiation of both (−) and (+) RNA synthesis will be inhibited. Our results showed that amiloride inhibited both CVB3 and PV1 VPgpUpU synthesis in a concentration-dependent manner in the membrane-associated replication complexes. Based on these findings, we concluded that amiloride inhibited the initiation of RNA synthesis by inhibiting the synthesis of VPgpUpU during CVB3 and PV1 RNA replication.
In structural studies with CVB3 3Dpol and VPg, a VPg binding site in the base of the thumb region at the back of 3Dpol was identified in the VPg-3Dpol co-crystal structure (Gruez et al., 2008). Using the co-crystal structure and a molecular docking analysis, we developed models which showed the interaction of full-length VPg with the VPg binding site on the back of CVB3 and PV1 3Dpol (Figs.6A and 7A). In this model, the UMP-linkage site in VPg, Y3, is shown to be remote from the nucleotide entry site and the catalytic site of the carrier 3Dpol (Figs.6A and 7A). This suggests that the VPg bound at this site in 3Dpol cannot be uridylylated by the carrier 3Dpol, which raises the possibility of an intermolecular uridylylation reaction in which two molecules of 3Dpol are required for VPgpUpU synthesis (Tellez et al., 2006;Gruez et al., 2008). In recent studies using EV-71, a similar model for intermolecular uridylylation has been proposed (Chen et al., 2013;Sun et al., 2012). In earlier studies, mutations in the VPg binding site on the back of PV1 3Dpol were shown to inhibit 3AB binding to 3Dpol (Hope et al., 1997;Lyle et al., 2002). These mutations were also shown to inhibit VPgpUpU synthesis in reconstituted assays without inhibiting polymerase elongation activity (Lyle et al., 2002; Boerner et al., 2005). These findings together with the co-crystal structure suggested that VPg binding to this site plays an important role during VPgpUpU synthesis.
Since our results demonstrated that amiloride inhibited VPgpUpU synthesis without inhibiting the catalytic activity of 3Dpol, it raised the possibility that amiloride inhibits VPg binding to the site on the back of 3Dpol and thereby inhibits VPgpUpU synthesis. To address this question, we used a molecular docking analysis to show that two potential binding sites for amiloride are located in the VPg binding site in both CVB3 and PV1 3Dpol. The calculated energies for amiloride binding at the two sites in 3Dpol were similar for both CVB3 and PV1 (Table 3). The docking analysis predicted that several amino acids in 3Dpol interact with both amiloride and VPg. This suggests that the amiloride binding sites overlap with the VPg binding site in 3Dpol (Tables 1 and 2). This finding appears to be analogous to the competitive inhibition of VPg uridylylation by 3AB in reconstituted assays, which was attributed to 3AB and VPg having overlapping binding sites on 3Dpol (Boerner et al., 2005). The presence of two putative amiloride binding sites that overlap with the VPg binding site on 3Dpol would be expected to increase the ability of amiloride to compete with VPg for binding to 3Dpol and inhibit VPg uridylylation. This would also provide an explanation for how amiloride inhibits VPgpUpU synthesis without inhibiting polymerase elongation activity.
In a previous study, an amiloride resistance mutation, A372V, in 3Dpol was identified in CVB3 infected cells (Harrison et al., 2008). This mutation was also shown to increase the fidelity of CVB3 3Dpol. It was proposed that the increase in fidelity may contribute to drug resistance by reducing the indirect mutagenic effect of amiloride (Levi et al., 2010). Interestingly, the A372 residue in CVB3 3Dpol is located close to amino acids, W369 and T370, which interact with VPg and amiloride in our docking models (Fig. 6 and Table 1). Although, a direct interaction of A372 with either amiloride or VPg was not observed in our docking model, it is possible that the A372V mutation alters the structure of the amiloride and/or VPg binding sites and contributes to the mechanism of amiloride resistance.
In summary, our results showed that amiloride inhibited CVB3 and PV1 RNA replication by inhibiting the synthesis of VPgpUpU, the primer used by 3Dpol to initiate viral RNA replication. Using a molecular docking analysis, we found two putative binding sites for amiloride that overlapped with the VPg binding site on the back of the viral polymerase. Based on these findings, we proposed a model in which amiloride inhibits CVB3 and PV1 VPgpUpU synthesis by competing with VPg for binding to 3Dpol. Importantly, these results support the functional importance of the VPg binding site on the back of 3Dpol during VPgpUpU synthesis and provide new insights into the mechanism of VPg uridylylation. Additionally, the results of this study show that amiloride can be used as a prototype to develop new drugs that target the VPg binding site on the polymerase and specifically inhibit VPg uridylylation and viral RNA replication.
Materials and methods
Coxsackievirus (CVB3) cDNA clones
The cDNA clone of Coxsackievirus B3 (CVB3) strain 28 was the parental cDNA clone used to construct the plasmids used in this study (Tracy et al., 2002). (i) Plasmids CVB3 pP23 and pRzP23 used in this study have been previously described (Sharma et al., 2009). Both plasmids pP23 and pRzP23 contain sequences for the 5′NTR, the P23 coding region, which encodes the replication proteins, the 3′NTR and associated poly(A) tail. CVB3 P23 RNA transcribed from the pP23 plasmid contains two non-viral G residues at the 5′ end which supports only (−) strand RNA synthesis. In contrast, CVB3 RzP23 RNA transcribed from the pRzP23 plasmid contains a 5′ hammerhead ribozyme (Rz) which upon cleavage generates an authentic 5′ terminus which supports both (−) and (+) strand synthesis.
Poliovirus (PV1) cDNA clones
A previously described cDNA clone of the Mahoney strain of type I poliovirus, designated pT7-PV1(A)80 (Barton et al., 2001), was used as the parent clone for the constructs used in this study. (i) PV1 pP23, is a previously described construct with a deletion of the P1 capsid coding region (Jurgens and Flanegan, 2003). PV1 P23 RNA transcribed from pP23 plasmid expresses all the replication proteins from the P2 and P3 regions of the viral genome and functions as an RNA replicon. PV1 P23 RNA contains two non-viral G residues at the 5′ end which supports only (−) strand RNA synthesis. (ii) PV1 pRzP23 was generated using PV1 pP23 plasmid by inserting a hammerhead ribozyme (Rz) downstream of the T7 promoter. PV1 RzP23 RNA transcribed from the pRzP23 plasmid contains a 5′ hammerhead ribozyme (Rz) which upon cleavage generates an authentic 5′ terminus which supports both (−) and (+) strand synthesis (Morasco et al., 2003;Sharma et al., 2005;Spear et al., 2008).
RNA transcript preparation
The plasmid DNAs described above were linearized by digestion with the restriction enzyme, Mlu1. In vitro transcription was performed in a 100 μl transcription reaction mixture containing bacteriophage T7 RNA polymerase and 1mM of each nucleoside triphosphate (NTP) (Barton et al., 1996). For PV1 RzP23 transcript RNA preparation, 0.4mM of each NTP was added. The 5X transcription buffer contained 200 mM Tris-HCl (pH7.9), 30 mM MgCl2 and 10 mM spermidine. For CVB3 P23 and RzP23 transcript RNA preparation an additional 4 mM MgCl2 was added. After 2 h incubation at 37°C, 0.5% SDS buffer [10 mM Tris-HCl (pH 7.5), 100 mm NaCl, 1mm EDTA, 0.5% sodium dodecyl sulfate] was added, and the reaction was extracted three times each with phenol-chloroform-isoamylalcohol followed by three extractions with chloroform-isoamylalcohol (Barton et al., 1996;Sharma et al., 2009). Transcript RNA was precipitated in three volumes of ethanol and purified by chromatography on a Sephadex G-50 gel filtration column.
HeLa S10 translation-replication reactions
HeLa S10 extracts and HeLa translation initiation factors were prepared as previously described (Barton et al., 1996). HeLa S10 translation-replication reactions contained 50% HeLa S10 extract, 20% HeLa translation initiation factors, 1X nucleotide reaction mix (1 mM ATP, 0.25 mM GTP, 0.25 mM UTP, 60 mM Potassium Acetate, 15.5 mM HEPES-KOH [pH 7.4], 30 mM creatine phosphate, 0.4 mg/ml creatine kinase). CVB3 and PV1 transcript RNAs (5 μg) were added to 100 μl of HeLa S10 translation-replication reactions containing 3 mM and 2mM guanidine hydrochloride (guanidine-HCl), respectively, and reactions were incubated at 34°C for 3 h. To confirm that equivalent levels of proteins were synthesized in each reaction, a 10 μl aliquot of each reaction was removed, [35S]-Met added and the reactions were incubated at 34°C for 3 h. The amount of labeled viral proteins synthesized in the reactions was measured by TCA precipitation as previously described (Barton et al., 1996).
Analysis of viral RNA synthesis in PIRCs
RNA replication was measured in preinitiation-replication complexes (PIRCs) isolated from HeLa S10 translation-replication reactions containing the indicated transcript RNAs and guanidine-HCl. The reactions were incubated at 34°C for the 3 h. PIRCs were isolated by centrifugation and were resuspended in a replication buffer containing [α-32P]CTP and 100 μg/ml of puromycin was added as previously described (Barton et al., 2001;Sharma et al., 2005;Spear et al., 2008;Sharma et al., 2009). Indicated concentrations of amiloride (purchased from Sigma-Aldrich) were added to the PIRCs and reactions were incubated at 37°C for 1 h. The resulting 32P-labeled product RNA was analyzed by denaturing CH3HgOH-1% agarose gel electrophoresis (Barton et al., 1996). In these denaturing gels, the RNA structure is completely disrupted, including RI and RF RNA, and the labeled product RNA runs as a single band in these gels. Equivalent loading of RNA was confirmed by ethidium bromide (Et-Br) staining of the gel to visualize the 28S and 18S rRNAs. The labeled product RNA synthesized in each reaction was detected by autoradiography and quantitated as PhosphorImager units (PI units) using a PhosphorImager (Molecular Dynamics). The PI units were used to calculate the percentage of amiloride inhibition. The 50% inhibitory concentration (IC50) of amiloride was calculated using the equation Y= 100/(1+10^((log IC50-X)*HillSlope)) (GraphPad Prism). It is important to note that the IC50 values determined for amiloride inhibition of viral replication is dependent on the assay conditions used and will vary depending on the enzyme and substrate concentrations used in the assay.
Analysis of (−) and (+) strand synthesis in PIRCs
P23 transcript RNA which contains two additional G residues at the 5′ terminus and supports only (−) strand synthesis, was used to measure (−) strand synthesis in PIRCs. (+) Strand synthesis was measured in PIRCS as previously described (Sharma et al., 2005;Sharma et al., 2009). To do this, PIRCs were isolated from independent reactions containing either P23 transcript RNA or RzP23 transcript RNA. RzP23 RNA contains an authentic 5′ terminus generated by the addition of a hammerhead ribozyme (Rz) upstream of the first viral nucleotide and supports both (−) and (+) strand synthesis. The total amount of (+) strand RNA synthesized was calculated by subtracting the amount of (−) strand RNA synthesized in the P23 RNA containing reaction from the total RNA synthesized in the RzP23 RNA containing reaction, which includes both (−) and (+) strand RNA. The ratio of (+)/(−) strand synthesis was calculated by dividing the amount of (+) strand RNA synthesized by the amount of (−) strand RNA synthesized (Sharma et al., 2005;Sharma et al., 2009).
Determination of elongation rates during nascent (+) strand RNA synthesis
To isolate preformed replication complexes, CVB3 RzP23 RNA or PV1 RzP23 RNA was added to HeLa S10 translation-replication reactions in the absence of guanidine-HCl. The reactions were incubated at 34°C for the 3 h. During this incubation period, normal replication complexes which contain (−) strand RNA and nascent (+) strand RNA chains are formed. These complexes were isolated at 3 h and incubated in replication assay buffer containing [α-32P]CTP in the absence or presence of 0.8 mM amiloride at 37°C for 5 min. The nascent (+) strands were elongated into full-length RNA during this time period (Fig. 4A). Aliquots were removed at 2, 3, 4 and 5 min after incubation and the resulting full-length 32P-labeled product RNA was analyzed by denaturing CH3HgOH-1% agarose gel electrophoresis (Barton et al., 1996). The amount of full-length product RNA synthesized at each time point was quantitated as PhosphorImager units (PI units) and plotted as a function of time. The elongation rate was determined as the amount of [α-32P]CMP incorporated per minute (PI units/min). The mean incorporation rate was calculated in the presence or absence of the drug.
Analysis of cre-dependent VPgpUpU synthesis in PIRCs
Synthesis of VPgpUpU was measured in reaction mixtures containing PIRCs. The uridylylation reactions were identical to those described above for the RNA replication assays, except the reaction mixtures contained [α-32P]UTP as previously described (Morasco et al., 2003). The indicated concentrations of amiloride were added to the PIRCs and the reactions were incubated at 37°C for 1 h. After incubation, the labeled VPgpUpU synthesized in the reactions were analyzed and quantitated as previously described (Morasco et al., 2003).
In silico modeling of VPg and binding energy calculations
Full-length models for VPg binding to CVB3 and PV1 3Dpol were generated utilizing the currently available X-ray crystallographic coordinates for a fragment of CVB3 VPg (residues 7–15) bound along the base of the thumb region of CVB3 3Dpol (PDB: 3CDW) (Gruez et al., 2008). A least-square fit of the Cα backbone of the X-ray crystallographic structure for an uncomplexed PV1 (PDB: 2ILZ) (Thompson and Peersen, 2004) was performed in Coot (Emsley and Cowtan, 2004) to establish the binding site of VPg in the back of 3Dpol. The coordinates for the CVB3 VPg were then merged into PV1 3Dpol, with any single amino acid differences between the two VPg’s (e.g., Q9K and R12N) were made by manual changes and rotamer selection to correct for any steric clashes. Any solvent, ligand and ion molecules (excluding the active site Mg2+ ions) located in the 3Dpol structures were removed prior to model building. Full-length models of VPg were generated via manual elongation in Coot (Emsley and Cowtan, 2004) utilizing available sequence details for each particular viral variant followed by subsequent rounds of simulated annealing for energy minimization and potential electrostatic interactions in DeepView (Guex and Peitsch, 1997). The interaction energies of VPg binding at this site in both CVB3 and PV1 3Dpol were calculated via PDBePISA (Krissinel and Henrick, 2007).
In silico docking of amiloride on 3Dpol and binding energy calculations
The inhibitor amiloride was docked in the VPg binding site on the back of the polymerase as well as in the active site of CVB3 and PV1 3Dpol using the DOCK 6 suite of programs (Lang et al., 2009) after being prepared for docking in Chimera (Pettersen et al., 2004). The 3Dpol enzyme was prepared for docking by deleting all solvent, ligand and ion molecules, but leaving in place the Mg2+ ions coordinated inside of the polymerase active site. Furthermore, hydrogen atoms along with charges were added to the amino acid residues utilizing AMBER (ff12SB) forcefields (Case et al., 2014), assuming a negative charge for Asp and Glu residues, a positive charge for Lys residues and an overall neutral charge for His residues. Amiloride was prepared for docking by adding hydrogens and was set to contain an overall zero charge (Gasteiger calculation method) using AMBER’s Antechamber module. The amiloride drug was positioned with no predefined bias for binding location or specific orientation along the VPg binding site on the back of 3Dpol as well as in the polymerase active site. The drug was allowed to sample over 5000 conformations around a 10 Å radius until the energy minimized conformation was obtained. The interaction energy of amiloride binding to both CVB3 and PV1 3Dpol was calculated via PEARLS (Han et al., 2006).
Supplementary Material
Highlights.
Both CVB3 and PV1 RNA replication was inhibited by amiloride
Amiloride inhibited initiation by inhibiting VPgpUpU synthesis
Amiloride had no effect on polymerase elongation activity
Amiloride docks in the VPg binding site on the viral polymerase
Model is proposed in which amiloride competes with VPg for binding to polymerase
Acknowledgments
We thank Nidhi Sharma, Allyn Spear and Joan Morasco for many helpful discussions, and Brian O’Donnell for excellent technical assistance. This work was supported by a Public Health Service Grant AI15539 from the National Institutes of Allergy and Infectious Diseases, a grant from the American Heart Association (AHA-11GRNT7990014) and a grant from the UF Research Opportunity Fund (Project #00108432).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Barton DJ, Flanegan JB. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J Virol. 1997;71:8482–8489. doi: 10.1128/jvi.71.11.8482-8489.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, Morasco BJ, Flanegan JB. Assays for poliovirus polymerase, 3Dpol, and authentic RNA replication in HeLa S10 extracts. Methods Enzymol. 1996;275:35–57. doi: 10.1016/s0076-6879(96)75005-x. [DOI] [PubMed] [Google Scholar]
- Barton DJ, O’Donnell BJ, Flanegan JB. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 2001;20:1439–1448. doi: 10.1093/emboj/20.6.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerner JE, Lyle JM, Daijogo S, Semler BL, Schultz SC, Kirkegaard K, Richards OC. Allosteric effects of ligands and mutations on poliovirus RNA-dependent RNA polymerase. J Virol. 2005;79:7803–7811. doi: 10.1128/JVI.79.12.7803-7811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- Chen C, Wang Y, Shan C, Sun Y, Xu P, Zhou H, Yang C, Shi PY, Rao Z, Zhang B, Lou Z. Crystal structure of enterovirus 71 RNA-dependent RNA polymerase complexed with its protein primer VPg: implication for a trans mechanism of VPg uridylylation. The J Virol. 2013;87:5755–5768. doi: 10.1128/JVI.02733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fogg MH, Teterina NL, Ehrenfeld E. Membrane Requirements for Uridylylation of the Poliovirus VPg Protein and Viral RNA Synthesis In Vitro. J Virol. 2003;77:11408–11416. doi: 10.1128/JVI.77.21.11408-11416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12:2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J Virol. 2000;74:2219–26. doi: 10.1128/jvi.74.5.2219-2226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina EV, Smidansky ED, Holien JK, Harrison DN, Cromer BA, Arnold JJ, Parker MW, Cameron CE, Petrou S. Amiloride is a competitive inhibitor of Coxsackievirus B3 RNA polymerase. J Virol. 2011;85:10364–10374. doi: 10.1128/JVI.05022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber K, Wimmer E, Paul AV. Biochemical and Genetic Studies of the Initiation of Human Rhinovirus 2 RNA Replication: Identification of a cis-Replicating Element in the Coding Sequence of 2A(pro) J Virol. 2001;75:10979–10990. doi: 10.1128/JVI.75.22.10979-10990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow I, Chaudhry Y, Richardson A, Meredith J, Almond JW, Barclay W, Evans DJ. Identification of a cis-acting replication element within the poliovirus coding region. J Virol. 2000;74:4590–600. doi: 10.1128/jvi.74.10.4590-4600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B. The crystal structure of Coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J Virol. 2008;82:9577–9590. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Han LY, Lin HH, Li ZR, Zheng CJ, Cao ZW, Xie B, Chen YZ. PEARLS: program for energetic analysis of receptor-ligand system. J Chem Inf Model. 2006;46:445–450. doi: 10.1021/ci0502146. [DOI] [PubMed] [Google Scholar]
- Harrison DN, Gazina EV, Purcell DF, Anderson DA, Petrou S. Amiloride derivatives inhibit coxsackievirus B3 RNA replication. J Virol. 2008;82:1465–1473. doi: 10.1128/JVI.01374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope DA, Diamond SE, Kirkegaard K. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J Virol. 1997;71:9490–9498. doi: 10.1128/jvi.71.12.9490-9498.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens C, Flanegan JB. Initiation of poliovirus negative-strand RNA synthesis requires precursor forms of p2 proteins. J Virol. 2003;77:1075–1083. doi: 10.1128/JVI.77.2.1075-1083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi LI, Gnadig NF, Beaucourt S, McPherson MJ, Baron B, Arnold JJ, Vignuzzi M. Fidelity variants of RNA dependent RNA polymerases uncover an indirect, mutagenic activity of amiloride compounds. PLoS Pathog. 2010;6:e1001163. doi: 10.1371/journal.ppat.1001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wimmer E, Paul AV. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim Biophys Acta. 2009;1789:495–517. doi: 10.1016/j.bbagrm.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle JM, Clewell A, Richmond K, Richards OC, Hope DA, Schultz SC, Kirkegaard K. Similar structural basis for membrane localization and protein priming by an RNA-dependent RNA polymerase. J Biol Chem. 2002;277:16324–16331. doi: 10.1074/jbc.M112429200. [DOI] [PubMed] [Google Scholar]
- Lyons T, Murray KE, Roberts AW, Barton DJ. Poliovirus 5′-Terminal Cloverleaf RNA is required in cis for VPg uridylylation and the initiation of negative-strand RNA synthesis. J Virol. 2001;75:10696–10708. doi: 10.1128/JVI.75.22.10696-10708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight KL, Lemon SM. The rhinovirus type 14 genome contains an internally located RNA structure that is required for viral replication. RNA. 1998;4:1569–1584. doi: 10.1017/s1355838298981006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morasco BJ, Sharma N, Parilla J, Flanegan JB. Poliovirus cre(2C)-dependent synthesis of VPgpUpU is required for positive-but not negative-strand RNA synthesis. J Virol. 2003;77:5136–5144. doi: 10.1128/JVI.77.9.5136-5144.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KE, Barton DJ. Poliovirus CRE-dependent VPg uridylylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. J Virol. 2003;77:4739–4750. doi: 10.1128/JVI.77.8.4739-4750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KE, Roberts AW, Barton DJ. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA. 2001;7:1126–1141. doi: 10.1017/s1355838201010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogram SA, Flanegan JB. Non-template functions of viral RNA in picornavirus replication. Curr Opin Virol. 2011;1:339–346. doi: 10.1016/j.coviro.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogram SA, Spear A, Sharma N, Flanegan JB. The 5′CL-PCBP RNP complex, 3′ poly(A) tail and 2A(pro) are required for optimal translation of poliovirus RNA. Virology. 2010;397:14–22. doi: 10.1016/j.virol.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Sharma N, O’Donnell BJ, Flanegan JB. 3′-Terminal sequence in poliovirus negative-strand templates is the primary cis-acting element required for VPgpUpU-primed positive-strand initiation. J Virol. 2005;79:3565–3577. doi: 10.1128/JVI.79.6.3565-3577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Ogram SA, Morasco BJ, Spear A, Chapman NM, Flanegan JB. Functional role of the 5′ terminal cloverleaf in Coxsackievirus RNA replication. Virology. 2009;393:238–249. doi: 10.1016/j.virol.2009.07.039. [DOI] [PubMed] [Google Scholar]
- Spear A, Sharma N, Flanegan JB. Protein-RNA tethering: the role of poly(C) binding protein 2 in poliovirus RNA replication. Virology. 2008;374:280–291. doi: 10.1016/j.virol.2007.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steil BP, Barton DJ. Cis-active RNA elements (CREs) and picornavirus RNA replication. Virus Research. 2009a;139:240–252. doi: 10.1016/j.virusres.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steil BP, Barton DJ. Conversion of VPg into VPgpUpUOH before and during poliovirus negative-strand RNA synthesis. J Virol. 2009b;83:12660–12670. doi: 10.1128/JVI.01676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wang Y, Shan C, Chen C, Xu P, Song M, Zhou H, Yang C, Xu W, Shi PY, Zhang B, Lou Z. Enterovirus 71 VPg uridylylation uses a two-molecular mechanism of 3D polymerase. J Virol. 2012;86:13662–13671. doi: 10.1128/JVI.01712-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takegami T, Kuhn RJ, Anderson CW, Wimmer E. Membrane-dependent uridylylation of the genome-linked protein VPg of poliovirus. Proc Natl Acad Sci USA. 1983a;80:7447–7451. doi: 10.1073/pnas.80.24.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takegami T, Semler BL, Anderson CW, Wimmer E. Membrane fractions active in poliovirus RNA replication contain VPg precursor polypeptides. Virology. 1983b;128:33–47. doi: 10.1016/0042-6822(83)90316-1. [DOI] [PubMed] [Google Scholar]
- Tellez AB, Crowder S, Spagnolo JF, Thompson AA, Peersen OB, Brutlag DL, Kirkegaard K. Nucleotide channel of RNA-dependent RNA polymerase used for intermolecular uridylylation of protein primer. J Mol Biol. 2006;357:665–675. doi: 10.1016/j.jmb.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Teterina NL, Egger D, Bienz K, Brown DM, Semler BL, Ehrenfeld E. Requirements for assembly of poliovirus replication complexes and negative-strand RNA synthesis. J Virol. 2001;75:3841–50. doi: 10.1128/JVI.75.8.3841-3850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Peersen OB. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 2004;23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Yang CF, Takeda N, Nomoto A, Wimmer E. Analysis of RNA synthesis of type 1 poliovirus by using an in vitro molecular genetic approach. J Virol. 1987;61:2816–2822. doi: 10.1128/jvi.61.9.2816-2822.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol. 2002;76:12097–12111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooij MJ, Vogt DA, Paul A, Castro C, Kuijpers J, van Kuppeveld FJ, Cameron CE, Wimmer E, Andino R, Melchers WJ. Structural and functional characterization of the coxsackievirus B3 CRE(2C): role of CRE(2C) in negative-and positive-strand RNA synthesis. J Gen Virol. 2006;87:103–113. doi: 10.1099/vir.0.81297-0. [DOI] [PubMed] [Google Scholar]
- Vogt DA, Andino R. An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010;6:e1000936. doi: 10.1371/journal.ppat.1000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.