

ABSTRACT
During biosynthesis of [NiFe]-hydrogenase 2 (Hyd-2) of Escherichia coli, a 15-amino-acid C-terminal peptide is cleaved from the catalytic large subunit precursor, pro-HybC. This peptide is removed only after NiFe(CN)2CO cofactor insertion by the Hyp accessory protein machinery has been completed, suggesting that it has a regulatory function during enzyme maturation. We show here that in hyp mutants that fail to synthesize and insert the NiFe cofactor, and therefore retain the peptide, the Tat (twin-arginine translocon) signal peptide on the small subunit HybO is not removed and the subunit is degraded. In a mutant lacking the large subunit, the Tat signal peptide was also not removed from pre-HybO, indicating that the mature large subunit must actively engage the small subunit to elicit Tat transport. We validated the proposed regulatory role of the C-terminal peptide in controlling enzyme assembly by genetically removing it from the precursor of HybC, which allowed assembly and Tat-dependent membrane association of a HybC-HybO heterodimer lacking the NiFe(CN)2CO cofactor. Finally, genetic transfer of the C-terminal peptide from pro-HyaB, the large subunit of Hyd-1, onto HybC did not influence its dependence on the accessory protein HybG, a HypC paralog, or the specific protease HybD. This indicates that the C-terminal peptide per se is not required for interaction with the Hyp machinery but rather suggests a role of the peptide in maintaining a conformation of the protein suitable for cofactor insertion. Together, our results demonstrate that the C-terminal peptide on the catalytic subunit controls biosynthesis, assembly, and membrane association of Hyd-2.
IMPORTANCE [NiFe]-hydrogenases are multisubunit enzymes with a catalytic subunit containing a NiFe(CN)2CO cofactor. Results of previous studies suggested that after synthesis and insertion of the cofactor by the Hyp accessory proteins, this large subunit changes conformation upon proteolytic removal of a short peptide from its C terminus. We show that removal of this peptide is necessary to allow the cleavage of the Tat signal peptide from the small subunit with concomitant membrane association of the heterodimer to occur. Genetic removal of the C-terminal peptide from the large subunit allowed productive interaction with the small subunit and Tat-dependent membrane insertion of a NiFe cofactor-free enzyme. Results based on swapping of C-terminal peptides between hydrogenases suggest that this peptide governs enzyme assembly via a conformational switch.
INTRODUCTION
Biosynthesis of complex metal cofactor-containing, multisubunit enzymes requires strict coordination of cofactor biosynthesis, subunit recruitment, and targeting of the protein to its final cellular location. This is particularly important for the numerous redox enzymes present in bacterial systems, many of which are membrane associated or indeed transported across the cytoplasmic membrane (1). Coordination of biosynthesis and assembly is important because cofactors are often highly complex and many of these membrane-associated enzymes are substrates of the twin-arginine transport (Tat) pathway, which transports only correctly preassembled, cofactor-containing protein complexes (2). How these events are orchestrated, at what stages control is exerted, and how this is achieved remain to be clearly elucidated for large multisubunit cofactor-containing enzymes. Major advances in our understanding of some of these biosynthetic and assembly processes have, however, been achieved for [NiFe]-hydrogenases (3–5), which therefore serve as excellent model systems to understand the maturation of modular enzymes.
[NiFe]-hydrogenases (Hyd) are among the most ancient enzymes, and they catalyze the reversible oxidation of hydrogen to protons and electrons (6, 7). The core of these enzymes comprises an αβ heterodimer that includes a catalytic large α subunit with a NiFe(CN)2CO (NiFe) cofactor and an electron-transferring small β subunit containing iron-sulfur (FeS) clusters (6). Frequently, these enzymes are membrane associated, and those involved in hydrogen uptake have to be transported across the cytoplasmic membrane in a fully folded, cofactor-loaded state, a feat performed by the Tat translocon (8–10). The αβ heterodimer is directed to the Tat translocon by a Tat signal peptide located on the small subunit, and once the heterodimer is transported across the membrane, it docks with other subunits to form the complete enzyme (9, 11). This series of steps must be tightly controlled and is dependent on the availability of the active site metals, as well as on the metabolic precursors of the carbon monoxide and cyanide ligands attached to the active site iron ion (3, 12, 13). If any one of these substrates becomes limiting, maturation stops and the subunit precursors are degraded (14).
Six Hyp accessory proteins, HypA, -B, -C, -D, -E, and -F, are involved in biosynthesis and insertion of the NiFe cofactor into the active site of the Hyd large subunit (3, 12, 13). Insertion of the Fe(CN)2CO moiety precedes nickel insertion (15–19), and after this has taken place, the large subunit of most Hyd (3) is C terminally processed by a hydrogenase-specific endoprotease. Processing occurs 3 amino acids C terminal to the fourth cysteine that coordinates the cofactor (20–23). Cleavage of the C-terminal peptide occurs only after biosynthesis and insertion of the NiFe cofactor have been completed (3). A first indication that this C-terminal peptide might be important during enzyme maturation was originally demonstrated for enzymes from both bacterial and archaeal sources (20, 21, 24). It was subsequently shown that the HypC protein, which is proposed to have an important role in delivering the completed Fe(CN)2CO moiety of the active site cofactor to the apo-large subunit (3, 25–27), interacts with the precursor of the HycE protein, the large subunit of Escherichia coli Hyd-3 (17, 18, 28). Similar observations were made for Hyd enzymes from Ralstonia eutropha (19, 29). It was also shown that the HypC paralog HybG binds to the E. coli Hyd-2 large subunit precursor, which we term here pro-HybC (30, 31). HybG is required for the maturation of hydrogen-oxidizing Hyd-2 and also functions in the maturation of the other hydrogen-oxidizing enzyme, Hyd-1, in the bacterium (30, 32).
Hyd-2 has an unusual architecture because it comprises a large- and small-subunit HybC-HybO heterodimer, together with two other subunits, HybA and HybB, which complete a heterotetrameric complex on the periplasmic side of the membrane (11). The HybA protein is a Tat-dependent polypeptide with four predicted FeS clusters, while HybB is an integral membrane protein with no known cofactors. It is still unknown how the assembly of the heterotetramer is coordinated subsequent to independent transport and membrane integration of the component parts (9, 11).
HybC and HyaB, the large subunits of Hyd-2 and Hyd-1, respectively, were identified in a study examining binding partners of HybG (31). The multiprotein complex that was identified also included the HypD and HypE accessory proteins, which are involved in NiFe cofactor biosynthesis. The HypE-HypF complex (33) synthesizes the CN− ligands of the cofactor (34, 35), while HypD forms the core of the maturation protein complex binding HypC (25–27) and HybG (30, 31). The importance of the C terminus on the HybC precursor (pro-HybC in Fig. 1A) of E. coli Hyd-2 was underscored when it was shown to be necessary to allow binding of another enzyme-specific chaperone, termed HybE (8). Notably, HybE also interacts with the Tat signal peptide on the precursor species of the enzyme's small subunit, pre-HybO (8). HybE is thought to mask the signal peptide on the subunit from the Tat translocon (8, 11). In the current study, pre-HybO refers to the precursor of HybO carrying the Tat signal peptide, while HybO refers to the mature subunit lacking the Tat signal peptide. Maturation of the HybO small subunit appears to occur in parallel with maturation of HybC, and pre-HybO receives its complement of FeS clusters from the Isc machinery prior to interaction with the mature large subunit (14). If maturation of the large subunit is impeded, then the small subunit is degraded (13, 14). Clearly, tight coordination of large and small subunit maturation is implied by all of these data, but the mechanisms underlying how this is achieved are not completely resolved. Here, we report on the maturation of the HybC-HybO heterodimer of E. coli Hyd-2 and in particular the key role of the cleavable C-terminal peptide. We provide evidence that this peptide coordinates biosynthesis and insertion of the bimetallic cofactor by the Hyp proteins with maturation of the pre-HybO small FeS-containing subunit, assembly of the HybC–pre-HybO heterodimer, and release of the active enzyme to the Tat translocon.
FIG 1.

Schematic representation of the C-terminal HybC constructs. (A) Schematic representation of N-terminally epitope-tagged HybC derivatives. Pro-HybC represents the unprocessed large subunit of HybC; HybCproc is the genetically truncated version lacking 15 amino acids at its C terminus and is equivalent to processed HybC present in the active Hyd-2 enzyme. Derivatives carrying an N-terminal Strep tag were prepared (not represented in the figure). (B) Schematic representation of the constructs in which the 15-amino-acid C terminus from HyaB was swapped onto HybC and, vice versa, the 15-amino-acid C-terminal peptide from HybC was introduced onto HyaB.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
All E. coli strains and plasmids used in this study are listed in Table 1.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype | Reference |
|---|---|---|
| Strains | ||
| MC4100 | F− araD139 α(argF-lac)U169 ptsF25 deoC1 relA1 flbB5301 rspL150 | 57 |
| DHPF2 | MC4100 ΔhypF aaa 59–629 | 34 |
| DHP-D | MC4100 ΔhypD | 58 |
| DHP-E | MC4100 ΔhypE | 58 |
| DHP-B | MC4100 ΔhypB | 58 |
| DHB-G | MC4100 ΔhybG | 30 |
| FTD147 | ΔhyaB ΔhybC ΔhycE | 45 |
| FTD150 | ΔhyaB ΔhybC ΔhycE ΔhyfB-R | 45 |
| CP793 | MC4100 ΔhyaB ΔhycE ΔhybO::Kanr | 39 |
| CP695 | MC4100 ΔhybC | 39 |
| CB10 | DHPF2 ΔhybC | This study |
| CB11 | MC4100 ΔhybG::Kanr | This study |
| CB13 | DADE ΔhybC::Kanr | This study |
| CB15 | FTD147 hybC+ ΔhybG::Kanr | This study |
| CB16 | FTD150 hyaB+ ΔhyaD::Kanr | This study |
| DADE | MC4100 ΔtatA-E | 59 |
| Plasmids | ||
| pASK-hyaB | hyaB in pASK-IBA5+, Ampr | 39 |
| pASK-hybC | hybC in pASK-IBA5+, Ampr | 39 |
| pASK-hybCproc | pASK-hybC, V553 stop (codon 553 converted to TAA), Ampr | This study |
| pASK-hyaBhybC | hyaB with hybC C-terminal sequence in pASK-IBA7, Ampr | This study |
| pASK-hybChyaB | hybC with hyaB C-terminal sequence in pASK-IBA7, Ampr | This study |
aa, amino acids.
Strains were grown anaerobically in glucose M9 minimal medium (36) at 37°C in Hungate tubes under a nitrogen gas atmosphere. When required, the growth medium was solidified with 1.5% (wt/vol) agar. The antibiotics kanamycin, chloramphenicol, and ampicillin were added to the medium at final concentrations of 50, 15, and 100 μg ml−1, respectively. Cells from small-scale cultures (10 ml) were harvested after the culture reached an optical density at 600 nm of over 1 (stationary-phase cultures) or between 0.6 and 0.8 (exponential-phase cultures). Optical density was measured in an Ultrospec 10 spectrophotometer (Amersham Biosciences). Cells were collected by centrifugation at 20,000 × g for 15 min at 4°C. The cell pellets were either used immediately or stored at −20°C until required.
Strain construction.
Strains were generally constructed by introduction of mutations from E. coli donor strains into MC4100 derivatives by P1kc phage-mediated transduction according to the method of Miller (37). Mutant alleles were from donor strains of the Keio collection (38).
Plasmid construction.
Plasmid pASK-hybC (39) served as the DNA template for the introduction of mutations into the hybC gene using the QuikChange site-directed mutagenesis strategy of Agilent Technologies, USA. The oligonucleotide primers used to introduce a stop codon at the position containing codon V553, generating pASK-hybCproc, were 5′-GCCTGTGCGGTACACTAAGTGGATGCTGACGGC-3′ and 5′-GCCGTCAGCATCCACTTAGTGTACCGCACAGGC-3′. The authenticity of the entire mutated hybC gene was verified by DNA sequencing.
To construct the plasmids pASK-hyaBhybC and pASK-hybChyaB, encoding pro-HyaBHybC and pro-HybCHyaB, respectively, the following procedure was used. To construct pASK-hyaBhybC, the megaoligonucleotide primers For-HybC-mega (5′ AAATTAGCCTCGCCTGTTCAACACACGTAGTGGATGCTGACGGCAACGAAGTGGTT 3′) and Rev-HybC-mega (5′CCCGGGGGTCTCCTATCATTACAGAACCTTCACTGAAACCACTTCGTTGCC 3′) were annealed and then treated with the Klenow fragment of DNA polymerase and deoxynucleoside triphosphates (dNTPs) to extend the single strands and to generate a double-stranded DNA product. The resulting DNA fragment was then used as an oligonucleotide megaprimer along with FOR-hyaB (5′CCCGGAGGTCTCCGCGCATGAGCACTCAGTACGAAAC) and pCAN-hyaB (38) as the DNA template in a PCR to amplify the hyaB gene encoding the 15-amino-acid C terminus of HybC. The complete DNA fragment was digested with BsaI and cloned into vector pASK-IBA7. A similar approach was used to construct pASK-hybChyaB. The primers that were used were For-HyaB-mega (5′ AAATTAGCATGGCCTGTGCGGTACACGTGCTGGGCGACGACGGTAGCGAGCTGATC 3′), Rev-HyaB-C-mega (5′AACGGGGGTCTCCTATCATTAACGCACCTGCACGGAGATCAGCTCGCTACC 3′), and FOR-hybC (5′ CCCCAAGGTCTCCGCGCATGAGCCAGAGAATTACTAT 3′). The DNA inserts of both plasmids were verified by DNA sequencing.
Preparation of subcellular fractions.
Cell pellets were resuspended in 50 mM morpholinepropanesulfonic acid (MOPS) buffer (pH 7.0) and were disrupted by sonication. The crude cell extract was obtained after removal of unbroken cells and cellular debris by a brief centrifugation step (19,000 × g, 4°C, for 45 min). When required, membrane and soluble fractions were prepared by a further centrifugation step (90,000 × g, 4°C, 1 h). Membrane fractions were washed twice with 50 mM MOPS buffer (pH 7.0) containing 200 mM NaCl and resuspended at a final protein concentration of 10 mg ml−1.
PAGE and Western blotting.
Aliquots of a defined amount of protein from a crude extract or a soluble or membrane fraction (between 30 and 100 μg of protein) were separated by denaturing SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% (wt/vol) polyacrylamide gels (40). Transfer of polypeptides to nitrocellulose membranes and Western blot analysis using anti-E. coli Hyd-2 (1:20,000) antiserum were performed as described previously (41, 42).
Nondenaturing PAGE was performed using 5% (wt/vol) polyacrylamide gels, pH 8.5, and included 0.1% (wt/vol) Triton X-100 in the gels. The buffer used for electrophoresis included 0.1 M Tris-HCl and 0.1 M glycine, pH 8. Protein samples were incubated with 5% (wt/vol) Triton X-100 immediately prior to application to the gels (Bio-Rad), and electrophoresis was carried out at 80 V for 2 to 3 h. Hydrogenase activity staining was done as described previously (41, 43), except that the buffer used was 50 mM MOPS, pH 7.0.
Determination of total hydrogenase enzyme activity.
Hydrogenase enzyme activity (H2-dependent reduction of benzyl viologen [BV]) determines the sum of the activities of Hyd-1, Hyd-2, and Hyd-3 and was measured according to the method in reference 41, except that the buffer used was 50 mM MOPS, pH 7.0. No detergent was added to extracts measured by this method. The wavelength used was 578 nm, and a molar extinction coefficient (EM) of 8,600 M−1 cm−1 was assumed for reduced BV. One milliunit of activity corresponded to the reduction of 1 nmol of hydrogen per min. Experiments were performed at least three times and each time in triplicate. Data are presented as standard deviations of the means.
RESULTS
Previous studies have shown that HypC can be isolated in a complex with the precursor form of HycE (28) while its paralog HybG interacts with pro-HybC (30, 31). To address whether the 15-amino-acid C-terminal peptide on pro-HybC is important for NiFe cofactor insertion, whether it controls the interaction of the large subunit with the small subunit pre-HybO, and whether it influences the subsequent Tat-dependent membrane translocation of the HybC-HybO heterodimer, two derivatives of N-terminally Strep-tagged HybC were constructed (see scheme in Fig. 1A). The first derivative was equivalent to the precursor of HybC (termed pro-HybC), while the second derivative lacked the final 15 amino acids at the C terminus and was a genetically processed species (termed HybCproc), equivalent in primary structure to the mature form of the subunit found in the active Hyd-2 enzyme (3, 12, 44).
The presence of the C-terminal peptide on pro-HybC prevents Tat-dependent cleavage of the signal peptide on pre-HybO.
Western blot analysis of the large and small subunits of Hyd-2 in an extract derived from a mutant that lacks the gene encoding HypF [the mutant fails to synthesize the Fe(CN)2CO moiety of the NiFe cofactor (14, 34)] exhibited only the precursor form (pre-HybO) of the small subunit in very low abundance (Fig. 2A). An apparent degradation product of pre-HybO was observed in a mutant lacking HybC. It should be noted that the appearance of pre-HybO in these mutants was always variable, and it was usually at a low level, particularly in stationary-phase cells. This is presumably because of rapid turnover due to proteolysis. It should be stressed that the processed form of HybO was never observed in either the hybC or the hypF mutant.
FIG 2.

NiFe-cofactor-free maturation of a hydrogenase large-small subunit heterodimer. (A) Western blot analysis using anti-Hyd-2 antibodies after denaturing SDS-PAGE of crude extracts (30 μg protein) of the precursor (pro-HybC) and mature forms of the Hyd-2 large and small subunits. wt, extract from the E. coli wild-type strain MC4100; hybC, extract from a ΔhybC mutant, CP695; hybO, extract from strain CP793 (ΔhyaB ΔhycE ΔhybO), which lacks the catalytic subunit of Hyd-1 and Hyd-3 but retains a chromosomal copy of hybC; hypF, extract from mutant DHPF2. The asterisk at the right indicates an unidentified cross-reacting polypeptide, and the migration positions of pro-HybC, HybCproc, pre-HybO, and HybO are also shown. (B) Effect of introducing multicopy pro-HybC or HybCproc into a hybC mutant strain or a hypF maturation-defective mutant strain. A Western blot assay is shown in which aliquots of crude extracts (30 μg protein) derived from the strains described above transformed with plasmid pASK-hybC or pASK-hybCproc were separated in a denaturing 10% (wt/vol) polyacrylamide gel and probed with antiserum raised against Hyd-2. (C) Hydrogenase activity-stained gel after nondenaturing PAGE of the same samples as those used for panel B. The gel was stained using H2 and the redox dyes benzyl viologen and triphenyl tetrazolium chloride. The migration position of the active Hyd-2 enzyme complex is indicated.
In the hypF mutant, only the precursor form of HybC (pro-HybC) was observed (Fig. 2A). Similar polypeptide profiles were observed in hypE and hybG mutants (see Fig. S1 in the supplemental material). Surprisingly, in hypD and hypB mutants pro-HybC was virtually undetectable, suggesting more rapid or complete degradation of this species when the respective gene products were missing. Processed HybO was never detected in extracts of these mutants (see Fig. S1; also data not shown).
Together, these findings indicate that if the 15-amino-acid oligopeptide at the C terminus of the large subunit is not proteolytically removed, normally signifying that maturation is complete (3), or if the large subunit is absent from the cells, then the Tat signal peptide is not removed from pre-HybO and it is degraded. Furthermore, these results suggest that direct physical contact between the large and small subunits likely stabilizes HybO against degradation.
Next, we tested what effect the genetic removal of the C-terminal peptide from pro-HybC (HybCproc) had on pre-HybO maturation in a hypF mutant. As a control, it was demonstrated that no processing of pro-HybC occurred in the mutant (Fig. 2B). Consequently, the Tat signal peptide on the pre-HybO small subunit was not removed (Fig. 2B), signifying that no Tat transport occurred (9). Although the genetically foreshortened HybCproc polypeptide also cannot be supplied with a NiFe cofactor in the hypF mutant, nonetheless, in this instance the pre-HybO polypeptide was processed to the mature HybO species (Fig. 2B). This result indicates that the C-terminal peptide on the large subunit prevents further maturation of pre-HybO and consequently assembly of the HybC-HybO heterodimer. To demonstrate the validity of this proposal, a similar result was obtained when the same experiment was performed in a ΔhybG genetic background (see Fig. S2 in the supplemental material), which also fails to complete NiFe cofactor biosynthesis specifically for Hyd-2 (30).
The C-terminal peptide on pro-HybC is required to generate a fully active hydrogenase enzyme.
We wished to determine whether the Hyd-2 enzyme synthesized with the genetically processed HybCproc polypeptide in a Hyp-competent genetic background exhibited hydrogenase enzyme activity. The plasmid encoding HybCproc was introduced into strain FTD147, which lacks the genes encoding the large subunit of Hyd-1, Hyd-2, and Hyd-3 (45). The in-gel hydrogenase activity of Hyd-2 in an extract derived from this strain was determined after separation of protein complexes by native PAGE (41). This hydrogenase activity was compared with that obtained when a plasmid encoding pro-HybC polypeptide was introduced into the strain (Fig. 2C). While the extract derived from the strain encoding pro-HybC resulted in active Hyd-2 (Fig. 2C), essentially no activity could be detected when FTD147 was transformed with a plasmid encoding HybCproc (Fig. 2C), despite the fact that the signal peptide on HybO had been removed (Fig. 2B). Quantitative determination of hydrogen-dependent benzyl viologen-reducing activity of Hyd-2 in the extracts derived from these strains revealed a specific activity of 180.2 ± 3.7 mU (mg protein−1) for the strain that synthesized pro-HybC. In contrast, the enzyme reconstituted with HybCproc had an activity that varied between an undetectable level and a maximal activity of 8.3 ± 0.4 mU (mg protein−1). As a negative control, the same plasmids transformed into the hypF mutant failed to result in any detectable Hyd-2 enzyme activity either after in-gel activity staining (Fig. 2C) or after hydrogen-dependent dye-reducing activity was measured in a cuvette assay (data not shown). Together, these results suggest that genetically preprocessed HybCproc lacking the 15-amino-acid, C-terminal peptide has a reduced ability to recruit the Hyp maturation machinery in vivo. Lack of Hyp recruitment would hinder efficient maturation of Hyd-2, supporting previous findings (17, 18, 28, 30).
Tat-dependent membrane association of the HybC-HybO heterodimer lacking the NiFe cofactor.
Next, we wished to determine whether the HybC-HybO heterodimer synthesized with an artificially processed HybCproc polypeptide was membrane associated and whether membrane association was Tat dependent. The plasmids encoding the pro-HybC and HybCproc variants were introduced into a strain lacking the hybC gene. After anaerobic growth and subcellular fractionation, the localization of Hyd-2 was determined. Regardless of whether the strain carried a plasmid encoding pro-HybC or HybCproc, processed HybC was detected in the membrane fractions and processed HybO was also associated with the membrane fraction in both strains (Fig. 3A). This result demonstrates that in a Hyp- and Tat-competent strain pro-HybC is processed and associates with pre-HybO and the heterodimer becomes associated with the membrane, as signified by the cleavage of the Tat signal peptide. HybCproc can also associate with pre-HybO and is introduced into the membrane, again based on the maturation of HybO. Significant amounts of both pro-HybC and HybCproc were also detected in the soluble cytoplasmic fraction, but no processed HybO small subunit was apparently present in this fraction (Fig. 3A). This probably results from overproduction of the large subunit, which could not be transported across the membrane due to a limitation in chromosomally encoded, Tat-dependent pre-HybO.
FIG 3.

Tat-dependent membrane association of NiFe-cofactor-free Hyd-2. (A) Western blot analysis using anti-Hyd-2 antiserum of crude extracts (30 μg protein) from CP695 (ΔhybC) or strain DADE (ΔtatA-E [deletion of tatA through tatE genes]) transformed either with a plasmid encoding pro-HybC or with a plasmid encoding artificially matured HybCproc. SF, soluble fraction; MF, membrane fraction. The asterisk denotes an unidentified membrane-associated cross-reacting polypeptide species. The migration positions of pro-HybC and HybC (equivalent to HybCproc) have been generically labeled HybC because the respective bands were not distinguished in the experiment. The migration position of mature HybO is shown. (B) Western blot analysis of crude extracts (30 μg protein) from anaerobically grown strain CB10 (ΔhypF ΔhybC) transformed with a plasmid encoding either pro-HybC or genetically matured HybCproc. Detection was performed using anti-Hyd-2 antiserum. The first lane shows a membrane fraction (MF) derived from strain FTD147, which lacks the large subunit of Hyd-1, Hyd-2, and Hyd-3. SF, soluble fraction. The asterisk denotes an unidentified cross-reacting species.
Notably, when either plasmid was introduced into strain CB10, lacking both hybC and hypF, and which is therefore unable to synthesize the NiFe cofactor, both HybCproc and processed HybO were nevertheless associated with the membrane, while pro-HybC was present in the soluble cytoplasmic fraction when the strain was transformed with plasmid pASK-hybC (Fig. 3B); no HybO polypeptides were detected in this soluble fraction, suggesting that they were degraded.
Proteolytic treatment of spheroplasts derived from strains with membrane-associated HybC plus HybO and HybCproc plus HybO indicated that the large subunits were sensitive to treatment with trypsin (Fig. 4), which strongly suggests that the catalytic subunit was exposed to the periplasmic side of the membrane (46). Together, these findings demonstrate that the HybC-HybO heterodimer lacking the NiFe cofactor can be integrated into the cytoplasmic membrane.
FIG 4.

The Hyd-2 enzyme species, including NiFe-cofactor-free HybCproc, is sensitive to trypsin in E. coli spheroplasts. Anaerobically grown E. coli cells (50-ml cultures) were harvested by centrifugation for 5 min at 4,000 rpm at room temperature. The cell pellet was resuspended in 1 ml of 25% (wt/vol) sucrose, 30 mM Tris, pH 8.0, at room temperature. A 20-μl aliquot of 250 mM EDTA (pH 8.0) and 20 μl of freshly prepared lysozyme solution (10 mg/ml) were added, and the suspension was incubated for 3 min at room temperature. The spheroplasts were harvested by centrifugation at 15,000 × g for 30 s at 4°C. The supernatant contains the periplasm. The following steps were carried out at 4°C except as noted otherwise. Spheroplasts (pellet) were gently resuspended in the above-described buffer, trypsin (0.025%) was added, and the suspension was incubated for 10 min at room temperature. Trypsin inhibitor was added to a final concentration of 0.5% (wt/vol). Spheroplasts were separated by centrifugation at 15,000 × g for 10 min at 4°C. The pellet was resuspended in the same buffer, and after sonication the soluble cytoplasmic fraction (SF) and the membrane fraction (MF) were prepared by ultracentrifugation (2 h at 120,000 × g). Aliquots of the cytoplasmic and membrane fractions (50 μg of protein) from strains MC4100 (wild type) (A), FTD147 carrying plasmid p-HybCproc (HybCproc) (B), and FTD147 carrying plasmid p-proHybC (pro-HybC) (C) were separated by 12.5% (wt/vol) PAGE, and after transfer to nitrocellulose membranes, the blots were challenged with anti-Hyd-2 antiserum. The plus and minus signs indicate treatment with trypsin and nontreatment, respectively. The arrow indicates the migration position of processed HybC.
Introduction of the plasmid encoding HybCproc into a hybC mutant that also carried a tat operon deletion (Table 1) revealed that neither HybCproc nor HybO was associated with the cytoplasmic membrane (Fig. 3A). This demonstrates that association of the genetically matured HybCproc-HybO heterodimer (lacking the NiFe cofactor) with the cytoplasmic membrane was Tat dependent.
A hybrid pro-HybC carrying a C-terminal 15-amino-acid peptide from HyaB is activated by the HybG-Hyp machinery.
Like pro-HybC, pro-HyaB, the catalytic subunit of Hyd-1, has a 15-amino-acid C-terminal extension that is proteolytically cleaved after successful insertion of the NiFe cofactor into the active site of the subunit (3). The C-terminal amino acid sequence on pro-HyaB is -VLGDDGSELISVQVR, while that on pro-HybC is -VVDADGNEVVSVKVL. In contrast to pro-HybC, which requires the cochaperone HybG for NiFe cofactor insertion, pro-HyaB can be activated either by HybG or by its paralog HypC (30). Therefore, to determine whether the C-terminal peptide potentially acts as a binding platform for the Hyp machinery, we swapped the 15-amino-acid peptide from pro-HyaB onto HybC and vice versa (schematically represented in Fig. 1B). Transformation of the plasmid encoding pro-HybCHyaB (pASK-hybChyaB in Table 1) into FTD147 (ΔhyaB ΔhybC ΔhycE) followed by determination of hydrogenase activity after native PAGE revealed that the hybrid pro-HybCHyaB variant delivered active Hyd-2 enzyme (Fig. 5A). The active enzyme migrated slightly more slowly than the wild-type enzyme because it also carries a Strep tag. Analysis of the extracts derived from this strain by Western blotting with anti-Hyd-2 antiserum revealed that processed HybC and processed HybO could both be detected (Fig. 5B).
FIG 5.

Introduction of the C-terminal peptide from HyaB onto pro-HybC delivers active Hyd-2 enzyme. (A) A native PAGE gel stained for hydrogenase activity using benzyl viologen-triphenyl tetrazolium chloride is shown. Samples of crude extracts (50 μg of protein) derived from MC4100 and FTD147 either lacking a plasmid or carrying a plasmid encoding pro-HyaBHybC or pro-HybCHyaB were loaded on the gel. (B) The same samples used for panel A were separated by 10% (wt/vol) SDS-PAGE and after transfer to nitrocellulose were challenged with anti-Hyd-2 antiserum. The migration positions of the respective polypeptides are indicated at the right. (C) The same samples used for panel A were stained for Hyd-1 enzyme activity with phenazine methosulfate-nitroblue tetrazolium dyes in the presence of H2.
In the reciprocal experiment, FTD147 transformed with a plasmid (pASK-hyaBhybC in Table 1) encoding pro-HyaBHybC, which carries the 15-amino-acid peptide derived from pro-HybC, also exhibited Hyd-1 activity, albeit significantly weaker than that for the wild type (Fig. 5C). The results of these experiments indicate that the C-terminal peptides from HyaB and HybC are exchangeable and retain functionality, allowing maturation of active enzymes.
To determine whether the respective HybG/HypC specificity of these hybrid proproteins was retained or also exchanged along with the C-terminal peptides, strain CB15 (ΔhyaB ΔhycE ΔhybG) was constructed and transformed with the appropriate plasmids, and after growth of the strains anaerobically, crude extracts were analyzed by native PAGE followed by hydrogenase activity staining. Neither native pro-HybC nor pro-HybCHyaB exhibited significant activity (data not shown). In Western blot analysis, extracts derived from these strains were challenged with anti-Hyd-2 antiserum, and the results confirmed that only the precursor forms of both HybC and HybO were detectable (Fig. 6). This result indicates that the hybrid pro-HybCHyaB variant retained its dependence on HybG. Together, these results strongly suggest that the function of the C-terminal peptide is to facilitate formation of the appropriate conformation of the apo-catalytic subunit for NiFe cofactor insertion and that it appears not to act as a specific binding site to recruit the Hyp machinery.
FIG 6.
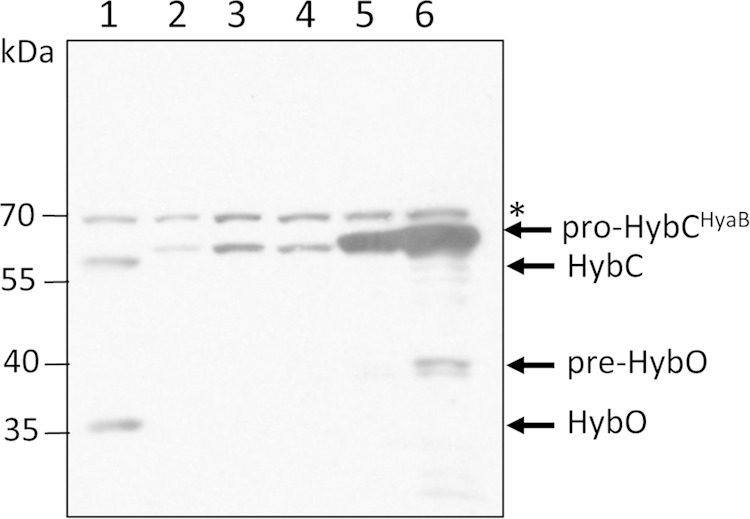
Pro-HybCHyaB carrying the 15-amino-acid C terminus from HyaB retains maturation specificity for HybG. A Western blot assay is shown in which crude extracts (50 μg of protein) derived from E. coli strain CB15 (ΔhyaB ΔhycE ΔhybG) carrying a plasmid encoding different HybC variants were challenged with anti-Hyd-2 antiserum. The migration positions of the polypeptides identified are indicated at the right. The asterisk denotes an unidentified cross-reacting species that acted as a loading control. Lane 1, MC4100 (wild type); lane 2, CB15 (ΔhyaB ΔhycE ΔhybG); lane 3, CB15 carrying pASK-hyaB; lane 4, CB15 carrying pASK-hyaBhybC, encoding pro-HyaBHybC; lane 5, CB15 carrying pASK-hybC; lane 6, CB15 carrying pASK-hybChyaB, encoding pro-HybCHyaB.
The hybrid pro-HybC carrying the 15-amino-acid C-terminal peptide from HyaB retains its dependence on HybD for proteolytic cleavage.
Insertion of the nickel ion into the active site during [NiFe]-hydrogenase maturation occurs after insertion of the Fe(CN)2CO moiety of the cofactor has taken place (3, 17, 19) and is the signal for proteolytic cleavage by a hydrogenase-specific protease. The protease specific for HybC is HybD, and that for HyaB is HyaD (44). To determine whether the C-terminal peptide of HybC and HyaB directs the recognition of the cleavage site by the respective protease, plasmid pASK-hybChyaB encoding pro-HybCHyaB was introduced into strain CB16 (ΔhyaD ΔhybC ΔhycE ΔhyfB-H [deletion of the hyfB through hyfH genes]). As controls, plasmids pASK-hybC, pASK-hyaB, and pASK-hyaBhybC were also introduced into the strain, and after anaerobic growth in minimal medium to stationary phase, crude extracts derived from cells were analyzed by nondenaturing PAGE (Fig. 7A). Hyd-2 activity was clearly observed for the wild-type strain MC4100 and CB16 transformed with pASK-hybC and with pASK-hybChyaB, while neither pASK-hyaB nor pASK-hyaBhybC delivered active Hyd-1 enzyme. To confirm HybD-dependent processing of pro-HybCHyaB, a Western blot assay of the same extracts using anti-Hyd-2 antiserum clearly revealed the processed forms of both the HybC and HybO polypeptides in strain CB16 transformed with pASK-hybChyaB (Fig. 7B). Together, these results demonstrate that the HybC hybrid carrying the 15-amino-acid C terminus from HyaB was recognized and cleaved by HybD, indicating that the C-terminal peptide is unlikely to be recognized directly by the HybD protease.
FIG 7.
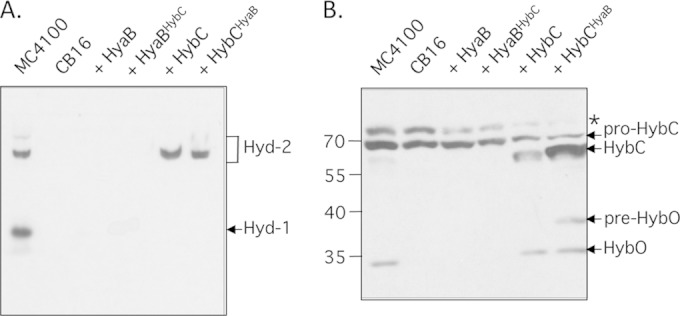
Pro-HybCHyaB retains dependence on the HybD protease. (A) A native PAGE gel stained for hydrogenase activity using benzyl viologen-triphenyl tetrazolium chloride is shown. Samples of crude extracts (50 μg of protein) derived from MC4100 and CB16 (ΔhyaD ΔhybC ΔhycE ΔhyfB-hyfH) either lacking a plasmid or, in the case of CB16, carrying a plasmid encoding pro-HyaBHybC or pro-HybCHyaB were loaded on the gel. (B) The same samples used for panel A were separated in a 12.5% (wt/vol) SDS gel and after transfer to nitrocellulose were challenged with anti-Hyd-2 antiserum. The migration positions of the respective polypeptides are indicated at the right.
DISCUSSION
A C-terminal peptide directs hydrogenase maturation.
In this study, we have shown that cleavage, or the lack thereof, of the 15-amino-acid C-terminal peptide on HybC, the large subunit of E. coli Hyd-2, determines whether the large subunit and electron-transferring small subunit assemble and whether this heterodimer is translocated across the cytoplasmic membrane by the Tat machinery. This agrees with earlier data that suggested that a change in conformation of the Hyd-3 large subunit HycE takes place after insertion of the NiFe cofactor and subsequent proteolytic removal of the C-terminal peptide (17). The findings presented here strongly support the hypothesis that simply the removal of the peptide is sufficient to cause the hypothesized conformational change, regardless of whether the cofactor has been inserted into the catalytic subunit or not. This is because genetic removal of the 15-amino-acid C-terminal peptide, delivering a polypeptide equivalent in primary structure to the processed large subunit in the mature enzyme, resulted in an inactive Hyd-2 enzyme that was nevertheless translocated across the membrane by the Tat machinery. Previous studies (28) have demonstrated that components of the Hyp machinery interact only with the unprocessed form of the large subunit, which led to the hypothesis that removal of the C-terminal peptide might cause a conformational change in the protein, thus closing the active site (17, 44). Notably, attempts to add back the C-terminal peptide in trans failed to restore maturation to the genetically foreshortened HybC subunit (C. Thomas and R. G. Sawers, unpublished observations), indicating that the peptide must be physically attached to the rest of HybC to effect NiFe cofactor insertion. This suggests that the C-terminal peptide somehow sterically prevents pro-HybC from adopting the conformation of the mature polypeptide.
Swapping of the 15-amino-acid peptide from HyaB, the Hyd-1 catalytic subunit, still allowed maturation of HybC into an active enzyme. Moreover, this peptide did not influence the requirement for HybG to allow NiFe cofactor incorporation, nor did it alter the specificity for the HybC-specific protease HybD. Pro-HybC is dependent on HybG for cofactor insertion, while pro-HyaB can use either HybG or its paralog HypC (30). HyaB fused with the 15-amino-acid C-terminal peptide derived from HybC was also partially functional, and the dependence on HypC or HybG was retained (data not shown). Despite sharing identity in 7 of the 15 amino acids, the C-terminal peptides are sufficiently different between HyaB and HybC to suggest that they do not act either as a direct interaction surface for the Hyp machinery or as a recognition sequence for the hydrogenase-specific protease. This agrees with previous data that showed that HypC from E. coli interacted with Cys241 within the active site cavity of pro-HycE, the apo-catalytic subunit of Hyd-3 from E. coli (17). That the Hyp proteins do not interact with the C-terminal peptide is also supported by the fact that glutathione S-transferase (GST) fused to the C-terminal 15 amino acids failed to interact with any Hyp protein (Thomas and Sawers, unpublished). Rather, the function of this C-terminal peptide appears to be that of an intramolecular chaperone, as has been previously suggested (3, 17), which prevents a conformational change in the polypeptide from occurring. How this peptide might be able to achieve this will require further extensive study.
It is also apparent that the conformation of the protein that is adopted with the C-terminal peptide attached prevents premature assembly of the αβ heterodimer complex in vivo. This also correlates with an early finding that HypC had to leave the pro-HycE protein before proteolytic cleavage could occur (18). Such a mechanism would ensure that in wild-type cells only a large subunit with a complete NiFe cofactor proceeds along the maturation pathway. After synthesis and insertion of the Fe(CN)2CO moiety into the apo-large subunit by the HybG-HypDEF machinery (26, 27), nickel insertion occurs (16, 19). This step signifies completion of cofactor biosynthesis. Nickel serves as the substrate recognition motif for the HybC-specific endoprotease HybD, ensuring transition metal fidelity (47, 48). Upon cleavage of the peptide (model in Fig. 8, scenario 1), the fully folded and cofactor-containing HybC large subunit engages with the FeS cluster-containing pre-HybO small subunit to displace the HybE private chaperone. HybE has been proposed to mask the Tat signal peptide (9, 10), releasing the heterodimer for transport by the Tat translocon. We could show that in a strain unable to synthesize HybC, no maturation of pre-HybO occurred and this precursor was degraded. This result indicates that the presence of the mature large subunit is somehow necessary to expose the Tat signal peptide on pre-HybO to allow interaction with the Tat translocon. If the 15-amino-acid C-terminal peptide of the large subunit cannot be cleaved off, for example, because of nickel limitation or incomplete Fe(CN)2CO biosynthesis, no maturation of pre-HybO occurs (Fig. 8, scenario 2). Together, these findings suggest that only the mature large subunit can displace HybE efficiently from pre-HybO. In support of this, it has been shown previously (9) that a strain lacking HybE targeted the pre-HybO protein prematurely to the membrane via the Tat translocon without the associated HybC large subunit. Moreover, it has been shown in R. eutropha that deletion of the gene encoding the HybE homolog, HoxO, also renders the precursor of the small subunit of the membrane-bound hydrogenase subject to degradation (49). Together, these data support a universal role for this class of private chaperone in masking the Tat signal sequence from the translocase.
FIG 8.
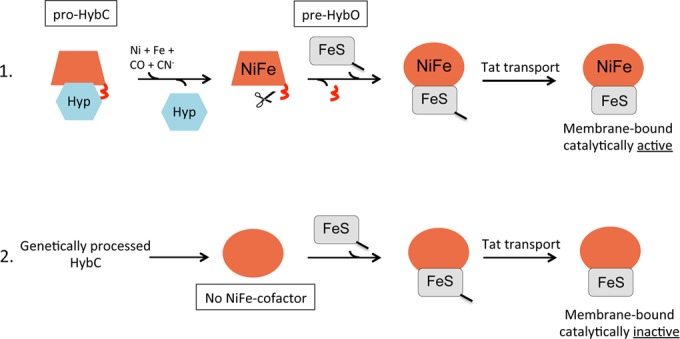
Model of the role of the C-terminal peptide in controlling [NiFe]-hydrogenase assembly. Scenario 1 depicts the situation in wild-type bacteria where the pro-HybC has received the completely synthesized NiFe cofactor, which leads to proteolytic cleavage (red squiggle) catalyzed by HybD protease (signified by the scissors). The hexagon labeled Hyp denotes the HypDEF complex plus HybG in the case of pro-HybC maturation. The line attached to pre-HybO signifies the Tat signal peptide. The change from the trapezoid to the ellipse signifies the hypothesized conformational change of pro-HybC to processed HybC. Scenario 2 depicts what happens when the C-terminal peptide of HybC is removed genetically, which enables interaction of HybCproc with pre-HybO and transport of the NiFe-cofactor-free heterodimer by the Tat translocon. Despite the lack of the NiFe cofactor, it is uncertain whether the active site of the large subunit is “empty.”
Tat-dependent translocation of a NiFe-cofactor-free hydrogenase heterodimer.
Our findings suggest that “proofreading” or “quality control” of the HybC-pre-HybO heterodimer occurs at the level of enzyme maturation and assembly, obviating the requirement to invoke an involvement of the Tat machinery directly in this process. The fact that we were able to dupe the Tat translocon into transporting a HybC-HybO heterodimer lacking a NiFe cofactor suggests that the Tat translocon might not necessarily need to “control” the folding quality of the delivered substrate. Furthermore, this finding suggests that the HybC-HybO heterodimer lacking the NiFe cofactor has the native conformation when delivered to the Tat translocon. Thus, for the Tat translocon, the substrate “appears” correctly folded. Proof that the Hyd-2 large subunit lacked the NiFe cofactor was delivered by demonstrating in a maturation-defective genetic background that the HybCproc-HybO heterodimer was associated with the cytoplasmic membrane and that this was Tat dependent (Fig. 8, scenario 2).
At this juncture, we cannot conclude that the active site of the NiFe-cofactor-free HybC subunit is “empty,” because it was shown previously (50) that overproduction of a pro-HybC variant, which cannot undergo proteolytic cleavage due to the presence of an additional 5 amino acids attached to its C terminus, can accept a [4Fe-4S] cluster. Future studies directed at determining the structure of the 15-amino-acid peptide, along with computer-based modeling, will provide important information as to how this short peptide is capable of influencing the conformation of pro-HybC. Interestingly, a recent study has demonstrated that the activity of the MCAK motor protein, which is involved in microtubule depolymerization, is regulated by a C-terminal conformational switch, perhaps suggesting that control of protein conformation by short C-terminal peptides is more widespread than previously considered (51).
Tat-dependent transport of an aberrantly matured enzyme has also been shown for a molybdenum-cofactor-free variant of dimethyl sulfoxide (DMSO) reductase in Rhodobacter sphaeroides WS8 (52). In this case, however, it was reported that insertion of a precursor of the cofactor might have facilitated folding sufficiently to allow Tat-mediated translocation. Whether another cofactor, such as a FeS cluster, can be temporarily inserted into the active site cavity of HybCproc in vivo in a Hyp− strain remains to be determined.
A common mechanism ensuring correct spatiotemporal assembly of modular oxidoreductases?
While a mechanism involving control of enzyme assembly probably holds true for most hydrogenase large subunits with a C-terminal extension, nevertheless, there are examples of hydrogenases whose large subunit lacks a C-terminal extension. These include the regulatory hydrogenase from R. eutropha (53) and the energy-converting hydrogenase (Ech) from extreme thermophiles and methanogens (54). Precisely how insertion of the NiFe cofactor and subunit assembly are coordinated in these enzymes remains to be established. Based on the findings presented here, however, it is possible that either the hydrogenase-specific Hyp machinery in these organisms recognizes a conformation similar to that of HybCproc or these large subunits retain a conformation similar to pro-HybC, even in the absence of the C-terminal peptide. A detailed biophysical analysis of the isolated apo-large subunits should resolve this question.
How do other modular oxidoreductases, which do not employ the same mechanism as [NiFe]-hydrogenases, coordinate the biosynthesis and insertion of their active site cofactor with enzyme assembly and membrane translocation? In a recent study by James et al. (55), it was shown that both the catalytic subunit TtrA and the electron transfer subunit TtrB of the heterotrimeric molybdoenzyme tetrathionate reductase from Salmonella enterica serovar Typhimurium carry a Tat signal sequence. Either of these Tat signal peptides could compensate for the absence of the other. However, in the absence of the TtrA subunit, the Tat signal peptide of the TtrB subunit also remained unprocessed (55), leading to the conclusion that processing of the TtrB Tat signal peptide depends on assembly of its partner protein, TtrA. This is analogous to the control of cofactor insertion and enzyme assembly that we have observed here for Hyd-2, but employing a slightly different mechanism. Together, these results suggest that modular membrane-associated oxidoreductases might all employ spatiotemporal control of cofactor synthesis, insertion, and subunit assembly. This can be achieved either through the use of a cleavable C-terminal peptide in the case of many [NiFe]-hydrogenases, through two separate Tat-targeting peptides in the case of tetrathionate reductase (55), or through private chaperones coupled with Tat proofreading and protein-folding quality control in the case of the molybdoenzymes periplasmic nitrate reductase, trimethylamine oxide (TMAO) reductase, and formate dehydrogenase (2).
Pro-HybC interacts with minimally three accessory proteins or protein complexes during hydrogenase assembly. These accessory proteins include the HybG-HypD-HypE (HypF) complex (31), HybE (8), and the HybD protease (44, 56). Moreover, it is likely that the nickel delivery complex of HypB, HybF, and SlyD (3, 12) also interacts with pro-HybC, as HypB has been shown to interact with the large subunit of the H2-sensing Hyd of R. eutropha (19). Future biochemical studies will be required to elucidate how and where on pro-HybC these proteins interact. Nonetheless, it is apparent from the findings of this study that the C-terminal peptide on the hydrogenase catalytic subunit governs the complete sequence of events during the maturation process by acting as a central checkpoint ensuring that only the cofactor-containing mature large subunit can continue on the assembly line to active [NiFe]-hydrogenase.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Basem Soboh, Matthias Boll, Thomas Brüser, and Frank Sargent for fruitful discussions; Frank Sargent for supplying anti-Hyd-2 antiserum and strains; and Monique Jaroschinsky also for providing strains.
This work was supported by grants from EFRE funds of the European Union and the Deutsche Forschungsgemeinschaft (SA-494-3-2).
All authors declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00437-15.
REFERENCES
- 1.Sargent F. 2007. Constructing the wonders of the bacterial world: biosynthesis of complex enzymes. Microbiology 153:633–651. doi: 10.1099/mic.0.2006/004762-0. [DOI] [PubMed] [Google Scholar]
- 2.Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol 10:483–496. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- 3.Böck A, King PW, Blokesch M, Posewitz MC. 2006. Maturation of hydrogenases. Adv Microb Physiol 51:1–71. doi: 10.1016/S0065-2911(06)51001-X. [DOI] [PubMed] [Google Scholar]
- 4.Fritsch J, Lenz O, Friedrich B. 2013. Structure, function and biosynthesis of O2-tolerant hydrogenases. Nat Rev Microbiol 11:106–114. doi: 10.1038/nrmicro2940. [DOI] [PubMed] [Google Scholar]
- 5.Soboh B, Sawers RG. 2013. [NiFe]-hydrogenase cofactor assembly, p 507–516. In Culotta V. (ed), Metals in cells. John Wiley & Sons Inc., Hoboken, NJ. [Google Scholar]
- 6.Vignais PM, Billoud B. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 107:4206–4272. doi: 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- 7.Lubitz W, Ogata H, Rüdiger O, Reijerse E. 2014. Hydrogenases. Chem Rev 114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 8.Dubini A, Sargent F. 2003. Assembly of Tat-dependent [NiFe] hydrogenases: identification of precursor-binding accessory proteins. FEBS Lett 549:141–146. doi: 10.1016/S0014-5793(03)00802-0. [DOI] [PubMed] [Google Scholar]
- 9.Jack RL, Buchanan G, Dubini A, Hatzixanthis K, Palmer T, Sargent F. 2004. Coordinating assembly and export of complex bacterial proteins. EMBO J 23:3962–3972. doi: 10.1038/sj.emboj.7600409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack RL, Dubini A, Palmer T, Sargent F. 2005. Common principles in the biosynthesis of diverse enzymes. Biochem Soc Trans 33:105–107. doi: 10.1042/BST0330105. [DOI] [PubMed] [Google Scholar]
- 11.Dubini A, Pye RL, Jack RL, Palmer T, Sargent F. 2002. How bacteria get energy from hydrogen: a genetic analysis of periplasmic hydrogen oxidation in Escherichia coli. Int J Hydrogen Energy 27:1413–1420. doi: 10.1016/S0360-3199(02)00112-X. [DOI] [Google Scholar]
- 12.Forzi L, Sawers RG. 2007. Maturation of [NiFe]-hydrogenases in Escherichia coli. Biometals 20:565–578. doi: 10.1007/s10534-006-9048-5. [DOI] [PubMed] [Google Scholar]
- 13.Pinske C, Sawers RG. 2014. The importance of iron in the biosynthesis and assembly of [NiFe]-hydrogenases. Biomol Concepts 5:55–70. doi: 10.1515/bmc-2014-0001. [DOI] [PubMed] [Google Scholar]
- 14.Pinske C, Sawers RG. 2012. Delivery of iron-sulfur clusters to the hydrogen-oxidizing [NiFe]-hydrogenases in Escherichia coli requires the A-type carrier proteins ErpA and IscA. PLoS One 7:e31755. doi: 10.1371/journal.pone.0031755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menon AL, Robson RL. 1994. In vivo and in vitro nickel-dependent processing of the [NiFe] hydrogenase in Azotobacter vinelandii. J Bacteriol 176:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maier T, Böck A. 1996. Generation of active [NiFe] hydrogenase in vitro from a nickel-free precursor form. Biochemistry 35:10089–10093. doi: 10.1021/bi960567l. [DOI] [PubMed] [Google Scholar]
- 17.Magalon A, Böck A. 2000. Analysis of the HypC-HycE complex, a key intermediate in the assembly of the metal center of the Escherichia coli hydrogenase. J Biol Chem 275:21114–21120. doi: 10.1074/jbc.M000987200. [DOI] [PubMed] [Google Scholar]
- 18.Magalon A, Böck A. 2000. Dissection of the maturation reactions of the [NiFe] hydrogenase 3 from Escherichia coli taking place after nickel incorporation. FEBS Lett 473:254–25818. doi: 10.1016/S0014-5793(00)01542-8. [DOI] [PubMed] [Google Scholar]
- 19.Winter G, Buhrke T, Lenz O, Jones AK, Forgber M, Friedrich B. 2005. A model system for [NiFe] hydrogenase maturation studies: purification of an active site containing hydrogenase large subunit without small subunit. FEBS Lett 579:4292–4296. doi: 10.1016/j.febslet.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 20.Gollin DJ, Mortenson LE, Robson RL. 1992. Carboxyl-terminal processing may be essential for production of active NiFe hydrogenase in Azotobacter vinelandii. FEBS Lett 309:371–375. doi: 10.1016/0014-5793(92)80809-U. [DOI] [PubMed] [Google Scholar]
- 21.Sorgenfrei O, Linder D, Karas M, Klein A. 1993. A novel very small subunit of a selenium containing [NiFe] hydrogenase of Methanococcus voltae is post-translationally processed by cleavage at a defined position. Eur J Biochem 213:1355–1358. doi: 10.1111/j.1432-1033.1993.tb17888.x. [DOI] [PubMed] [Google Scholar]
- 22.Menon NK, Robbins J, Vartanian M, Patil D, Peck HD Jr, Menon AL, Robson RL, Przybyla AE. 1993. Carboxy-terminal processing of the large subunit of [NiFe] hydrogenases. FEBS Lett 331:91–95. doi: 10.1016/0014-5793(93)80303-C. [DOI] [PubMed] [Google Scholar]
- 23.Rossmann R, Sauter M, Lottspeich F, Böck A. 1994. Maturation of the large subunit (HycE) of Escherichia coli hydrogenase 3 requires nickel incorporation followed by C-terminal processing at Arg537. Eur J Biochem 220:377–384. doi: 10.1111/j.1432-1033.1994.tb18634.x. [DOI] [PubMed] [Google Scholar]
- 24.Kortlüke C, Friedrich B. 1992. Maturation of membrane-bound hydrogenase of Alcaligenes eutrophus H16. J Bacteriol 174:6290–6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blokesch M, Böck A. 2002. Maturation of [NiFe]-hydrogenases in Escherichia coli: the HypC cycle. J Mol Biol 324:287–296. doi: 10.1016/S0022-2836(02)01070-7. [DOI] [PubMed] [Google Scholar]
- 26.Bürstel I, Siebert E, Winter G, Hummel P, Zebger I, Friedrich B, Lenz O. 2012. A universal scaffold for synthesis of the Fe(CN)2(CO) moiety of [NiFe] hydrogenase. J Biol Chem 287:38845–38853. doi: 10.1074/jbc.M112.376947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soboh B, Stripp ST, Muhr E, Granich C, Braussemann M, Herzberg M, Heberle J, Sawers RG. 2012. [NiFe]-hydrogenase maturation: isolation of a HypC-HypD complex carrying diatomic CO and CN− ligands. FEBS Lett 586:3882–3887. doi: 10.1016/j.febslet.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 28.Drapal N, Böck A. 1998. Interaction of the hydrogenase accessory protein HypC with HycE, the large subunit of Escherichia coli hydrogenase 3 during enzyme maturation. Biochemistry 37:2941–2948. doi: 10.1021/bi9720078. [DOI] [PubMed] [Google Scholar]
- 29.Jones AK, Lenz O, Strack A, Buhrke T, Friedrich B. 2004. NiFe hydrogenase active site biosynthesis: identification of Hyp protein complexes in Ralstonia eutropha. Biochemistry 43:13467–13477. doi: 10.1021/bi048837k. [DOI] [PubMed] [Google Scholar]
- 30.Blokesch M, Magalon A, Böck A. 2001. Interplay between the specific chaperone-like proteins HybG and HypC in maturation of hydrogenases 1, 2, and 3 from Escherichia coli. J Bacteriol 183:2817–2822. doi: 10.1128/JB.183.9.2817-2822.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butland G, Zhang JW, Yang W, Sheung A, Wong P, Greenblatt JF, Emili A, Zamble DB. 2006. Interactions of the Escherichia coli hydrogenase biosynthetic proteins: HybG complex formation. FEBS Lett 580:677–681. doi: 10.1016/j.febslet.2005.12.063. [DOI] [PubMed] [Google Scholar]
- 32.Soboh B, Lindenstrauss U, Granich C, Javed M, Herzberg M, Thomas C, Stripp ST. 2014. [NiFe]-hydrogenase maturation in vitro: analysis of the roles of the HybG and HypD accessory proteins. Biochem J 464:169–177. doi: 10.1042/BJ20140485. [DOI] [PubMed] [Google Scholar]
- 33.Shomura Y, Higuchi Y. 2012. Structural basis for the reaction mechanism of S-carbamoylation of HypE by HypF in the maturation of [NiFe]-hydrogenases. J Biol Chem 287:28409–28419. doi: 10.1074/jbc.M112.387134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paschos A, Bauer A, Zimmermann A, Zehelein E, Böck A. 2002. HypF, a carbamoylphosphate-converting enzyme involved in [NiFe] hydrogenase maturation. J Biol Chem 277:49945–49951. doi: 10.1074/jbc.M204601200. [DOI] [PubMed] [Google Scholar]
- 35.Reissmann S, Hochleiter E, Wang H, Paschos A, Lottspeich F, Glass RS, Böck A. 2003. Taming of a poison: biosynthesis of the NiFe-hydrogenase cyanide ligands. Science 299:1067–1070. doi: 10.1126/science.1080972. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 37.Miller JA. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299. [DOI] [PubMed] [Google Scholar]
- 39.Pinske C, Krüger S, Soboh B, Ihling C, Kuhns M, Braussemann M, Jaroschinsky M, Sauer C, Sargent F, Sinz A, Sawers RG. 2011. Efficient electron transfer from hydrogen to benzyl viologen by the [NiFe]-hydrogenases of Escherichia coli is dependent on the coexpression of the iron-sulfur cluster-containing small subunit. Arch Microbiol 193:893–903. doi: 10.1007/s00203-011-0726-5. [DOI] [PubMed] [Google Scholar]
- 40.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Ballantine SP, Boxer DH. 1985. Nickel-containing hydrogenase isoenzymes from anaerobically grown Escherichia coli K-12. J Bacteriol 163:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinske C, Jaroschinsky M, Sargent F, Sawers RG. 2012. Zymographic differentiation of [NiFe]-hydrogenase 1, 2 and 3 of Escherichia coli K-12. BMC Microbiol 12:134. doi: 10.1186/1471-2180-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Theodoratou E, Huber R, Böck A. 2005. [NiFe]-hydrogenase maturation endopeptidase: structure and function. Biochem Soc Trans 33:108–111. doi: 10.1042/BST0330108. [DOI] [PubMed] [Google Scholar]
- 45.Redwood MD, Mikheenko IP, Sargent F, Macaskie LE. 2008. Dissecting the roles of Escherichia coli hydrogenases in biohydrogen production. FEMS Microbiol Lett 278:48–55. doi: 10.1111/j.1574-6968.2007.00966.x. [DOI] [PubMed] [Google Scholar]
- 46.Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. 1998. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J 17:3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Theodoratou E, Paschos A, Magalon A, Fritsche E, Huber R, Böck A. 2000. Nickel serves as a substrate recognition motif for the endopeptidase involved in hydrogenase maturation. Eur J Biochem 267:1995–1999. doi: 10.1046/j.1432-1327.2000.01202.x. [DOI] [PubMed] [Google Scholar]
- 48.Magalon A, Blokesch M, Zehelein E, Böck A. 2001. Fidelity of metal insertion into hydrogenases. FEBS Lett 499:73–76. doi: 10.1016/S0014-5793(01)02525-X. [DOI] [PubMed] [Google Scholar]
- 49.Schubert T, Lenz O, Krause E, Volkmer R, Friedrich B. 2007. Chaperone specific for the membrane-bound [NiFe]-hydrogenase interact with the Tat signal peptide of the small subunit precursor in Ralstonia eutropha H16. Mol Microbiol 66:453–467. doi: 10.1111/j.1365-2958.2007.05933.x. [DOI] [PubMed] [Google Scholar]
- 50.Soboh B, Kuhns M, Braussemann M, Waclawek M, Muhr E, Pierik AJ, Sawers RG. 2012. Evidence for an oxygen-sensitive iron-sulfur cluster in an immature large subunit species of Escherichia coli [NiFe]-hydrogenase 2. Biochem Biophys Res Commun 424:158–163. doi: 10.1016/j.bbrc.2012.06.096. [DOI] [PubMed] [Google Scholar]
- 51.Talapatra SK, Harker B, Welburn JP. 2015. The C-terminal region of the motor protein MCAK controls its structure and activity through a conformational switch. eLife 4:e06421. doi: 10.7554/eLife.06421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchanan G, Kuper J, Mendel RR, Schwarz G, Palmer T. 2001. Characterisation of the mob locus of Rhodobacter sphaeroides WS8: mobA is the only gene required for molybdopterin guanine dinucleotide synthesis. Arch Microbiol 176:62–68. doi: 10.1007/s002030100291. [DOI] [PubMed] [Google Scholar]
- 53.Friedrich B, Buhrke T, Burgdorf T, Lenz O. 2005. A hydrogen-sensing multiprotein complex controls aerobic hydrogen metabolism in Ralstonia eutropha. Biochem Soc Trans 33:97–101. doi: 10.1042/BST0330097. [DOI] [PubMed] [Google Scholar]
- 54.Soboh B, Linder D, Hedderich R. 2004. A multisubunit membrane-bound [NiFe]-hydrogenase and an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tengcongensis. Microbiology 150:2451–2463. doi: 10.1099/mic.0.27159-0. [DOI] [PubMed] [Google Scholar]
- 55.James MJ, Coulthurst SJ, Palmer T, Sargent F. 2013. Signal peptide etiquette during assembly of a complex respiratory enzyme. Mol Microbiol 90:400–414. doi: 10.1111/mmi.12373. [DOI] [PubMed] [Google Scholar]
- 56.Fritsche E, Paschos A, Beisel HG, Böck A, Huber R. 1999. Crystal structure of the hydrogenase maturating endopeptidase HybD from Escherichia coli. J Mol Biol 288:989–998. doi: 10.1006/jmbi.1999.2719. [DOI] [PubMed] [Google Scholar]
- 57.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 58.Jacobi A, Rossmann R, Böck A. 1992. The hyp operon gene products are required for the maturation of catalytically active hydrogenase isoenzymes in Escherichia coli. Arch Microbiol 158:444–451. [DOI] [PubMed] [Google Scholar]
- 59.Wexler M, Sargent F, Jack RL, Stanley NR, Bogsch EG, Robinson C, Berks BC, Palmer T. 2000. TatD is a cytoplasmic protein with DNase activity: no requirement for TatD family proteins in Sec-dependent protein export. J Biol Chem 275:16717–16722. doi: 10.1074/jbc.M000800200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.