

ABSTRACT
Exponentially growing cultures of Bacillus subtilis (PY79) are composed primarily of nonmotile, chained cells. The alternative sigma factor, SigD, promotes the phenotypic switch from nonmotile, chained cells to unchained, motile cells. In the present work, we investigated the role of the GTP-sensing protein CodY in the regulation of SigD. Deletion of codY resulted in a significant increase in SigD accumulation and activity and shifted the proportion of unchained cells up from ∼15% to ∼75%, suggesting that CodY is an important regulator of SigD. CodY was previously shown to bind to the PD3 and Pfla/che promoters located upstream of the first gene in the sigD-containing fla/che operon. Using electrophoretic mobility shift assays, we found that CodY also binds to two other previously uncharacterized sites within the fla/che operon. Mutations in any one of the three binding sites resulted in SigD levels similar to those seen with the ΔcodY mutant, suggesting that each site is sufficient to tip cells toward a maximal level of CodY-dependent SigD accumulation. However, mutations in all three sites were required to phenocopy the ΔcodY mutant's reduced level of cell chaining, consistent with the idea that CodY binding in the fla/che operon is also important for posttranslational control of SigD activity.
IMPORTANCE One way that bacteria adapt quickly and efficiently to changes in environmental quality is to employ global transcriptional regulators capable of responding allosterically to key cellular metabolites. In this study, we found that the conserved GTP-sensing protein CodY directly regulates cell motility and chaining in B. subtilis by controlling expression and activity of SigD. Our results suggest that B. subtilis becomes poised for cell dispersal as intracellular GTP levels are depleted.
INTRODUCTION
Genetically identical populations of bacteria often exhibit phenotypic heterogeneity, possessing cells that switch between distinct physiological and/or developmental states. Phenotypic switching is most widely understood in the context of highly regulated responses to changes in environmental cues, such as nutrient status and cell density. However, phenotypic switching can also occur spontaneously, and stochastic switching between two stable phenotypes at the subpopulation level is termed phenotypic bistability (1, 2). Bistability likely confers a selective advantage to a species under unpredictable environmental conditions by increasing the chance that at least one alternative cell state will survive under any given adverse condition (2–4). For example, in a given population of antibiotic-sensitive bacteria, small subpopulations of genetically identical and yet nongrowing and antibiotic-tolerant bacteria known as “persisters” are detectable (5, 6). Since the persister subpopulation is nongrowing, it is able to survive a sudden onslaught of antibiotic selection.
Exponentially growing populations of Bacillus subtilis are comprised of both chained, nonmotile cells and unchained, motile cells, the latter of which are presumably poised for dispersal (7). The two cell types differ from each other at the level of gene expression. More specifically, motile cells have higher levels of active SigD, an alternative sigma factor that promotes expression of the genes responsible for flagellar assembly and cell separation (7, 8). During exponential growth, the switch between the SigD “on” and SigD “off” states is stochastic and exhibits bistability (9, 10). SigD levels also increase as cells exit exponential growth, reaching a maximum during the transition phase, suggesting that SigD expression is tightly linked to nutrient status and/or cell density cues (11).
In a previous study, we found that the proportion of chained and nonmotile versus unchained and motile cells in exponentially growing B. subtilis cultures can be manipulated by introducing genetic backgrounds with altered GTP levels (12). More specifically, we found that a ΔrelA mutant harboring reduced levels of intracellular GTP (13, 14) is trapped in a SigD on state, with 100% of the population exhibiting the unchained, motile phenotype. Mutants with GTP levels intermediate to the wild-type and the ΔrelA mutant levels exhibited intermediate chaining and motility phenotypes (12). These results suggested that GTP might be an important intracellular signal controlling SigD regulation. In this study, we investigated the idea that the GTP-sensing protein, CodY (15), previously shown to bind to promoters that regulate flagellin expression (Phag) as well as promoters upstream of the fla/che operon (PD3 and Pfla/che) (16), might be responsible for mediating the shift between nonmotile and motile cell states that we observed in the ΔrelA mutant background.
In the present work, we found that deletion of codY increases the proportion of unchained, motile cells in an exponentially growing population from ∼15% to ∼75%. We also found that SigD levels in the ΔcodY mutant are comparable to those produced by the ΔrelA mutant, suggesting that CodY represses sigD expression. Using electrophoretic mobility shift assays (EMSAs), we found that CodY binds to three sites located in the sigD-containing fla/che operon and that point mutations in any one of the three binding sites result in production of SigD protein at levels comparable to those seen with the ΔrelA and ΔcodY mutants. These results suggest that the fla/che operon may be finely tuned to detect even small changes in GTP status. Lastly, we found that mutations in all three CodY binding sites were required to phenocopy the primarily unchained phenotype of the ΔcodY mutant, suggesting that CodY binding in the fla/che operon may also be important for the posttranslational regulation of SigD activity.
MATERIALS AND METHODS
General methods.
The B. subtilis strains, plasmids, and oligonucleotides used in this study are listed in Tables S1, S2, and S3 in the supplemental material. All strains were grown in 250-ml baffled flasks in a shaking water bath (280 rpm) at 37°C in casein hydrolysate (CH) medium (42) or LB-Lennox broth (10 g/liter tryptone, 5 g/liter yeast extract, 5 g/liter NaCl), as indicated. To make solid-medium plates, CH and LB-Lennox media were supplemented with 1.5% (wt/vol) Bacto agar. All Escherichia coli strains were grown in LB-Lennox medium. E. coli DH5α was used for isolation of plasmid DNA and cloning. Plasmids used to generate point mutations were isolated from E. coli TG-1. E. coli BL21(λDE3) was used for protein overexpression. For B. subtilis natural transformation, cells were grown in MC medium (10.7 g K2HPO4, 5.2 g KH2PO4, 20 g dextrose, 0.88 g sodium citrate dehydrate, 2.2 g l-glutamic acid monopotassium salt, 1 ml of [2.2%] ferric ammonium citrate, and 1 g casein hydrolysate per 1,000 ml). When selection was required, the following concentrations of antibiotics were used for Bacillus strains: 100 μg/ml spectinomycin, 7.5 μg/ml chloramphenicol, 10 μg/ml tetracycline, and 1 μg/ml erythromycin plus 25 μg/ml lincomycin (to select for macrolide-lincosamide-streptogramin B [MLS] resistance). E. coli harboring the plasmids used in this study was grown in the presence of 100 μg/ml ampicillin.
Strains and plasmid construction.
PY79 genomic DNA was used as a template to amplify PCR products for cloning. All marked deletion strains were confirmed by PCR. To mutate the CodY binding sites, the region upstream of each binding site was PCR amplified using the following primer pairs: for the PD3 site, oQA150/oQA151; for fliE, oQA154/oQA155; and for fliK, oQA158/oQA159. Similarly, the region downstream of each binding site was PCR amplified using the following primer pairs: for the PD3 site, oQA152/oQA153; for fliE, oQA156/oQA157; and for fliK, oQA160/oQA161. The two PCR products for each binding site were used as the template for overlap extension PCR using the following primer pairs: for the PD3 site, oQA150/oQA153; for fliE, oQA154/oQA157; and for fliK, oQA158/oQA161. The amplified fragments were gel purified, digested with EcoRI and KpnI, and cloned into pMiniMAD cut with the same enzymes. The resulting plasmids, pQA023 (Dmu), pQA024 (Emu), and pQA025 (Kmu), were introduced into the PY79 background by single-crossover integration, propagated in the absence selection, and plated on LB agar. Colonies were patched to identify MLS-sensitive colonies, and the corresponding region was PCR amplified and sequenced to identify strains harboring the appropriate mutations.
Plasmids pJW053, pJW054, pJW055, pJW058, pJW063, pJW064, pQA014, pQA015, and pQA20 were constructed as described previously (12). Plasmid pQA021 was constructed by two-way ligation with a PCR product containing the DNA sequence upstream of the PD3 promoter in the fla/che operon (primer pair oQA100/oQA106). The PCR product was digested with EcoRI and BamHI and cloned into pDG1661 digested with the same enzymes. pDG1661 [amyE::lacZ (cat)] is an ectopic integration vector. Plasmid pQA022 was constructed by two-way ligation with a PCR product containing a DNA sequence upstream of the PA promoter of the fla/che operon (primer pair oQA103/oQA107). The PCR product was digested with EcoRI and BamHI and cloned into pDG1661 cut with the same enzymes. Plasmid pQA026 was generated by amplifying the codY gene with primer pair oQA130/oQA131. The PCR product was digested with NheI and XhoI and cloned into pET-24b(+) digested with the same enzymes. Plasmid pQA027 was generated by overlap extension PCR. The promoter region of the cod operon was PCR amplified using primer pair oQA166/oQA167, and the coding region of codY was PCR amplified using primer pair oQA168/oQA169. The two PCR products were used as the template for overlap extension PCR with primer pair oQA166/oQA169. The amplified fragment was cut with EcoRI and BamHI and ligated to pDR111 cut with the same enzymes.
Swim plate assay.
Cultures (25 ml) of B. subtilis strains were grown at 37°C in CH medium until the optical density (OD600) reached approximately 0.5. Cultures were centrifuged, and cells were concentrated to an OD600 of 10 in phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 [pH 7.4]) containing 0.5% (wt/vol) India ink (Higgins). The cell suspensions (10 μl) were spotted on top of 150-by-15-mm petri plates filled with ∼100 ml of LB fortified with 0.25% Bacto agar, dried for 30 min at room temperature to allow time for absorption, and incubated at 37°C inside a covered glass dish containing water-soaked paper towels to generate humid conditions. Plates were scanned after 5 h of incubation.
SDS-PAGE and Western blotting.
B. subtilis strains were grown to the mid-logarithmic phase, the optical density was measured (for equivalent loading levels) at 600 nm, and 1-ml samples were harvested at an OD600 near 0.5. Cells were pelleted by centrifugation and resuspended in 50 μl lysis buffer (20 mM Tris [pH 7.0], 10 mM EDTA, 10 mg/ml lysozyme, 10 μg/ml DNase I, and 100 μg/ml RNase A, with 1 mM phenylmethylsulfonyl fluoride [PMSF]) and incubated for 15 min at 37°C. A 50-μl volume of sample buffer (0.25 M Tris [pH 6.8], 4% SDS, 20% glycerol, 10 mM EDTA) containing 10% 2-mercaptoethanol was added to an equal volume of lysate and the sample, and the sample was boiled for 5 min prior to loading. Sample loads were normalized to OD600 values. Proteins were separated by SDS-PAGE on 4% to 20% mini-Protean TGX polyacrylamide gels (Bio-Rad), transferred onto a nitrocellulose membrane (Pall), and blocked in 5% (wt/vol) nonfat milk–PBS–0.5% (vol/vol) Tween 20. Membranes were probed with an anti-SigD antibody at a 1:5,000 dilution or anti-SigA antibody at a 1:20,000 dilution (Fujita Masaya, University of Houston, Houston, TX), followed by a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary antibody (Bio-Rad). Washed membranes were incubated with SuperSignal West Femto chemiluminescent substrate (Thermo) according to the manufacturer's instructions.
Purification of 6His-CodY.
E. coli BL21(λDE3) was transformed with pQA26. To overexpress 6His-CodY protein, cultures were grown in 25 ml of Cinnabar high-yield protein expression medium (Teknova) supplemented with 25 μg/ml kanamycin and 0.1% (wt/vol) glucose in a shaking water bath at 300 rpm at 37°C. When the cell density of the cultures reached an OD600 of 2.5, protein expression was induced by the addition of 1 mM (final concentration) IPTG (isopropyl-β-d-thiogalactopyranoside). Cultures were grown to an OD600 of 7 and harvested by centrifugation at 15,000 × g. The cell pellets were stored at −80°C until needed. For lysis, pellets were resuspended in 25 ml of lysis buffer (50 mM Tris-HCl [pH 8.5], 300 mM NaCl, 50 μl of 1 mg/ml DNase I, and 50 μl of protease inhibitor cocktail [Sigma]) and passed three times through a French pressure cell at 10,000 lb/in2. The cells lysate was centrifuged at 15,000 × g for 30 min at 4°C to remove cell debris. The supernatant was collected and loaded onto a 1-ml bed volume of preequilibrated nickel-nitrilotriacetic acid (Ni-NTA) (Qiagen) and washed with 5 ml wash buffer (50 mM Tris-HCl [pH 8.5], 300 mM NaCl, 20 mM imidazole, and 10% glycerol). Protein was eluted with 2 ml elution buffer (50 mM Tris-HCl [pH 8.5], 300 mM NaCl, 250 mM imidazole, and 10% glycerol) and collected in 250-μl fractions. All fractions were subjected to SDS-PAGE to assess purity, and peak fractions were pooled and dialyzed against binding buffer (see below). Aliquots were frozen at −80°C prior to use.
Electrophoretic mobility shift assays.
The PCR products containing the CodY binding sites upstream of promoter PD3 and the coding sequences of fliE and fliK were amplified using primer pairs oQA136/oQA137, oQA140/oQA143, and oQA146/oQA149, respectively. PCR products were cleaned using a DNA concentrator kit (Zymogen).
The DNA binding reaction mixtures contained 5 nM DNA, 25 mM 2-(N-morpholino) ethanesulfonic acid [pH 6.0], 200 mM sodium acetate, 1 mM dithiothreitol (DTT), 1 mM Tris (2-carboxyethyl) phosphine, 1 mM EDTA, 0.05% n-octylglucoside, 5% glycerol, 100 μg/ml bovine serum albumin, 2 mM GTP, and 10 mM (each) valine, leucine, and isoleucine, as described previously (17). The reaction mixtures contained various amounts of CodY and were incubated for 15 to 20 min at room temperature and separated on 5% nondenaturing polyacrylamide gels containing 50 mM bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane [pH 6.0] and 10 mM isoleucine. The running buffer was composed of 25 mM 2-(N-morpholino) ethanesulfonic acid plus 25 mM bis(2-hydroxyethyl)amino-tris(hydroxymethyl) methane running buffer [pH 6.0]. The gels were run at 175 V for 3 h and then stained with SYBR green electrophoretic mobility shift assay (EMSA) gel stain (Life Technologies) for 10 min. The gels were washed with distilled H2O, and DNA was visualized with a Typhoon Trio fluorescence imager (GE Healthcare) following excitation at 488 nm with a 520-nm band pass emission filter.
Microscopy.
Samples (1 ml) were collected at an OD600 of 0.5, and cells were pelleted by centrifugation at room temperature and 6,010 × g in a tabletop centrifuge. To stain membranes, cell pellets were resuspended in PBS containing 0.02 TMA-DPH [1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate] and mounted on glass slides with polylysine-treated coverslips. Fluorescence microscopic analysis was performed with a Nikon Ti-E microscope equipped with a CFI Plan Apo lambda DM 100× objective, a Prior Scientific Lumen 200 illumination system, a UV-2E/C DAPI (4′,6-diamidino-2-phenylindole) filter cube, and a CoolSNAP HQ2 monochrome camera. All images were captured with NIS Elements Advanced Research software (version 4.10) and processed with ImageJ64 software (18).
β-Galactosidase assays.
To assay expression from lacZ transcriptional fusions, B. subtilis strains were grown at 37°C in baffled flasks containing CH medium. Samples (1 or 2 ml) were harvested at an OD600 of 0.5 by centrifugation at room temperature and 21,130 × g in a tabletop centrifuge, and cell pellets were frozen at −80°C. The pellets were thawed on ice and resuspended in 500 μl of Z-buffer (40 mM NaH2PO4, 60 mM Na2HPO4, 1 mM MgSO4, 10 mM KCl, 38 mM β-mercaptoethanol, 0.2 mg ml−1 lysozyme) and incubated for 15 min at 30°C. To start the reaction, 100 μl of 4 mg ml−1 O-nitrophenyl β-d-galactopyranoside (in Z-buffer) was added to the resuspended pellet. The reaction was stopped with the addition of 250 μl of 1 M Na2CO3 after a yellow color developed. The reaction mixtures were centrifuged at 21,130 × g in a tabletop centrifuge at room temperature for 1 min, and the supernatants were transferred to a cuvette. The optical density of the supernatant was measured at 420 and 550 nm, and the resulting values were used to calculate the β-galactosidase-specific activity according to the following equation: 1,000 × {[OD420 − (1.75 × OD550)]/(time × OD600)}.
Statistical analysis.
To determine significance between independent samples, unpaired Student t tests were performed.
RESULTS
A ΔcodY mutant grows primarily as unchained cells.
In a previous study, we found that a ΔrelA mutant, which possesses decreased GTP levels, grows as a homogenous population of unchained, swimming cells during exponential growth (12). We hypothesized that the GTP-sensing global regulator CodY, previously implicated in repression of flagellar gene expression (16), might become derepressed in the ΔrelA mutant due to the lower levels of GTP. In this model, CodY derepression could be responsible for mediating the shift toward the unchained, motile cell state that we observed in the ΔrelA mutant. To test this idea, we first examined the cell-chaining phenotype in a ΔcodY mutant during exponential growth, where the wild-type (PY79) cells grew as a mixed population of 85% chained and 15% unchained cells (7). We found that the ΔcodY mutant is less chained than the wild type during exponential growth but, unlike the ΔrelA mutant, is comprised of both chained and unchained cells (Fig. 1). Quantitation revealed that the proportion of unchained cells increased from 15% in the wild type to ∼75% in the ΔcodY mutant. The cell chaining was complemented by introducing Pcod-codY at an ectopic locus on the chromosome (see Fig. S1A in the supplemental material). We conclude that CodY has a significant role in preventing the switch from chained cells to unchained cells during exponential growth. This result suggests that derepression of CodY-regulated genes could account for most of the unchained cells observed in the ΔrelA mutant. The result also indicates that CodY derepression alone cannot account for the 100% unchained phenotype observed in the ΔrelA mutant, since 25% of ΔcodY mutant cells remained chained.
FIG 1.
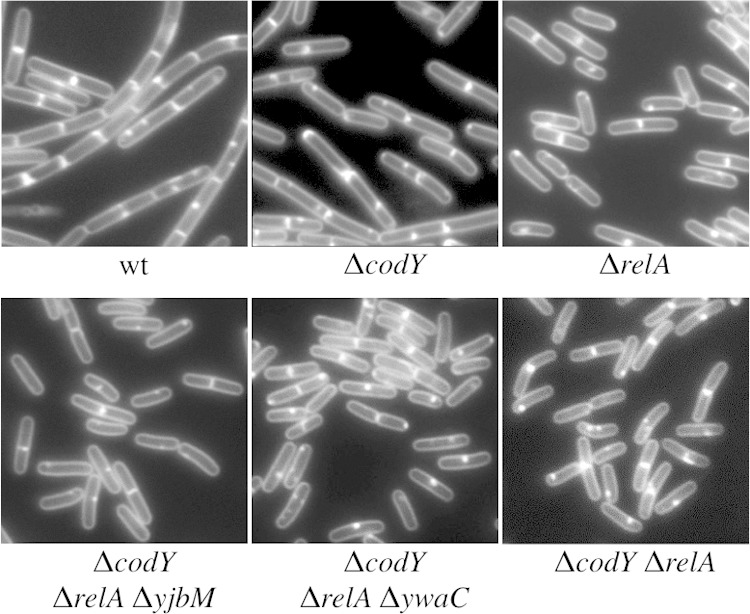
The ΔrelA mutant is less chained than the wild type. Representative images of wild-type (wt) (BJH001) and ΔrelA (BQA009), ΔcodY (BQA090), ΔcodY ΔrelA ΔyjbM (BQA096), ΔcodY ΔrelA ΔywaC (BQA097), and ΔrelA ΔcodY (BQA091) cells grown to the mid-log phase in CH medium are shown. Membranes were stained with TMA.
We hypothesized that reduced GTP levels in the ΔrelA mutant could contribute to the CodY-independent unchained phenotype observed. In this case, strains harboring higher levels of GTP than the ΔrelA mutant but lower levels than the wild type should shift the ΔcodY mutant toward a more unchained phenotype. To test this hypothesis, we introduced the codY deletion into the ΔrelA ΔywaC and ΔrelA ΔyjbM mutant backgrounds, previously shown to have intermediate GTP levels (14) and to be 66% and 22% unchained during exponential growth, respectively (12). In comparison to the codY deletion alone (∼75% unchained), the ΔcodY ΔrelA ΔywaC and ΔcodY ΔrelA ΔyjbM triple mutants grew exclusively as unchained populations (Fig. 1). These results suggest that the switch of the remaining chained cells to unchained cells could be accounted for by a GTP-dependent but CodY-independent mechanism. The ΔrelA ΔywaC and ΔrelA ΔyjbM mutant backgrounds likely harbor increased (p)ppGpp levels (12–14), so we do not rule out the possibility that (p)ppGpp levels contribute to the phenotypic switch to unchained cells that we observed.
The ΔcodY mutant strain exhibits increased mobility on swim plates.
Cell chaining and cell motility are inversely correlated; unchained cells possess flagella and are motile, while chained cells are mainly aflagellate and nonmotile (7). Since the ΔcodY mutant population is less chained than the wild type, we expected that the ΔcodY mutant would also produce a more motile population than the wild type. To assess the motility behavior of a ΔcodY mutant population, we performed a swim plate assay (12). Cultures were grown to the mid-exponential phase, and equal cell densities were spotted on semisolid media. In the swim plate assay, motile cells migrate away from the original spot and produce a halo, the outer edge of which is referred to as the swimming front. As expected, the ΔcodY mutant swimming front migrated further than the swimming front seen with the wild type but more slowly than the ΔrelA mutant swimming front (Fig. 2). This result is consistent with the proportions of unchained cells observed in the ΔcodY and ΔrelA mutants (Fig. 1, upper panels). The ΔsigD mutant population, which is composed exclusively of chained, nonmotile cells (7), did not migrate appreciably on the swim plate. The ΔcodY swimming phenotype could be complemented by introducing Pcod-codY at an ectopic locus on the chromosome (see Fig. S1B in the supplemental material). We conclude that the ΔcodY mutant population not only is more unchained but also is composed of more swimming cells than the wild type.
FIG 2.
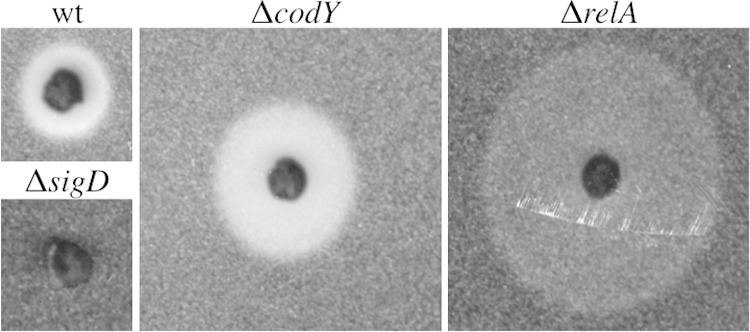
The ΔcodY mutant strain exhibits increased mobility on swim plates. Wild-type (wt; BJH001), ΔrelA (BQA009), ΔcodY (BQA090), and ΔsigD (BQA022) strains were inoculated on LB fortified with 0.25% agar. After 5 h of incubation at 37°C, the plates were scanned against a black background. The images shown were cropped from the same petri plate and are representative of the swimming behavior of each strain for at least three biologically independent replicates. The diameter values for the swimming fronts are as follows: wild-type strain, 80 mm; ΔrelA strain, 190 mm; ΔcodY strain, 120 mm; ΔsigD strain, 40 mm. The density of cells around the ΔrelA mutant is less than the density seen with the wild-type and ΔcodY strains due to the lower growth rate and increased mobility of the strain (12).
SigD levels and activity are elevated in the codY mutant.
The phenotypic switch to the unchained, motile cell phenotype is regulated by both SigD levels and SigD activity, and we previously found that SigD levels and activity are elevated in the ΔrelA mutant (12). Since the ΔcodY mutant is less chained than the wild type, we hypothesized that the ΔcodY mutant would also produce higher levels of SigD than the wild type. Western blot analysis on cell lysates collected from exponentially growing cultures showed that the ΔcodY mutant exhibited higher levels of SigD than the wild type (Fig. 3A and B). Wild-type levels of SigD could be restored in the ΔcodY mutant by introducing Pcod-codY at an ectopic locus on the chromosome (see Fig. S1C in the supplemental material). Surprisingly, the levels of SigD protein in the ΔcodY mutant were not statistically significantly different from those in the ΔrelA mutant (Fig. 3B), which exhibits a completely SigD on state phenotypically (12). The ΔcodY ΔrelA ΔywaC and ΔcodY ΔrelA ΔyjbM triple mutants phenocopied the ΔrelA mutant with respect to the SigD protein produced (Fig. 3A and B).
FIG 3.
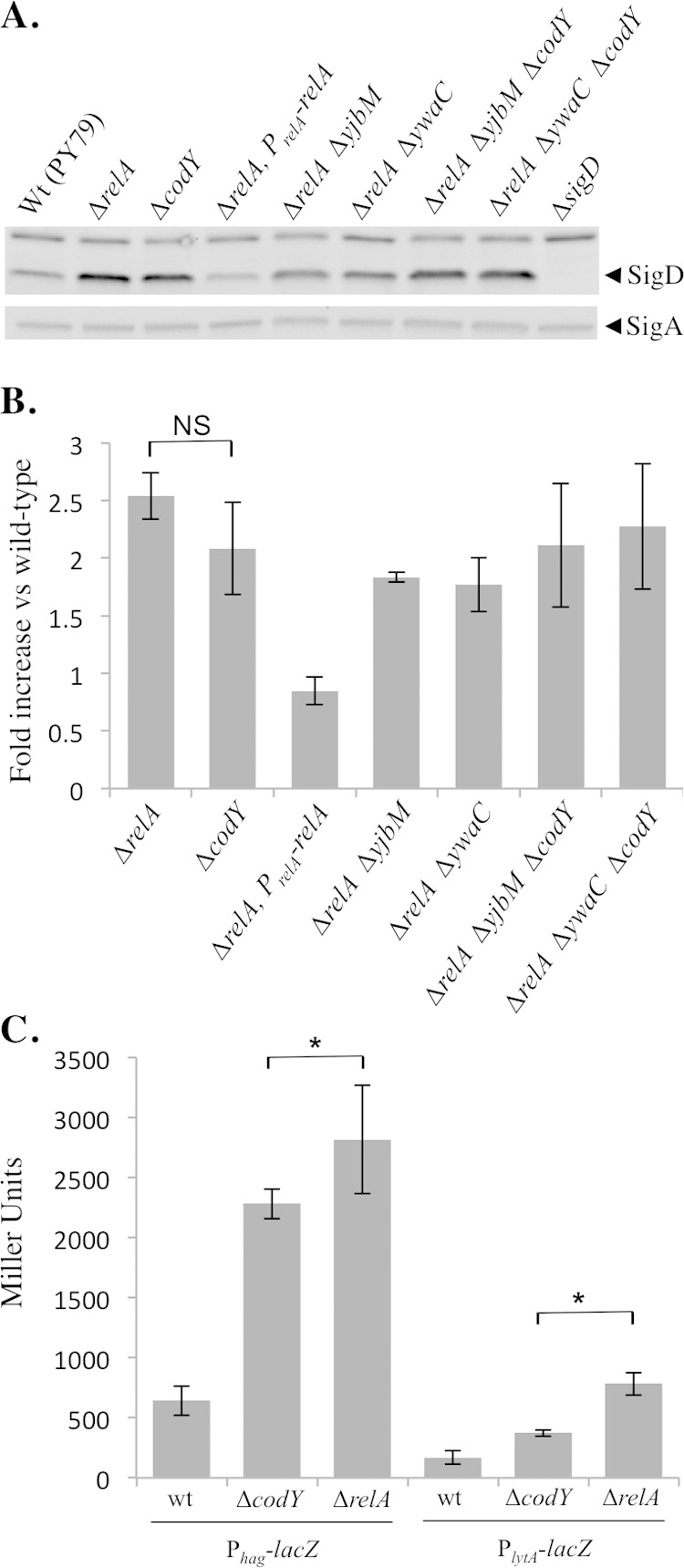
SigD levels and activity are elevated in the ΔcodY mutant. (A) Comparison of SigD protein levels in the wild-type (Wt; BJH001), ΔrelA (BQA009), ΔrelA PrelA-relA (BQA068), ΔcodY (BQA090), ΔrelA ΔyjbM (BQA081), ΔrelA ΔywaC (BQA082), ΔcodY ΔrelA ΔyjbM (BQA096), ΔcodY ΔrelA ΔywaC (BQA097), and ΔsigD (BQA022) strains. Samples were collected from mid-log cultures grown in CH medium. Proteins from cell lysates were separated by SDS-PAGE and analyzed by Western blotting by probing with anti-SigD antibodies. SigA was used as a loading control. (B) Quantification of SigD protein from the strains described for panel A. Five independent biological and experimental replicates were analyzed. NS, nonsignificant. (C) β-Galactosidase assays of Phag-lacZ and PlytA-lacZ transcriptional activities conducted on the wild-type (BJH046 and BJH047), ΔrelA (BQA050 and BQA051), and ΔcodY (BQA102 and BQA103) strains. Samples were collected from mid-log-phase cultures grown in CH medium. Data shown are the means of the results of three independent biological replicates with standard deviations. Significant (P < 0.05) differences between the strain ΔcodY and strain ΔrelA results are indicated by asterisks.
The ΔrelA and ΔcodY mutants accumulated similar levels of SigD during exponential growth (Fig. 3A and B). Although the average SigD levels were consistently lower in the ΔcodY mutant than in the ΔrelA mutant, densitometric analysis of five independent biological replicates indicated that the observed difference was not significant within a 95% confidence interval. The ΔcodY mutant is more chained and less motile than the ΔrelA mutant, suggesting that SigD protein levels alone may not account for the differences in phenotypes. To examine SigD activity, we analyzed expression from two SigD-dependent promoters (Phag and PlytA) using transcriptional fusions to a lacZ reporter gene. CodY was previously shown to bind the hag promoter and to repress hag transcription (11, 16). Consistent with the data from a prior study performed in another B. subtilis strain (JH642) under different growth conditions (16), we observed that Phag became approximately 3.5-fold derepressed in the absence of codY in wild-type PY79 grown in CH medium (Fig. 3C). We also observed that expression from PlytA was increased 2.2-fold compared to the wild-type level, suggesting that CodY also regulates expression from PlytA (Fig. 3C), possibly indirectly through SigD. As expected based on the phenotypic differences that we observed, expression from Phag and PlytA in the ΔcodY mutant was lower than in a ΔrelA mutant (Fig. 3C). Since SigD protein levels do not differ significantly between the ΔrelA and ΔcodY strains (this study), these results may suggest that the ΔcodY mutant harbors a higher level of inactive SigD than the ΔrelA mutant. Alternatively, it is also possible that there are small differences in SigD levels between the two strains that fall outside the limit of detection for Western blot analysis.
CodY has three putative binding sites in the fla/che operon region.
CodY is a DNA binding protein that globally regulates the expression of over 100 gene products in response to changes in nutrient status (19). The DNA binding domain of CodY recognizes a 15-bp, AT-rich motif, AATTTTCWGAAAATT (20–22). To determine if CodY could be directly regulating the accumulation of SigD by directly regulating sigD expression from the fla/che operon (23, 24), we searched the literature for previously reported CodY binding sites in the region. Three putative CodY binding sites were identified. One site, found upstream of the SigD-dependent promoter PD3 (referred to as site D in this study), was previously shown to be bound by CodY directly (16). We also identified two additional untested, intragenic sites in the fla/che operon in fliE (site E) and fliK (site K) within a data set from a CodY in vitro DNA affinity purification sequencing (IDAP-Seq) study (19). The locations and sequences of the three sites are shown in Fig. 4. The site D motif sequence has three mismatches with respect to the CodY binding consensus motif, while sites E and K have six and five mismatches, respectively (Fig. 4).
FIG 4.

CodY has three binding sites in the fla/che operon. The genetic organization of the fla/che operon genes is shown. Transcription of Pfla/che is driven by SigA, while SigD directs transcription from PD-3 and PylxF3. Open reading frames are represented by arrows (not to scale). Components essential for FlgM secretion are shaded gray. The CodY consensus sequence, CodY binding sites, and point mutations made to the mutated versions of the sites are shown. Nucleotides that differ between the consensus and the sites are indicated by bold type.
To determine if CodY interacts directly with sites E and K, we PCR amplified DNA fragments from the chromosome centered at each site and performed electrophoretic mobility gel shift assays with 6His-CodY in the presence of GTP and branched-chain amino acids. As a positive control, we included the previously identified site, site D, in our analysis (16). In vitro, CodY bound to all three sites, showing an apparent preference for sites E and D compared to site K (Fig. 5). DNA lacking the predicted binding motifs did not show altered mobility at any of the protein concentrations tested (data not shown), suggesting that the binding of CodY to each of these sites is specific.
FIG 5.
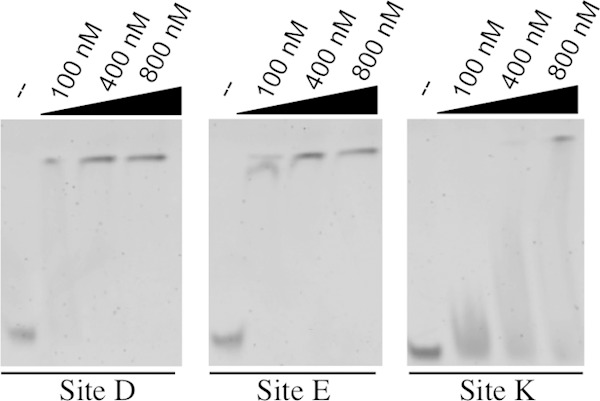
CodY binds directly to three sites in the fla/che operon. Results of electrophoretic mobility gel shift analysis of DNA fragments (5 nM) centered on the putative CodY binding motif incubated with the indicated concentrations of 6His-CodY are shown. Gel shifts were performed in the presence of the CodY cofactors GTP, isoleucine, leucine, and valine.
Mutating the three CodY binding sites in the fla/che operon phenocopies the ΔcodY mutation.
CodY negatively regulates transcription by binding to promoter regions and preventing transcription initiation (22). CodY has also been shown to prevent synthesis of full-length transcripts by acting as a roadblock to RNA polymerase transit (25). Since CodY binding site D is located upstream of the SigD-dependent and SigA-dependent promoters, PD3 and Pfla/che, respectively (Fig. 4), it could function to prevent initiation of transcription from PD3 and/or Pfla/che. On the other hand, sites E and K lie within open reading frames (Fig. 4). If CodY is disrupting transcription from sites E and K, then CodY is most likely acting through a roadblock mechanism, preventing transcription of RNA downstream of each binding site (25).
To determine the contribution of each of the CodY binding sites to the unchained phenotype and increased SigD levels and activity that we observed in the ΔcodY mutant, we created point mutations within each of the sites on the chromosome (Fig. 4). Since sites E and K lie within genes, we introduced point mutations that did not disrupt the open reading frames of fliE and fliK, respectively. To determine the contribution of each binding site to the chaining phenotype, we determined the proportion of unchained versus chained cells in strains harboring all six possible combinations of mutant CodY binding sites. The proportion of unchained cells was higher than that seen with the wild type by at least double for each of the mutants tested (Fig. 6A). The relative contribution of each single site to cell chaining showed the following pattern: site D → site E → site K. Moreover, the double mutants that included Dmu were most similar to the ΔcodY mutant. These results suggest that site D has the strongest control of SigD activity but that sites E and K are also independently important. The triple-binding-site mutant most closely phenocopied the ΔcodY strain (Fig. 6A). The ΔcodY mutant harbors reduced levels of cellular GTP relative to the wild type (26). To assess how much of the shift toward unchained cells in the ΔcodY mutant could be attributed to reduced GTP levels in the strain background, we compared the percentage of unchained cells in the ΔcodY mutant to the percentage in a double-mutant strain harboring both a deletion of codY and point mutations in all three CodY binding sites. The double mutant was 78% (±2.4%) unchained compared to 76% (±7.8%) for the ΔcodY mutant and 75% (±5.3%) for the CodY binding site mutant. These results suggest that CodY acts in the same pathway as the CodY binding sites in the regulation of SigD activity and also support the idea that the ΔcodY mutant phenotype (more unchained cells) occurs independently of the reduced cellular GTP levels present in the ΔcodY mutant background.
FIG 6.
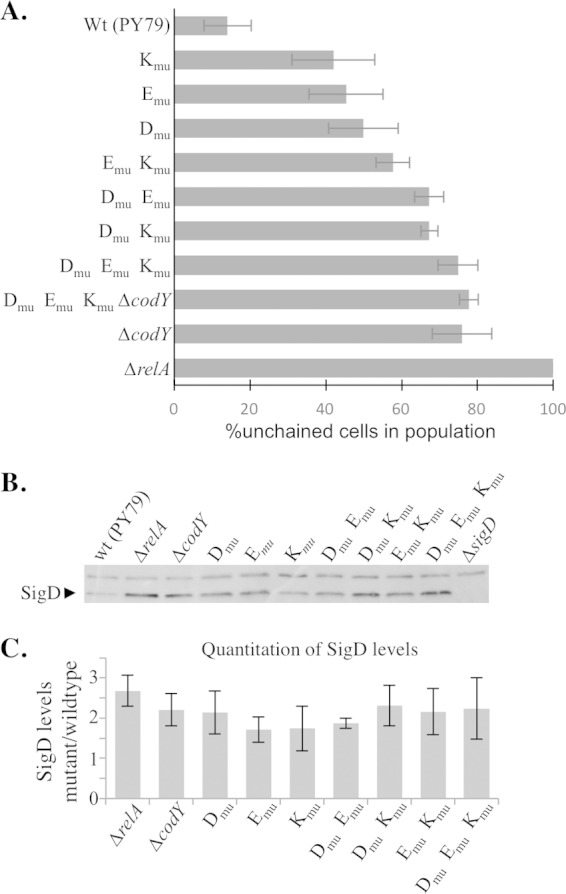
Mutating three CodY binding sites in the fla/che operon phenocopies ΔcodY. (A) Frequency of chained cells (three or more cells linked) and unchained cells (single and doublet) across a population. Cells in images from at least three independent cultures were counted, and no fewer than 1,000 cells were quantitated for each strain represented in the graph. The standard deviations for the independent biological samples are shown. Wt, wild type. (B) Western blot analysis of SigD protein levels from samples collected from mid-log-phase cultures grown in CH medium. (C) Quantification of SigD protein across five biologically and experimentally independent samples. The averages and standard deviations for the five independent replicates are shown normalized to the wild-type strain (PY79). None of the CodY binding site mutants or mutant combinations differed significantly from the ΔcodY mutant at the 95% confidence level.
We then examined SigD levels in backgrounds harboring all possible combinations of the mutant CodY binding sites. Surprisingly, mutating any one of the CodY binding sites caused a significant increase in SigD levels compared to the wild-type results, with each strain reaching SigD levels statistically indistinguishable from those seen with the ΔcodY mutant (Fig. 6B and C). Strains in which either two of the binding sites or all three of the binding sites were mutated also showed SigD levels comparable to the ΔcodY mutant levels (Fig. 6B and C). We conclude that each of sites D, E, and K contributes to preventing the accumulation of SigD during exponential growth.
To assess whether the increase in SigD levels observed in the ΔcodY mutant could be attributed to the reduced GTP levels present in the strain background, we compared the SigD levels of the ΔcodY mutant to those seen with a double mutant harboring both a deletion of codY and point mutations in the three CodY binding sites (Dmu Emu Kmu). The levels of SigD in the ΔcodY mutant also harboring mutations in the three CodY binding sites were indistinguishable from those seen with the ΔcodY single mutant (see Fig. S2A in the supplemental material). The swimming behavior of the double mutant was also similar to the swimming behavior of the ΔcodY single mutant (see Fig. S2B). These results further suggest that the ΔcodY SigD on phenotype occurs independently of the reduced cellular GTP levels present in the ΔcodY mutant background.
DISCUSSION
RelA is a dual-function (p)ppGpp synthetase and hydrolase enzyme which, along with two additional (p)ppGpp synthetases, YjbM (also known as RelQ and SasB) and YwaC (also known as RelP and SasA), modulates (p)ppGpp levels in B. subtilis (14). (p)ppGpp synthesis by YjbM and YwaC in the absence of RelA (p)ppGpp hydrolysis activity is sufficient to bias cells toward the 100% motile, unchained phenotype (12).
Accumulation of (p)ppGpp induces a wide range of physiological alterations that affect many important cellular processes such as DNA replication, transcription, translation, and metabolism (27). In E. coli, (p)ppGpp and a coregulator, DksA, bind directly to RNA polymerase to inhibit or activate transcription (28, 29). (p)ppGpp has also been shown be important for the association of alternative sigma factors with the RNA polymerase core (30). In the Gram-positive bacterium B. subtilis, (p)ppGpp's effects on transcriptional regulation occur primarily through modulation of GTP levels (27). GTP is one of the substrates for (p)ppGpp synthesis, so as (p)ppGpp levels rise, GTP becomes depleted. In addition, (p)ppGpp inhibits hypoxanthine-guanine phosphoribosyltransferase (HprT) and guanylate kinase (GMK), enzymes involved in the biosynthesis of GTP biosynthesis (26). GTP levels affect transcription in B. subtilis through at least two mechanisms. First, GTP is an important cofactor for the global transcriptional regulator CodY (31), so reductions in cellular GTP levels lead to derepression of CodY-repressed target genes (15, 32). Second, decreased GTP levels reduce transcription from promoters that use GTP as the initiating nucleotide for transcription (33–37).
In this study, we investigated the role of CodY in regulating the switch between the SigD on and off states. We found that CodY represses expression of SigD by binding directly to three sites within the fla/che operon. To our surprise, cells harboring mutations in any one of the CodY binding sites produced SigD levels that were indistinguishable from those seen with the ΔcodY mutant, suggesting that SigD expression in PY79 is largely a function of whether or not CodY is bound within the fla/che operon. We found that the SigD protein levels were not necessarily predictive of the phenotypic consequence of mutating each of the CodY binding sites. For example, a site D mutant (Dmu) and a ΔcodY mutant produced similar SigD levels on average (Fig. 6C); however, Dmu possessed two times more chained cells than a ΔcodY mutant (Fig. 6A). It is possible that subtle differences (outside the linear range of detecting quantitative differences) in SigD levels explain the observed differences in cell chaining. Alternatively, the phenotypic differences could be explained by posttranslational regulation of SigD activity (see below).
One model suggests that CodY's binding to various sites within the chromosome is hierarchical (38). In other words, when GTP is abundant, CodY-GTP would bind to all sites, but as GTP levels drop, CodY would be gradually released from lower-affinity sites. Based on our electrophoretic gel mobility shift data, CodY has a higher affinity for sites D and site E than for site K (Fig. 5). If the same relative affinities are present in vivo, this would suggest that CodY is released from site K first, allowing transcription from the PylxF promoter to the end of the operon to occur (Fig. 4) (10). A further drop in GTP levels would allow transcription through site K from the Pfla/che and PD3 promoters, respectively.
Our analysis of the CodY binding sites within the fla/che operon revealed that the newly characterized CodY binding sites (sites E and K) (Fig. 4) are positioned just upstream of flagellar apparatus genes important for FlgM secretion (Fig. 4) (39). FlgM is an anti-sigma factor that inhibits SigD activity by preventing its association with RNA polymerase (10, 40, 41). Upon completion of the hook-basal body complex, FlgM is secreted outside the cell, where it is no longer competent to antagonize SigD (39). The minimal set of flagellar genes in the fla/che operon required for FlgM secretion consists of fliF, fliG, fliK, fliO, fliP, fliQ, fliR, flhB, and flhA (39) (Fig. 4, shaded arrows). Therefore, in addition to regulating SigD levels, CodY may exert some control on SigD activity by specifically regulating expression of genes important for FlgM secretion.
We found that a ΔcodY mutant is ∼75% unchained, suggesting that CodY derepression could contribute to the unchained, motile phenotype observed in the ΔrelA mutant. We were unable to determine whether CodY is required for the 100%-unchained phenotype of the ΔrelA mutant because the double mutant is itself 100% unchained (Fig. 1). However, we were able to ascertain that adjusting the GTP levels in the ΔcodY mutant background through the inactivation of relA and ywaC or relA and yjbM was sufficient to phenocopy the 100%-unchained phenotype of the ΔrelA mutant (Fig. 1). Our results are consistent with the idea that both CodY-dependent and CodY-independent mechanisms contribute to the SigD on state in the ΔrelA mutant background and also suggest that each of these mechanisms is responsive to the cellular levels of GTP and/or (p)ppGpp. Finally, our results also support the idea that global transcriptional regulators such as CodY are capable of finely tuning gene expression in response to even small changes in the concentrations of important intracellular signaling molecules.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Center for Phage Technology and the members of the Herman laboratory for helpful feedback and suggestions.
This work was supported by start-up funding to J.K.H. from the Center for Phage Technology and the Department of Biochemistry and Biophysics at Texas A&M University.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00288-15.
REFERENCES
- 1.Dubnau D, Losick R. 2006. Bistability in bacteria. Mol Microbiol 61:564–572. doi: 10.1111/j.1365-2958.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- 2.Veening JW, Smits WK, Kuipers OP. 2008. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol 62:193–210. doi: 10.1146/annurev.micro.62.081307.163002. [DOI] [PubMed] [Google Scholar]
- 3.Maisonneuve E, Castro-Camargo M, Gerdes K. 2013. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 4.Losick R, Desplan C. 2008. Stochasticity and cell fate. Science 320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 6.Jayaraman R. 2008. Bacterial persistence: some new insights into an old phenomenon. J Biosci 33:795–805. doi: 10.1007/s12038-008-0099-3. [DOI] [PubMed] [Google Scholar]
- 7.Kearns DB, Losick R. 2005. Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev 19:3083–3094. doi: 10.1101/gad.1373905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen R, Guttenplan SB, Blair KM, Kearns DB. 2009. Role of the sigmaD-dependent autolysins in Bacillus subtilis population heterogeneity. J Bacteriol 191:5775–5784. doi: 10.1128/JB.00521-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norman TM, Lord ND, Paulsson J, Losick R. 2013. Memory and modularity in cell-fate decision making. Nature 503:481–486. doi: 10.1038/nature12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cozy LM, Kearns DB. 2010. Gene position in a long operon governs motility development in Bacillus subtilis. Mol Microbiol 76:273–285. doi: 10.1111/j.1365-2958.2010.07112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirel DB, Estacio WF, Mathieu M, Olmsted E, Ramirez J, Marquez-Magana LM. 2000. Environmental regulation of Bacillus subtilis sigma(D)-dependent gene expression. J Bacteriol 182:3055–3062. doi: 10.1128/JB.182.11.3055-3062.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ababneh QO, Herman JK. 2015. RelA inhibits Bacillus subtilis motility and chaining. J Bacteriol 197:128–137. doi: 10.1128/JB.02063-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Natori Y, Tagami K, Murakami K, Yoshida S, Tanigawa O, Moh Y, Masuda K, Wada T, Suzuki S, Nanamiya H, Tozawa Y, Kawamura F. 2009. Transcription activity of individual rrn operons in Bacillus subtilis mutants deficient in (p)ppGpp synthetase genes, relA, yjbM, and ywaC. J Bacteriol 191:4555–4561. doi: 10.1128/JB.00263-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nanamiya H, Kasai K, Nozawa A, Yun CS, Narisawa T, Murakami K, Natori Y, Kawamura F, Tozawa Y. 2008. Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol Microbiol 67:291–304. doi: 10.1111/j.1365-2958.2007.06018.x. [DOI] [PubMed] [Google Scholar]
- 15.Ratnayake-Lecamwasam M, Serror P, Wong KW, Sonenshein AL. 2001. Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev 15:1093–1103. doi: 10.1101/gad.874201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergara F, Ibarra C, Iwamasa J, Patarroyo JC, Aguilera R, Marquez-Magana LM. 2003. CodY is a nutritional repressor of flagellar gene expression in Bacillus subtilis. J Bacteriol 185:3118–3126. doi: 10.1128/JB.185.10.3118-3126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wray LV Jr, Fisher SH. 2011. Bacillus subtilis CodY operators contain overlapping CodY binding sites. J Bacteriol 193:4841–4848. doi: 10.1128/JB.05258-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasband WS. 1997–2014. ImageJ. U.S. National Institutes of Health, Bethesda, MD. [Google Scholar]
- 19.Belitsky BR, Sonenshein AL. 2013. Genome-wide identification of Bacillus subtilis CodY-binding sites at single-nucleotide resolution. Proc Natl Acad Sci U S A 110:7026–7031. doi: 10.1073/pnas.1300428110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.den Hengst CD, van Hijum SA, Geurts JM, Nauta A, Kok J, Kuipers OP. 2005. The Lactococcus lactis CodY regulon: identification of a conserved cis-regulatory element. J Biol Chem 280:34332–34342. doi: 10.1074/jbc.M502349200. [DOI] [PubMed] [Google Scholar]
- 21.Guédon E, Sperandio B, Pons N, Ehrlich SD, Renault P. 2005. Overall control of nitrogen metabolism in Lactococcus lactis by CodY, and possible models for CodY regulation in Firmicutes. Microbiology 151:3895–3909. doi: 10.1099/mic.0.28186-0. [DOI] [PubMed] [Google Scholar]
- 22.Belitsky BR, Sonenshein AL. 2008. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J Bacteriol 190:1224–1236. doi: 10.1128/JB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmann JD, Marquez LM, Chamberlin MJ. 1988. Cloning, sequencing, and disruption of the Bacillus subtilis sigma 28 gene. J Bacteriol 170:1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Márquez LM, Helmann JD, Ferrari E, Parker HM, Ordal GW, Chamberlin MJ. 1990. Studies of sigma d-dependent functions in Bacillus subtilis. J Bacteriol 172:3435–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belitsky BR, Sonenshein AL. 2011. Roadblock repression of transcription by Bacillus subtilis CodY. J Mol Biol 411:729–743. doi: 10.1016/j.jmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kriel A, Bittner AN, Kim SH, Liu K, Tehranchi AK, Zou WY, Rendon S, Chen R, Tu BP, Wang JD. 2012. Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol Cell 48:231–241. doi: 10.1016/j.molcel.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu K, Bittner AN, Wang JD. 2015. Diversity in (p)ppGpp metabolism and effectors. Curr Opin Microbiol 24:72–79. doi: 10.1016/j.mib.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul BJ, Berkmen MB, Gourse RL. 2005. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci U S A 102:7823–7828. doi: 10.1073/pnas.0501170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haugen SP, Ross W, Gourse RL. 2008. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol 6:507–519. doi: 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jishage M, Kvint K, Shingler V, Nystrom T. 2002. Regulation of sigma factor competition by the alarmone ppGpp. Genes Dev 16:1260–1270. doi: 10.1101/gad.227902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handke LD, Shivers RP, Sonenshein AL. 2008. Interaction of Bacillus subtilis CodY with GTP. J Bacteriol 190:798–806. doi: 10.1128/JB.01115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brinsmade SR, Sonenshein AL. 2011. Dissecting complex metabolic integration provides direct genetic evidence for CodY activation by guanine nucleotides. J Bacteriol 193:5637–5648. doi: 10.1128/JB.05510-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krásný L, Gourse RL. 2004. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J 23:4473–4483. doi: 10.1038/sj.emboj.7600423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krásný L, Tiserova H, Jonak J, Rejman D, Sanderova H. 2008. The identity of the transcription +1 position is crucial for changes in gene expression in response to amino acid starvation in Bacillus subtilis. Mol Microbiol 69:42–54. doi: 10.1111/j.1365-2958.2008.06256.x. [DOI] [PubMed] [Google Scholar]
- 35.Sojka L, Kouba T, Barvik I, Sanderova H, Maderova Z, Jonak J, Krasny L. 2011. Rapid changes in gene expression: DNA determinants of promoter regulation by the concentration of the transcription initiating NTP in Bacillus subtilis. Nucleic Acids Res 39:4598–4611. doi: 10.1093/nar/gkr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tojo S, Kumamoto K, Hirooka K, Fujita Y. 2010. Heavy involvement of stringent transcription control depending on the adenine or guanine species of the transcription initiation site in glucose and pyruvate metabolism in Bacillus subtilis. J Bacteriol 192:1573–1585. doi: 10.1128/JB.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tojo S, Hirooka K, Fujita Y. 2013. Expression of kinA and kinB of Bacillus subtilis, necessary for sporulation initiation, is under positive stringent transcription control. J Bacteriol 195:1656–1665. doi: 10.1128/JB.02131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brinsmade SR, Alexander EL, Livny J, Stettner AI, Segre D, Rhee KY, Sonenshein AL. 2014. Hierarchical expression of genes controlled by the Bacillus subtilis global regulatory protein CodY. Proc Natl Acad Sci U S A 111:8227–8232. doi: 10.1073/pnas.1321308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvo RA, Kearns DB. 2015. FlgM is secreted by the flagellar export apparatus in Bacillus subtilis. J Bacteriol 197:81–91. doi: 10.1128/JB.02324-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caramori T, Barilla D, Nessi C, Sacchi L, Galizzi A. 1996. Role of FlgM in sigma d-dependent gene expression in Bacillus subtilis. J Bacteriol 178:3113–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertero MG, Gonzales B, Tarricone C, Ceciliani F, Galizzi A. 1999. Overproduction and characterization of the Bacillus subtilis anti-sigma factor FlgM. J Biol Chem 274:12103–12107. doi: 10.1074/jbc.274.17.12103. [DOI] [PubMed] [Google Scholar]
- 42.Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. Wiley, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.