

ABSTRACT
Iron availability functions as an environmental cue for enteropathogenic bacteria, signaling arrival within the human host. As enterotoxigenic Escherichia coli (ETEC) is a major cause of human diarrhea, the effect of iron on ETEC virulence factors was evaluated here. ETEC pathogenicity is directly linked to production of fimbrial colonization factors and secretion of heat-labile enterotoxin (LT) and/or heat-stable enterotoxin (ST). Efficient colonization of the small intestine further requires at least the flagellin binding adhesin EtpA. Under iron starvation, production of the CFA/I fimbriae was increased in the ETEC H10407 prototype strain. In contrast, LT secretion was inhibited. Furthermore, under iron starvation, gene expression of the cfa (CFA/I) and etp (EtpBAC) operons was induced, whereas transcription of toxin genes was either unchanged or repressed. Transcriptional reporter fusion experiments focusing on the cfa operon further showed that iron starvation stimulated cfaA promoter activity in ETEC, indicating that the impact of iron on CFA/I production was mediated by transcriptional regulation. Evaluation of cfaA promoter activity in heterologous E. coli single mutant knockout strains identified IscR as the regulator responsible for inducing cfa fimbrial gene expression in response to iron starvation, and this was confirmed in an ETEC ΔiscR strain. The global iron response regulator, Fur, was not implicated. IscR binding sites were identified in silico within the cfaA promoter and fixation confirmed by DNase I footprinting, indicating that IscR directly binds the promoter region to induce CFA/I.
IMPORTANCE Pathogenic enterobacteria modulate expression of virulence genes in response to iron availability. Although the Fur transcription factor represents the global regulator of iron homeostasis in Escherichia coli, we show that several ETEC virulence factors are modulated by iron, with expression of the major fimbriae under the control of the iron-sulfur cluster regulator, IscR. Furthermore, we demonstrate that the apo form of IscR, lacking an Fe-S cluster, is able to directly fix the corresponding promoter region. These results provide further evidence implicating IscR in bacterial virulence and suggest that IscR may represent a more general regulator mediating the iron response in enteropathogens.
INTRODUCTION
Iron is an essential nutrient for almost all organisms due to its ability to switch between two oxidative states. Implicated in cell processes including DNA replication, metabolism, and the response to oxidative stress, iron levels are tightly regulated to ensure cellular function while limiting production of damaging free radicals via the Fenton reaction. Within the human host, iron availability is limited due to insolubility at physiological pH and sequestration by iron binding proteins. To circumvent this challenge, enteropathogenic bacteria secrete high-affinity siderophores capable of scavenging both free and complexed ferric iron, among other strategies (1). However, bacteria can also take advantage of low-iron conditions to sense arrival within the host environment and to trigger virulence. Toxin gene expression in particular is induced in many bacteria, including Escherichia coli (α-hemolysin and Shiga-like toxin 1) and Vibrio cholerae (hemolysin), at low iron concentrations (2). As secreted toxins can induce cell lysis, this may represent an additional mechanism to increase iron availability. Indeed, several studies have found an increased risk of bacterial infection in pathologies with iron overload (3). This likely results from a combination of lowered intestinal barrier integrity and enhanced bacterial growth in the presence of iron, as seen for Vibrio vulnificus and Yersinia enterocolitica (4, 5). Recently, Salmonella enterica serovar Typhimurium growth and adhesion to epithelial cells were also shown to be enhanced in the presence of iron in vitro (6).
Enterotoxigenic Escherichia coli (ETEC) represents a major cause of diarrhea in children under 5 years of age in developing countries and is the leading cause of travelers' diarrhea (7). Virulence within the small intestine is characterized by the presence of one or more colonization factors (CFs), mediating adherence to the intestinal epithelium, and the secretion of heat-labile enterotoxin (LT) and/or heat-stable enterotoxin (ST), inducing aqueous diarrhea. Although the majority of studies have been performed on the prototype fimbrial colonization factor CFA/I, recent works have identified several additional factors that appear to be involved in host colonization by ETEC, including the EtpA protein, which localizes at the flagellar tip following secretion (8).
The cfaABCE (cfa) and eltAB operons, encoding the CFA/I fimbriae and the LT, are regulated in response to various environmental signals, including temperature and glucose, via the H-NS and cyclic AMP receptor protein (CRP) transcriptional regulators, respectively (9–13). cfa expression is further activated by the specific virulence regulator CfaD, although it remains unknown whether CfaD activation is dependent on any external cues (9). Production of CFA/I also responds to iron concentration in vitro (14). In contrast, the effect of iron on production of other virulence factors, including LT and ST, remains unclear.
Regulation of bacterial iron homeostasis is most commonly mediated either directly or indirectly by the ferric uptake regulator, Fur. In the presence of its cofactor Fe(II), Fur forms a homodimer capable of binding to a 19-bp A/T-rich palindromic motif or Fur box, thereby blocking gene expression (15). In the absence of iron, apo-Fur dissociates, permitting expression of target genes associated with iron capture and virulence. In contrast to Fur-mediated repression, gene activation is indirect in most bacterial species, including E. coli. In the presence of iron, Fur represses transcription of the small RNA RyhB, which would otherwise lead to targeted RNA degradation via recruitment of the degradosome (16).
However, other regulators are also implicated in the iron response. OxyR and SoxR mediate the oxidative stress response following exposure to hydrogen peroxide and superoxide, respectively. Both OxyR and SoxR activate fur expression to inhibit iron acquisition and thereby minimize oxidative stress, indicating a strong interplay between the iron and oxidative stress responses (17). Furthermore, SoxR possesses a [2Fe-2S] cluster for redox sensing and is dependent on Fe-S cluster biogenesis controlled by the iron-sulfur cluster regulator IscR (18). In contrast to the E. coli Fur repressor, IscR is capable of binding promoter regions in both holo and apo forms, based on the presence or absence of an internal [2Fe-2S] cluster (19). IscR can therefore also respond to both iron limitation and oxidative stress.
Both Fur and IscR are implicated in bacterial pathogenesis. A V. cholerae fur mutant shows reduced colonization in a murine model, as does a Campylobacter jejuni fur mutant in the chick model (20, 21). Similarly, a V. vulnificus iscR mutant has reduced virulence in vivo (22, 23), while a Yersinia pseudotuberculosis iscR mutant shows lower effector secretion through the type III secretion system (H. K. Miller, L. Kwuan, L. Schwiesow, D. L. Bernick, H. A. Ramirez, J. M. Ragle, P. P. Chan, T. M. Lowe, and V. Auerbuch, presented at the West Coast Bacterial Physiologists Annual Asilomar Conference, Pacific Grove, CA, 13 to 15 December 2013). In E. coli, apo-IscR activates transcription of the fimE recombinase gene under low-iron conditions, leading to decreased type I fimbrial expression and reduced biofilm formation (25).
Given the critical role of iron in enterobacterial infection, the goal of this study was to further explore its effect on virulence gene expression in the ETEC H10407 prototype strain. Interestingly, we show that iron starvation reduces LT secretion while activating cfaA promoter activity. cfaA promoter activity was further examined in ETEC H10407 Δfur and ΔiscR mutants and in heterologous E. coli strains inactivated for transcriptional regulators connected with iron homeostasis, including Fur, IscR, OxyR, and SoxR. The impact of iron starvation on the cfaA promoter was found to be mediated by IscR.
MATERIALS AND METHODS
Strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. The ETEC H10407 prototype strain (provided by the Culture Collection at the University of Göteborg) and ETEC E24377A (provided by S. Savarino) were chosen for analysis, as they have been fully sequenced. Strains were grown in Luria-Bertani (LB) broth for introduction of plasmids and generation of stocks or in CFA broth (1% Casamino Acids [Difco], 0.15% yeast extract [Difco], 0.4 mM MgSO4, 0.04 mM MnCl2, pH 7.4) for all other analyses (26). Cultures were performed at 37°C with aeration. For iron chelation, the culture medium was supplemented with 1 to 50 μM deferoxamine mesylate (Sigma). Antibiotics were used as needed at the following concentrations: 100 μg ml−1 ampicillin, 37 μg ml−1 chloramphenicol, and 50 μg ml−1 kanamycin.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| H10407 | O78:H11:K80 CFA/I STh STp LT | 67 |
| H10407 Δfur | H10407 fur::kan | 54 |
| H10407 ΔiscR | H10407 iscR::kan | This study |
| H10407 ΔiscR-C | H10407 iscR::kan pCA24N-iscR | This study |
| E24377A | O139:H28 CS1 CS3 LT STh | 68 |
| BW25113 | F− Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514 | 33 |
| JW0669 | BW25113 fur::kan | 55 |
| JW5714 | BW25113 zur::kan | 55 |
| JW3933 | BW25113 oxyR::kan | 55 |
| JW4024 | BW25113 soxR::kan | 55 |
| JW2515 | BW25113 iscR::kan | 55 |
| JW2514 | BW25113 iscS::kan | 55 |
| JW2513 | BW25113 iscU::kan | 55 |
| JW2512 | BW25113 iscA::kan | 55 |
| JW1674 | BW25113 sufA::kan | 55 |
| JW5273 | BW25113 sufB::kan | 55 |
| H10407-cfaA | H10407 pPROBE-cfaAp | This study |
| H10407-cfaD | H10407 pPROBE-cfaDp | This study |
| H10407-eltA | H10407 pPROBE-eltAp | This study |
| H10407-fepA | H10407 pPROBE-fepAp | This study |
| H10407fur-cfaA | H10407Δfur pPROBE-cfaAp | This study |
| H10407fur-cfaD | H10407Δfur pPROBE-cfaDp | This study |
| H10407fur-eltA | H10407Δfur pPROBE-eltAp | This study |
| H10407fur-fepA | H10407Δfur pPROBE-fepAp | This study |
| SH200 | BW25113 pBAD pPROBE-cfaAp | This study |
| SH201 | BW25113 pBAD-cfaD pPROBE-cfaAp | This study |
| SH202 | JW0669 pBAD pPROBE-cfaAp | This study |
| SH203 | JW0669 pBAD-cfaD pPROBE-cfaAp | This study |
| SH204 | JW5714 pBAD pPROBE-cfaAp | This study |
| SH205 | JW5714 pBAD-cfaD pPROBE-cfaAp | This study |
| SH206 | JW3933 pBAD pPROBE-cfaAp | This study |
| SH207 | JW3933 pBAD-cfaD pPROBE-cfaAp | This study |
| SH208 | JW4024 pBAD pPROBE-cfaAp | This study |
| SH209 | JW4024 pBAD-cfaD pPROBE-cfaAp | This study |
| SH210 | JW2515 pBAD pPROBE-cfaAp | This study |
| SH211 | JW2515 pBAD-cfaD pPROBE-cfaAp | This study |
| SH212 | JW2514 pBAD-cfaD pPROBE-cfaAp | This study |
| SH213 | JW2513 pBAD-cfaD pPROBE-cfaAp | This study |
| SH214 | JW2512 pBAD-cfaD pPROBE-cfaAp | This study |
| SH215 | JW1674 pBAD-cfaD pPROBE-cfaAp | This study |
| SH216 | JW5273 pBAD-cfaD pPROBE-cfaAp | This study |
| SH301 | BW25113 pCA24N pPROBE-cfaAp | This study |
| SH302 | BW25113 pCA24N-iscR pPROBE-cfaAp | This study |
| SH303 | JW2515 pCA24N pPROBE-cfaAp | This study |
| SH304 | JW2515 pCA24N-iscR pPROBE-cfaAp | This study |
| Plasmids | ||
| pMA-T | ColEI Ampr | GeneArt (Life Technologies) |
| pMK | ColEI Kanr | GeneArt (Life Technologies) |
| pMA-T-cfaA | cfaA promoter (−223 to +77), Ampr | This study |
| pSUB7 | kan cassette template | 69 |
| pKD46 | λ Red recombinase, Ampr | 33 |
| pBAD33 | PBAD Cmr | 34 |
| pBAD33-cfaD | cfaD under PBAD control, Cmr | This study |
| pPROBE-AT | Promoterless GFP reporter, Ampr | 32 |
| pPROBE-cfaD | cfaD promoter (−274 to + 285), Ampr | This study |
| pPROBE-cfaA | cfaA promoter (−223 to +77), Ampr | This study |
| pPROBE-fepA | fepA promoter (−125 to +175), Ampr | This study |
| pPROBE-eltA | eltA promoter (−134 to +166), Ampr | This study |
| pCA24N (gfp-) | T7 promoter, GFP− Cmr | 33 |
| pCA24N-iscR | iscR under T7 promoter control, GFP− Cmr | 36 |
| pSP401 | ColEI Kanr, T7 promoter | This study |
| pSP401-iscR | iscR under T7 promoter control, Kanr | This study |
p, promoter; Cmr, Ampr, and Kanr, chloramphenicol, ampicillin, and kanamycin resistance, respectively.
Western blotting.
Electrophoresed whole-cell bacterial lysate was transferred onto a 0.2-μm nitrocellulose membrane using a Novex semidry blotter (Invitrogen). The membrane was blocked with 5% skim milk in phosphate-buffered saline (PBS) (pH 7.4) prior to incubation with rabbit sera raised against the purified CFA/I fimbriae (1:2,000) (27) or GroEL (1:40,000; Sigma). Membranes were then incubated with goat anti-rabbit IgG coupled to IRDye800CW (1:5,000; Rockland) for fluorescent revelation. After incubation with the primary and secondary antibodies, membranes were washed in PBS–0.1% Tween 20. Revelation was performed with the Odyssey infrared imaging system (LiCor).
Dot blotting.
Bacteria were pelleted and resuspended in 50 mM Tris-HCl (pH 8.0) at 2.5 × 108 CFU ml−1. Whole-cell suspensions were serially diluted in a 96-well plate with 50 μl dotted on a wet 0.45-μm nitrocellulose membrane. After drying for 5 min, the membrane was blocked with TBS (50 mM Tris-HCl, 150 mM NaCl, pH 8.0) containing 1% (wt/vol) skim milk (Difco), followed by incubation with rabbit serum raised against the purified CFA/I fimbriae (1:1,000 in TBS). After washing in TBS, the membrane was incubated with goat anti-rabbit secondary antibody coupled to horseradish peroxidase (1:1,000 in TBS). All incubations were performed for 1 h at 37°C with agitation. CFA/I surface expression was visualized by colorimetry with the Opti4CN kit (Bio-Rad).
GM1 ELISA for quantification of LT.
Following overnight growth in CFA broth, culture supernatant was treated with antiprotease (Roche), filter sterilized, concentrated using an Amicon Ultra-4 filter (Millipore), and frozen at −70°C until quantification of secreted LT. Periplasmic LT was released from bacterial pellets as previously described (28). The suspension underwent at least 4 freeze-thaw cycles before recovery of the cleared lysate. LT quantification was performed by GM1 enzyme-linked immunosorbent assay (ELISA) as previously described and normalized to bacterial density at 600 nm (A600) (29).
RNA extraction and real-time quantitative PCR (RT-qPCR) analysis.
Bacteria were treated with RNA Later (Ambion) and pellets frozen at −70°C. Total RNA was extracted on-column using the NucleoSpin RNA II kit (Macherey-Nagel) according to the manufacturer's instructions, treated with DNase I (Invitrogen) at 37°C for 30 min, and repurified via on-column cleanup using the NucleoSpin RNA II kit. Total RNA quantity was determined via NanoDrop and quality evaluated via the 2100 Agilent Bioanalyzer (Agilent Technologies).
cDNA was generated from 500 ng of total RNA with the ThermoScript RT-PCR system for first-strand cDNA synthesis with random hexamers according to the manufacturer's instructions (Invitrogen). For ETEC H10407, specific target amplification was performed on cDNA to selectively amplify transcripts of interest using the TaqMan Preamp master mix (Applied Biosystems) and target primers, followed by treatment with ExoSAP-IT (Affymetrix). Primers were designed and validated by Fluidigm (San Francisco, CA) and are listed in Table S1 in the supplemental material. Samples were diluted 1:5 in TE-4 buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) and analyzed by RT-qPCR in a 96.96 integrated fluidic circuit chip using the BioMark system (Fluidigm) with the TaqMan Gene Expression master mix (Applied Biosystems) and EvaGreen (Biotium). A thermal mix was performed with incubation at 50°C for 2 min, 70°C for 30 min, and 25°C for 10 min to enhance sample and primer diffusion within the chip, followed by 50°C for 2 min to ensure uracil-DNA glycosylase protection and 95°C for 10 min for hot-start PCR. Amplification was then performed for 35 cycles at 95°C for 15 s followed by 60°C for 1 min. A melting curve was generated at the end of each run to verify primer specificity. Expression was normalized against the three most stable housekeeping genes from the set consisting of bglA, gapA, hns, rplD, rpoA, and tufB, as determined by GeNorm (30). For supplementary analysis of virulence genes expressed in ETEC E24377A or within the cfa operon in H10407, RT-qPCR was performed on a Stratagene Mx3000P with SYBR green and data normalized against the 16S housekeeping gene. Relative gene expression was calculated using the 2−ΔΔCT method. Fold changes represent the ratio of expression in treated versus untreated cultures.
Construction of transcriptional fusions.
The promoter and partial coding region of cfaD (−274 to +285 relative to the transcription start site) (31) was amplified from ETEC H10407 genomic DNA with primers cfaDF (GCTGGATCCAAAATATAATTCGTCAAAG) and cfaDR (TAAGAATTCGCAACATATGGCCTTTCTGA). BamHI and EcoRI restriction sites were added, as shown by underlined sequences, and the PCR product directionally cloned into the pPROBE-AT vector. pPROBE-AT is a low-copy-number plasmid with a promoterless green fluorescent protein (GFP) reporter gene used for the generation of transcriptional fusions (32).
The fepA (−125 to +175), eltA (−134 to +166), and cfaA (−223 to +77) promoters were assembled from synthetic nucleotides with addition of HindIII and KpnI sites to the 5′ and 3′ ends, respectively (GeneArt; Life Technologies). The fepA promoter was inserted into the pMA-T cloning vector using SfiI sites, while the eltA and cfaA promoters were inserted into the pMK cloning vector using SacI and KpnI sites (GeneArt; Life Technologies). Vectors were then digested with HindIII/KpnI for directional cloning into pPROBE-AT.
ETEC H10407 iscR mutant construction.
The iscR deletion mutant was generated by PCR amplification of a kanamycin cassette flanked by FLP recombination target (FRT) sites from pSUB7 with 50-bp flanking regions homologous to the regions surrounding the iscR gene (underlined) using the primers IscR-FRT-FW (5′-ATACCCCCACTTTTACAATAAAAAACCCCGGGCAGGGGCGAGTTTGAGGTGAAGTAAGACTGTAGGCTGGAGCTGCTTCG-3′) and IscR-FRT-RV (5′-GGATGTACGACCGTGTTTACGAAGTATTTAGCACTCCGGCCTGATTCTGAATTCTTTTTACATATGAATATCCTCCTTAG-3′) (Recombina). The PCR product was introduced into ETEC strain H10407 previously transformed with pKD46, carrying the phage λ red recombinase system under the control of an inducible promoter, as described previously (33). Successful recombination was verified by PCR amplification of the region using the primers IscR-Verif-FW 5′-GAATGTCAGACTTGACCCTGC-3′ and IscR-Verif-RV 5′-CACTCAATGCAAGGAATCAGG-3′ (Recombina). The H10407 ΔiscR strain was complemented with the pCA24N-iscR overexpression plasmid.
Construction of cfaD and iscR expression vectors.
The cfaD coding sequence was assembled from synthetic oligonucleotides with addition of XbaI and HindIII sites to the 5′ and 3′ ends, respectively, and inserted into the pMA-T cloning vector at the SfiI site (GeneArt; Life Technologies). The gene was then directionally cloned into the pBAD33 expression vector using the XbaI and HindIII sites to place cfaD under the control of the araBAD promoter (pBAD-cfaD) for arabinose-inducible expression (34).
The iscR gene was assembled from synthetic nucleotides with addition of NcoI and HindIII sites to the 5′ and 3′ ends, respectively, and inserted into the pMA-T cloning vector using SfiI sites (GeneArt; Life Technologies). The vector was digested with NcoI/HindIII for directional cloning into pSP401, placing iscR under the control of the T7 promoter.
GFP reporter assay.
pPROBE transcriptional fusions were electroporated into the ETEC H10407 and E. coli BW25113 strains as previously described (35). The empty pBAD vector or pBAD-cfaD was electroporated into BW25113 strains in addition to pPROBE constructs to evaluate the effect of CfaD on promoter activity. The pCA24N empty vector or pCA24N-iscR lacking the GFP gene was electroporated into BW25113 wild-type (WT) and ΔiscR strains for complementation (36). Following electroporation, plasmid maintenance in H10407 was validated by colony PCR using CFA/I and LT primers (37). Cultures were grown to stationary phase with shaking overnight in the presence of 0.66 μM arabinose for pBAD induction or 50 μM IPTG (isopropyl-β-d-thiogalactopyranoside) for pCA24N induction. Two hundred microliters of bacterial suspension was distributed in duplicate in a black 96-well microplate (ThermoFisher). GFP fluorescence was quantified by fluorometry using a Varioskan Flash microplate reader with an excitation wavelength of 490 nm and an emission wavelength of 510 nm ± 1 nm. Fluorescence was expressed in relative fluorescence units (RFU) normalized against the A600.
In silico analysis of IscR binding sites.
IscR binding motifs were generated by MEME using a training set based on sites previously validated by footprinting for both apo- and holo-IscR in E. coli K-12 (38). IscR binding site locations within the defined cfaA promoter region were predicted by MAST (39).
IscR purification.
IscR was purified from E. coli BL21 transformed with pSP401-iscR following growth at 37°C under aerobic conditions in LB broth. iscR expression was induced with 500 μM IPTG for 1 h from A600 0.4 to 0.6. Purification was performed under aerobic conditions as previously described (40). Briefly, bacteria were harvested by centrifugation and resuspended in buffer A (50 mM Tris-HCl [pH 8.0], 10% glycerol, 1 mM dithiothreitol [DTT], 5 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride) supplemented with 0.5 M KCl and lysed by passage through a French press. Cell debris was removed by centrifugation at 20,000 × g at 4°C for 15 min. The supernatant was further diluted by adding 3 volumes of buffer A to decrease the KCl concentration and loaded on a 5-ml HiTrap heparin column (GE Healthcare) equilibrated in buffer A containing 0.1 M KCl at a flow rate of 1 ml min−1. The column was washed with 5 volumes (25 ml) of buffer A containing 0.1 M KCl, and IscR was eluted with a linear gradient of 0.1 to 1.0 M KCl in buffer A. Fractions containing IscR were pooled, diluted by adding 2 volumes of buffer A to decrease the KCl concentration, and loaded onto a 5-ml HiTrap Q FF column (GE Healthcare) at 1.4 ml min−1, and IscR was eluted as described above. Purified IscR was dialyzed against conservation buffer (20 mM Tris-HCl [pH 8.0], 50% glycerol, 400 mM KCl, 1 mM DTT, 5 mM EDTA), and residual iron levels were determined by inductively coupled plasma mass spectrometry (ICP-MS) (41). No iron could be detected, indicating that IscR was in the apo form.
DNase I footprinting.
The cfaA promoter region (300 bp) was isolated from pMA-T by digestion with AflIII and KpnI and the coding strand labeled by incorporation of [α-32P]dCTP (3,000 Ci mmol−1; Amersham) at the AflIII site with the Klenow fragment. The labeled fragment was isolated on a 2% agarose gel and purified with the QIAquick gel extraction kit (Qiagen). DNase I footprinting was performed as described previously (42), with the modifications listed below. Labeled DNA (100,000 cpm; at least 2 nM) was incubated with IscR under aerobic conditions for 20 min at 37°C in buffer containing 20 mM Tris-HCl (pH 8.0), 70 mM KCl, 100 μg ml−1 bovine serum albumin (BSA), 1 mM DTT and Nonidet P-40 (Roche Applied Science) at a final concentration of 0.1% (vol/vol). The buffer was adjusted to include 10 mM MgCl2 and 5 mM CaCl2 prior to the addition of 0.01U DNase I (Roche Applied Science) and incubated for 20 s at 37°C. The digestions were blocked by the addition of 25 μl of stop solution (50 mM EDTA [pH 8.0], 40 μg glycogen), and 100 μl of ice-cold Tris-EDTA (TE) (pH 8.0) was added to increase the volume of the mixture. After phenol-chloroform extraction, DNA fragments were ethanol precipitated, resuspended in a formamide dye mixture, and separated by electrophoresis on a 6% polyacrylamide sequencing gel. Bands were visualized by autoradiography.
Statistical analyses.
Statistical analyses were performed on fold change data using the one-sample t test and on comparisons between groups using the Student t test (SigmaPlot 11.0; Systat Software Inc.). P values of <0.05 were considered significant.
RESULTS
Effect of iron starvation on CFA/I fimbriae and LT.
ETEC H10407 was grown to stationary phase overnight in CFA medium supplemented or not with the iron chelator deferoxamine. CFA/I production was increased in the presence of deferoxamine from 5 μM as shown by detection of the CFA/I major subunit, CfaB, by Western blotting (Fig. 1A). Evaluation of CFA/I surface expression by dot blotting of intact bacteria confirmed this result, indicating that deferoxamine increased both total and surface-exposed CFA/I levels (Fig. 1B). As CFA broth contains approximately 5 μM iron (43), 50 μM deferoxamine was chosen for future analyses to ensure complete iron chelation. In contrast to the results obtained for CFA/I, LT levels in the periplasm were significantly decreased in the presence of 50 μM deferoxamine, as shown by GM1 ELISA (Fig. 1C). Furthermore, the amount of secreted LT detected in the culture supernatant dropped to undetectable levels under iron starvation. As LT production occurs early during infection (12), periplasmic LT levels were also quantified in exponentially growing cells to determine if the in vitro growth phase influenced the toxin response. Although the total amount of LT was higher during exponential growth phase, a similar decrease in LT was seen in cultures treated with deferoxamine (data not shown).
FIG 1.

CFA/I production and LT secretion under iron starvation. ETEC H10407 was cultured overnight in the presence or absence of 50 μM deferoxamine (Def) unless otherwise indicated. (A) Western blot of bacterial lysate, showing protein levels of the CFA/I major subunit CfaB using anti-CFA/I serum. GroEL was detected with anti-GroEL serum and served as the loading control. (B) Dot blot of surface-expressed CFA/I using anti-CFA/I sera, with an initial concentration of 2.5 × 10−8 bacteria/ml (lane 1) followed by 2-fold serial dilutions (lanes 2 to 9). Circled blots indicate the last bacterial dilution with detectable CFA/I under each condition. (C) Periplasmic LT and secreted LT were quantified by GM1 ELISA via analysis of the periplasmic fraction and culture supernatant, respectively. LT was normalized against bacterial density. ND, not detected. Means from at least three independent replicates are shown, with error bars representing the standard error of the mean (SEM). Statistical analyses were performed using the Student t test. **, P < 0.01.
These results indicate that iron starvation increased CFA/I production, as expected (14), but inhibited LT secretion.
Temporal gene expression analysis under iron starvation.
To determine if cfaABCE (CFA/I) or eltAB (LT) operon expression was modified in response to iron starvation, RT-qPCR analysis was performed. Gene expression was evaluated in both exponential and early stationary growth phases, as several ETEC virulence genes have previously been shown to be growth phase regulated (44). As this is the first study to evaluate the impact of iron starvation on gene expression in ETEC, we first analyzed the expression levels of iron-regulated genes (fur, ryhB, fepA, fyuA, and ompW) previously characterized in other E. coli strains (16, 45, 46).
Although the iron response regulator Fur (fur product) is moderately autoregulated in E. coli (47), we found no major change in gene expression under iron starvation in either the exponential or early stationary growth phase compared to control cultures (Fig. 2). However, genes known to be repressed by Fur in E. coli were induced in the presence of deferoxamine in ETEC H10407, as expected. The small RNA ryhB and the fepA and fyuA genes, encoding siderophore receptors, were upregulated under iron starvation (16, 45, 46), with the strongest expression in exponential phase. In contrast, the gene encoding the outer membrane protein OmpW, implicated in resistance to environmental stress, was inhibited under iron starvation, as shown previously in E. coli (45, 48), with the strongest repression in early stationary phase. Thus, although fur gene expression was unchanged in exponential phase, Fur responded to changes in iron availability with derepression of Fur-regulated genes in the presence of deferoxamine.
FIG 2.
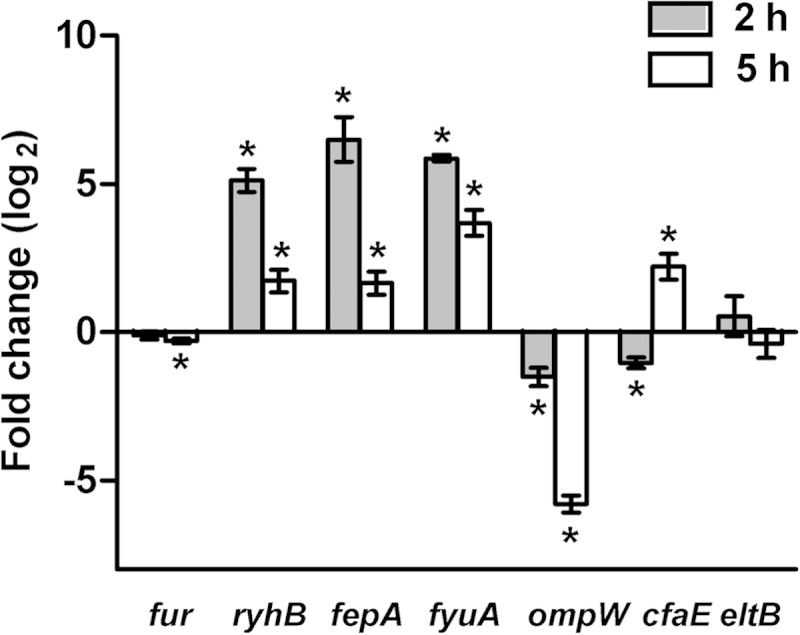
Temporal expression of iron-regulated genes and two major ETEC virulence genes under iron starvation. Gene expression analysis of ETEC H10407 was performed following growth to exponential phase (2 h) or early stationary phase (5 h) in CFA medium alone or supplemented with 50 μM deferoxamine. Data are represented as the fold change ratio of expression in treated versus untreated cultures, calculated from relative expression values using the 2−ΔΔCT method. Values are log2 transformed. Means from at least three independent replicates are shown, with error bars representing the SEM. Statistical analyses were performed using the one-sample t test. *, P ≤ 0.05.
We next examined the expression of the major ETEC virulence operon cfaABCE, represented here by the adhesin gene cfaE. Under iron starvation, expression of cfaE was weakly repressed in exponential growth phase, while its transcription was increased nearly 4-fold in early stationary phase (Fig. 2). Further expression analysis of the cfaABCE operon showed upregulation of all four genes under iron starvation in early stationary phase, indicating that they behave in a similar manner (see Fig. S1 in the supplemental material). The increase in CFA/I production under this condition was therefore associated with mRNA levels. In contrast to that of cfaE, expression of eltB, encoding the LT B subunit, was unchanged under iron starvation during both exponential and early stationary growth phases. The reduced amount of LT found in the periplasm and in the culture supernatant was therefore independent of eltAB transcription.
Effect of iron starvation on expression of additional ETEC virulence genes.
The impact of iron starvation on additional genes implicated in ETEC adhesion and toxicity was further investigated. As expression of the cfa operon appears to be growth phase dependent, with induction in early stationary phase, the global virulence response was evaluated in this growth phase. In addition to cfaE and eltB, genes selected for this experiment were cfaD, cexE, aatA, etpB, sta3, sta1, astA, clyA, leoA, tia, csgA, and csgE.
Expression of the major virulence regulator gene cfaD was significantly increased under iron starvation compared to expression in control cultures, as was expression of several other plasmid-based virulence genes. These included etpB (encoding the outer membrane protein of the Etp two-partner secretion system), cexE (encoding the small secreted protein CexE), and aatA (encoding an outer membrane protein of the Aat type I secretion system [T1SS]) (Fig. 3). Like CFA/I, the EtpBAC two-partner secretion system is implicated in early colonization, secreting the highly glycosylated protein EtpA, which then localizes to the tip of the flagellum to mediate attachment to epithelial cells (8). Based on homology to enteroaggregative E. coli, the Aat T1SS may be responsible for CexE secretion, although the function of CexE in ETEC has not yet been described (49, 50). Thus, it is not surprising that they showed similar expression profiles here. Of note, cfaD, cfaABCE, and cexE are all known members of the CfaD regulon (9, 49, 51).
FIG 3.

Effect of iron starvation on ETEC virulence gene expression. Gene expression analysis of ETEC H10407 was performed in early stationary phase (5 h) in CFA medium alone or treated with 50 μM deferoxamine. Data are represented as the fold change ratio of expression in treated versus untreated cultures, calculated from relative expression values using the 2−ΔΔCT method. Values are log2 transformed. The median is indicated as a band within the box plot. Whiskers represent minimum and maximum values. Statistical analyses were performed using the one-sample t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In contrast to these plasmid-based genes, chromosomal genes encoding the Tia adhesin and curli fimbriae were repressed. Tia was previously shown to interact with host cell surface proteoglycans to promote adherence and invasion, while curli are implicated in adherence and biofilm formation (52). As the curli genes are divided into two operons (csgBA and csgDEFG), we evaluated the effect of iron starvation on both operons and found them to be similarly repressed under iron starvation.
The toxin gene encoding EAST1 (astA) showed no significant change in expression, as shown for eltB. However, genes encoding the STp (sta1), STh (sta3), and cytolysin A (clyA) toxins were all repressed under iron starvation in early stationary phase. Furthermore, leoA, encoding a GTPase involved in LT secretion from the periplasm (28, 53), was also repressed under iron starvation. As expression of the eltB gene was not affected by iron starvation, the repression of leoA could explain the absence of LT in culture supernatant.
Promoter activity in response to iron starvation in ETEC H10407 WT and Δfur strains.
We hypothesized that the Fur repressor might be implicated in cfaABCE regulation, as it is the central regulator of iron homeostasis in enterobacteria. To determine if the iron response was mediated by Fur at the transcriptional level, the activity of promoter-GFP transcriptional fusions was evaluated in the ETEC H10407 wild-type (WT) strain and its isogenic fur mutant (Δfur) (54). Analysis of transcriptional fusions incorporated directly into the ETEC strain allowed us to evaluate the iron response under native conditions, in the presence of the virulence plasmids and the CfaD regulator, which is responsible for activation of the cfaABCE operon (9). Activity of the fepA, eltA (eltAB operon), cfaA (cfaABCE operon), and cfaD promoters was measured after overnight growth in the presence or absence of deferoxamine.
As expected, the activity of the control promoter, fepA, was derepressed in the WT strain in the presence of deferoxamine and in the Δfur strain in iron-replete medium (Fig. 4). No further significant increase in promoter activity occurred in the Δfur strain under iron starvation, indicating that promoter activity was already at the maximum level in the absence of Fur. In contrast, eltA promoter activity was unchanged under all conditions, as expected based on the earlier RT-qPCR analysis, confirming that it is not transcriptionally regulated in response to iron (Fig. 4). Evaluation of the activity of the fepA and eltA promoters validated the use of the pPROBE reporter system in ETEC.
FIG 4.

Promoter activity of selected genes in the ETEC H10407 wild-type strain and its isogenic fur mutant in response to iron starvation. Transcriptional fusions of fepA, eltA (LT operon), cfaA (CFA/I operon), and cfaD promoter regions to the GFP reporter gene were introduced into the ETEC H10407 wild-type (WT) strain and its isogenic fur mutant (Δfur). GFP fluorescence was measured following overnight growth in CFA broth in the presence or absence of 50 μM deferoxamine (Def). Background fluorescence was subtracted, with fluorescence normalized against the bacterial density at 600 nm and expressed as relative fluorescent units (RFU). Means from at least three independent replicates are shown, with error bars representing the SEM. Statistical analyses were performed using the Student t test. **, P < 0.01; ***, P < 0.001. The difference in fluorescence of the fepA-GFP gene fusion in the fur mutant was not significant.
Like that of fepA, promoter activity of both cfaA and cfaD was increased under iron starvation compared to activity in untreated cultures (Fig. 4). Basal levels of cfaD promoter activity were approximately 10-fold lower than those seen for cfaA in CFA medium. Furthermore, under iron starvation, cfaA promoter activity increased by nearly 5-fold, whereas cfaD promoter activity increased by only 2-fold. Therefore, the cfaA promoter appears to be more sensitive to iron starvation than the cfaD promoter.
However, no difference in promoter activity could be seen between the WT and Δfur strains, indicating that Fur does not regulate cfaA or cfaD at all, including through direct control. Thus, both genes are induced in response to iron starvation but are Fur independent. Further transcript quantification in ETEC WT and Δfur strains by qPCR also showed no difference in cfaE and cfaD mRNA levels under iron-replete conditions, in contrast to results obtained for ryhB, which was upregulated as expected in the absence of fur (see Fig. S2 in the supplemental material). These results eliminated a possible modification in the stability of the cfaE or cfaD transcripts in the Δfur background.
Overall, these data indicate that Fur does not modulate cfa or cfaD expression in response to iron starvation.
Modulation of cfaA promoter activity in response to transcriptional regulators implicated in iron homeostasis.
As the cfa operon undergoes transcriptional regulation in response to iron starvation with no implication of the Fur repressor, the effect of other transcriptional regulators was evaluated. The E. coli KEIO collection, including single-gene deletion mutants for nearly all chromosomal genes in strain BW25113, represents a powerful screening tool (55), enabling us to circumvent the need to generate individual knockout mutants of ETEC H10407. We took advantage of this heterologous system to evaluate cfaA promoter activity in strains lacking transcriptional regulators that may be associated with iron homeostasis, including Fur, OxyR, SoxR, IscR, and Zur. Both OxyR and SoxR respond to oxidative stress, which is associated with iron concentration due to the generation of hydroxyl radicals via the Fenton reaction (17). IscR regulates iron-sulfur cluster biogenesis (40). A bioinformatic analysis identified Zur as the only known Fur homolog in ETEC H10407 (data not shown).
cfaA promoter activity was evaluated in the absence or presence of the CfaD regulator, as it is a key activator of cfa expression in ETEC but absent in E. coli BW25113. To this end, the cfaD gene was placed under the control of an arabinose inducible promoter in the pBAD vector and introduced into BW25113 in addition to the pPROBE-cfaA plasmid. Given the inability of E. coli BW25113 to metabolize arabinose and the low glucose concentration in CFA medium, 0.66 μM arabinose was sufficient to induce cfaD overexpression.
In the E. coli BW25113 WT strain, cfaA promoter activity was significantly increased under iron starvation, as seen previously in ETEC H10407. CfaD was not required, as promoter activity was increased in both the absence and presence of the activator under iron starvation (Fig. 5). However, cfaA promoter activity was 7- to 10-fold higher in the presence of CfaD than in the control cultures (empty pBAD vector), indicating that CfaD did indeed increase cfaA promoter activity in E. coli BW25113 (Fig. 5B). The effects of iron starvation and CfaD activation on cfaA promoter activity were additive, with the highest activity in the presence of both factors.
FIG 5.

cfaA promoter activity in E. coli deletion mutants. E. coli BW25113 wild-type and mutant strains containing the pPROBE-cfaA transcriptional fusion were cultivated in CFA medium in the presence or absence of 50 μM deferoxamine (Def). GFP reporter fluorescence was measured following overnight growth in the presence of the empty pBAD vector (A) or in the presence of pBAD-cfaD (B). Cultures were performed in the presence of 0.66 μM l-arabinose for pBAD induction. GFP fluorescence was normalized against bacterial density at 600 nm and expressed as relative fluorescent units (RFU). Means from at least three independent replicates are shown, with error bars representing the SEM. Statistical analyses were performed using the Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
cfaA promoter activity was significantly increased under iron starvation in all mutant strains tested except the ΔiscR strain (Fig. 5). cfaA promoter activity in the Δfur, Δzur, and ΔsoxR strains showed profiles similar to that in the WT strain in both the absence and presence of the CfaD activator, although promoter activity was slightly reduced in the Δfur strain in the presence of CfaD (Fig. 5). The ΔoxyR strain showed higher promoter activity than the WT strain in the absence of CfaD in both treated and untreated cultures, indicating that this effect was not specific to the iron response.
Promoter activity was unchanged in the ΔiscR strain in both the presence and absence of CfaD, suggesting that the effect of IscR is independent of CfaD. The phenotype associated with the iscR mutant was successfully restored to wild-type levels in the presence of the IscR expression vector, pCA24N-iscR (see Fig. S3 in the supplemental material). Moreover, overexpression of IscR in the WT strain further increased cfaA promoter activity under iron starvation (see Fig. S3 in the supplemental material).
Based on these data, we infer that IscR is the major regulator responsible for mediating the impact of iron starvation on the cfaA promoter.
We next investigated the action of IscR in ETEC H10407 by generating an isogenic iscR deletion mutant. While cfaE gene expression was induced in the WT strain under iron starvation, this effect was abolished in the ΔiscR strain (Fig. 6). Complementation of the ΔiscR strain with a plasmid overexpressing IscR permitted recovery of cfaE expression, indicating that the effect of IscR was specific. In contrast, eltB (LT) expression remained unchanged under iron starvation in all strains, consistent with previous results (Fig. 6).
FIG 6.
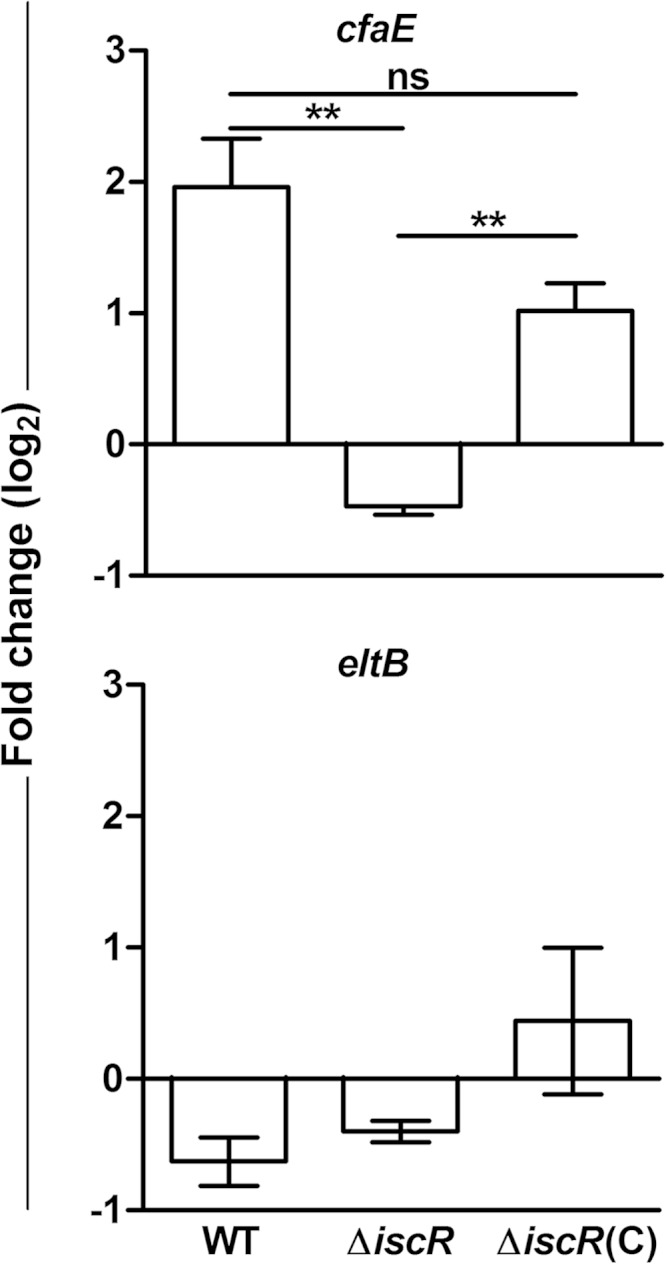
Effect of iron starvation on gene expression in ETEC H10407 ΔiscR. Gene expression analysis was performed following growth to late log phase in CFA medium alone or treated with 50 μM deferoxamine. cfaE (top) or eltB (bottom) gene expression was evaluated in the H10407 wild-type strain (WT), the isogenic ΔiscR deletion mutant (ΔiscR), or the mutant complemented with an iscR overexpression plasmid and induced with 50 μM IPTG [ΔiscR(C)]. Data are represented as the fold change ratio of expression in treated versus untreated cultures, calculated from relative expression values using the 2−ΔΔCT method. Values are log2 transformed. Means from three independent replicates are shown, with error bars representing the SEM. Statistical analyses were performed using the one-sample t test. **, P < 0.01. ND, not detected; ns, not significant.
As IscR transcriptionally regulates the iscRSUA and sufABCDE operons, which are associated with Fe-S cluster biogenesis, cfaA promoter activity may depend on these downstream elements rather than directly on IscR. cfaA promoter activity was therefore evaluated in E. coli isc and suf deletion mutant strains (ΔiscS, ΔiscU, ΔiscA, ΔsufA, and ΔsufB) in the presence of CfaD (see Fig. S4 in the supplemental material). cfaA promoter activity was induced in all mutant strains under iron starvation, in contrast to the ΔiscR mutant, as shown previously (Fig. 5; see Fig. S4 in the supplemental material). This suggests that cfaA promoter activity is directly dependent on IscR rather than Fe-S cluster biogenesis.
Although IscR can regulate gene expression in both its holo (presence of an internal [2Fe-2S] cluster) and apo (absence of the [2Fe-2S] cluster) forms, cfaA promoter activity was increased in a ΔiscU strain under iron-rich conditions (see Fig. S4 in the supplemental material). As iscU encodes the scaffold protein essential for generation of Fe-S clusters via the Isc pathway and maturation of IscR to its holo form (56), these data further suggest that the effect of IscR may be specifically mediated by its apo form.
Identification of IscR binding sites within the cfaA promoter.
To determine if IscR could function by directly binding to the cfaA promoter, we searched the cfaA promoter for IscR binding site motifs. IscR can directly bind DNA using two consensus binding motifs (19, 42, 57). Although holo-IscR binds both type 1 and type 2 motifs, apo-IscR can bind only the type 2 motif (57). Here, type 1 and type 2 motifs were generated by MEME (Fig. 7A), based on IscR binding sites previously identified and validated by DNase I footprinting in E. coli (42). These motifs were then used to identify potential binding sites within the cfaA promoter by MAST. No binding sites similar to the type 1 motif were identified within the cfaA promoter. Two putative binding sites similar to the type 2 motif were identified, from position −164 to −137 (site A, 5′-ATATCCACAGGAACTGCATATTGGA-3′) and from position −40 to −16 (site B, 5′-TTTTTATCTCATTTTTTTTTGTTTT-3′) relative to the start of transcription (Fig. 7B). Furthermore, both site A and site B showed 80% identity to a previously published type 2 motif generated with a different algorithm (5′-WWWWCCXYAXXXXXXXTRXGGWWWW-3′, where W is A or T, R is A or G, Y is C or T, and X is any base) (57). Site B overlapped with a previously identified CfaD binding site and the −35 site, whereas site A was located upstream of all previously identified regulator binding sites (49).
FIG 7.

Identification of IscR binding sites within the cfaA promoter. (A) Type 1 or type 2 IscR binding site motifs generated by MEME were used to identify potential IscR binding sites within the cfaA promoter. (B) Two type 2 binding motifs (site A and site B, boxed) were identified in silico by MAST. Key features of the cfaA promoter are shown, including Rns/CfaD binding sites (underlined) (46), possible −10 and −35 sites (bold), the transcription start site (arrow), and the translation initiation codon of the cfaA gene (ATG, bold). (C) Apo-IscR binding sites were identified in vitro by DNase I footprinting from positions −7 to −48 and −138 to −165 under aerobic conditions. Lanes 1 and 6 do not contain IscR. Lanes 2 to 5 contain 50, 100, 200, and 500 nM IscR, respectively. The sequence is numbered relative to the transcription start site, indicated by an arrow. Protected regions within the coding strand of the cfaA promoter are indicated by bars, and hypersensitive regions induced by binding of apo-IscR are indicated by closed circles. These sites are also shown in panel B with the same symbol. To localize bands generated by DNase I digestion, sequencing reactions run in parallel were used as a ladder.
As the putative binding sites identified in silico were similar to motif 2, IscR may be capable of interacting with the cfaA promoter in both holo and apo forms. However, as promoter activity was induced under iron starvation conditions, which lead to increased apo-IscR levels, we hypothesized that apo-IscR may interact with the cfaA promoter. DNase I footprinting studies on the coding strand of the cfaA promoter confirmed that apo-IscR bound the cfaA promoter. Complete protection of site A (−138 to −165) could be seen from 50 nM apo-IscR, indicating high affinity (Fig. 7C), while apo-IscR bound site B (−7 to −48) with moderate affinity from 200 nM. The hypersensitivity that occurs at the −10 region upon IscR binding indicates a DNA structural modification that may facilitate binding of the RNA polymerase or promoter opening. In contrast, no sites were identified on the noncoding strand (data not shown).
These data therefore suggest that apo-IscR functions as a direct activator of cfaABCE operon expression under iron starvation, leading to increased presentation of the CFA/I fimbriae.
DISCUSSION
Within the human gastrointestinal tract, pathogenic bacteria are exposed to extreme, rapidly changing environmental conditions such as pH, oxidative stress, and nutrient availability. Therefore, production of virulence factors and the programs associated with virulence must be tightly coordinated for appropriate disease development in response to diverse environmental factors. Iron availability represents a key factor in initiation of bacterial pathogenesis, signaling arrival within the small intestine. Indeed, dietary iron acquisition occurs in the duodenum and upper jejunum of the small intestine to generate a longitudinal gradient in mammals (58).
In ETEC H10407, we found that the major virulence factors CFA/I and LT showed a differential response to iron starvation in the CFA medium. Iron starvation led to increased production of the CFA/I fimbriae but inhibited LT secretion, supporting the hypothesis that ETEC-specific fimbriae and toxins may be differentially regulated. Indeed, differential expression between CFs and the LT has been previously observed in vitro in the presence of glucose or bile salts (10, 13, 44).
Quantification of transcripts and measurement of promoter activity further revealed that iron starvation induced the expression of several genes encoding ETEC virulence factors at the transcriptional level. These included genes encoding the CFA/I fimbriae (cfa operon), the transcriptional regulator CfaD (cfaD), the small secreted protein CexE (cexE), the Aat T1SS (aat), and the Etp two-partner secretion system responsible for secretion of the EtpA adhesin (etp operon). All upregulated genes were located on the major virulence plasmid p948, with CfaD responsible for activation of cfaD, the cfa operon, and cexE (49, 51). The similar expression profiles seen for cexE, aatA, and etpB suggest that these genes may be expressed in tandem with cfa in ETEC to permit early adhesion to epithelial cells upon arrival in the ileum (59). Following initial epithelial cell contact, intimate adhesion events may then be mediated by outer membrane adhesins, such as EaeH, which were not evaluated here (60). In contrast to the known roles of the cfa and etp operons in pathogenesis, the role of cexE remains unknown (49). However, in enteroaggregative E. coli, the Aat secretion system secretes dispersin, a CexE homolog that modifies outer membrane surface charge to permit fimbrial extension (61). An analogous role for Aat in CexE secretion in ETEC has been suggested (50, 62). The similar gene expression profile seen here for both cexE and the aat operon supports this hypothesis.
In contrast to the induction of these plasmid-based genes, genes encoding both curli and the Tia adhesin were repressed under iron starvation, suggesting that they are not required during early colonization by ETEC. Indeed, a recent study of ETEC virulence factors found that Tia is not associated with disease (63). We hypothesize that repression of these nonspecific adhesins may enhance presentation of ETEC-specific colonization factors within the small intestine.
Preliminary analyses of virulence gene expression in the heterologous ETEC strain E24377A also show induction of CF expression under iron starvation for both cooA (CS1) and cstA (CS3), as seen for cfaE (CFA/I) in H10407 (see Fig. S5 in the supplemental material). This suggests that the virulence response to iron starvation may be more widespread among ETEC strains than seen for other environmental factors, including glucose and bile, which are strain dependent (44, 64).
Although iron starvation has a positive effect on toxin expression in other pathogens (2), we found that it did not positively influence the expression of any toxin gene considered here. Similar results were seen for eltB (LT) transcription in E24377A (see Fig. S5 in the supplemental material) and are consistent with previous data showing that ferrous iron has no effect on LT synthesis in ETEC (65). Interestingly, an H10407 ΔleoA mutant was previously shown to have impaired LT secretion from the periplasm (28). LeoA is a dynamin-like GTPase encoded within a chromosomal pathogenicity island, with a major role in LT secretion in H10407 (53). As leoA expression was significantly inhibited with iron starvation, reduced LeoA levels may in turn decrease the amount of secreted LT seen here. Finally, it is important to note that although LT and ST have previously been shown to undergo differential expression (11), neither was induced under iron starvation here. Consistent with recently published data (12), our data support the hypothesis that LT and ST secretion may occur in tandem, responding in a similar manner to key environmental cues.
Although an early study suggested that Fur might repress expression of CFA/I (14), our results indicate that Fur has no major influence on either gene expression or promoter activity of the cfa operon under iron starvation. In addition, cfaA promoter activity was induced under iron starvation in all E. coli deletion mutants evaluated except the ΔiscR mutant, identifying IscR as a possible regulator of the cfa operon. Moreover, RT-qPCR analyses indicate that the iscR gene is subject to upregulation under iron starvation in both ETEC H10407 and E24377A, suggesting that IscR induction may lead to the upregulation of colonization factors (Fig. 6; see Fig. S5 in the supplemental material).
Apo-IscR has been previously implicated in modulating type 1 fimbrial phase variation in E. coli via activation of the fimE recombinase under iron starvation (25). Indeed, a type 2 motif has been identified in the fimE promoter from position −178 to −153 (25). Apo-IscR also binds two sites with high affinity within the suf promoter, from −60 to −26 and from −164 to −132 (66). In the cfaA promoter, we also identified two sites with high affinity in similar locations, overlapping the −35 hexamer and from −163 to −124. Thus, based on site localization, IscR likely functions in a similar manner at the fim, suf, and cfa promoters.
While the regulatory role of Fur in both the iron response and bacterial virulence has been largely documented (2), the role of IscR in virulence regulation continues to be elucidated. The results presented here provide further support for the involvement of IscR in modulation of bacterial virulence and should motivate further investigation concerning the possible coordination of virulence programs in pathogenic E. coli.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Culture Collection at the University of Göteborg for kindly providing ETEC H10407, David Lee (University of Birmingham) for providing H10407 Δfur, and Stephen Savarino (Naval Medical Research Center, Silver Spring, MD) for providing anti-CFA/I serum and ETEC strain E24377A. We also thank Jean-Marc Ghigo (Pasteur Institute, Paris, France) and Stéphan Lacour (Jean Roget Institute, Grenoble, France) for providing E. coli strains, Christophe Beloin (Pasteur Institute, Paris, France) for providing pCA24N constructs, Philippe Telouk (CNRS, ENS Lyon, France) for ICP-MS analyses, and Laurent Aussel (Aix-Marseille University-CNRS) for helpful discussions.
The project described here was supported by Sanofi Pasteur and EZUS.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00214-15.
REFERENCES
- 1.Skaar EP. 2010. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog 6:e1000949. doi: 10.1371/journal.ppat.1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carpenter BM, Whitmire JM, Merrell DS. 2009. This is not your mother's repressor: the complex role of fur in pathogenesis. Infect Immun 77:2590–2601. doi: 10.1128/IAI.00116-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan FA, Fisher MA, Khakoo RA. 2007. Association of hemochromatosis with infectious diseases: expanding spectrum. Int J Infect Dis 11:482–487. doi: 10.1016/j.ijid.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Bullen JJ, Spalding PB, Ward CG, Gutteridge JM. 1991. Hemochromatosis, iron and septicemia caused by Vibrio vulnificus. Arch Intern Med 151:1606–1609. [PubMed] [Google Scholar]
- 5.Robins-Browne RM, Prpic JK. 1985. Effects of iron and desferrioxamine on infections with Yersinia enterocolitica. Infect Immun 47:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kortman GA, Boleij A, Swinkels DW, Tjalsma H. 2012. Iron availability increases the pathogenic potential of Salmonella typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS One 7:e29968. doi: 10.1371/journal.pone.0029968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qadri F, Svennerholm AM, Faruque AS, Sack RB. 2005. Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin Microbiol Rev 18:465–483. doi: 10.1128/CMR.18.3.465-483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy K, Hilliard GM, Hamilton DJ, Luo J, Ostmann MM, Fleckenstein JM. 2009. Enterotoxigenic Escherichia coli EtpA mediates adhesion between flagella and host cells. Nature 457:594–598. doi: 10.1038/nature07568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jordi BJ, Dagberg B, de Haan LA, Hamers AM, van der Zeijst BA, Gaastra W, Uhlin BE. 1992. The positive regulator CfaD overcomes the repression mediated by histone-like protein H-NS (H1) in the CFA/I fimbrial operon of Escherichia coli. EMBO J 11:2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karjalainen TK, Evans DG, Evans DJ, Graham DY, Lee C-H. 1991. Catabolite repression of the colonization factor antigen I (CFA/I) operon of Escherichia coli. Curr Microbiol 23:307–313. doi: 10.1007/BF02104131. [DOI] [Google Scholar]
- 11.Bodero MD, Munson GP. 2009. Cyclic AMP receptor protein-dependent repression of heat-labile enterotoxin. Infect Immun 77:791–798. doi: 10.1128/IAI.00928-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzales L, Ali ZB, Nygren E, Wang Z, Karlsson S, Zhu B, Quiding-Jarbrink M, Sjoling A. 2013. Alkaline pH is a signal for optimal production and secretion of the heat labile toxin, LT in enterotoxigenic Escherichia Coli (ETEC). PLoS One 8:e74069. doi: 10.1371/journal.pone.0074069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haycocks JR, Sharma P, Stringer AM, Wade JT, Grainger DC. 2015. The molecular basis for control of ETEC enterotoxin expression in response to environment and host. PLoS Pathog 11:e1004605. doi: 10.1371/journal.ppat.1004605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karjalainen TK, Evans DG, Evans DJ Jr, Graham DY, Lee CH. 1991. Iron represses the expression of CFA/I fimbriae of enterotoxigenic E. coli. Microb Pathog 11:317–323. doi: 10.1016/0882-4010(91)90017-5. [DOI] [PubMed] [Google Scholar]
- 15.de Lorenzo V, Wee S, Herrero M, Neilands JB. 1987. Operator sequences of the aerobactin operon of plasmid ColV-K30 binding the ferric uptake regulation (fur) repressor. J Bacteriol 169:2624–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masse E, Gottesman S. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci U S A 99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng M, Doan B, Schneider TD, Storz G. 1999. OxyR and SoxRS regulation of fur. J Bacteriol 181:4639–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crack JC, Green J, Thomson AJ, Le Brun NE. 2012. Iron-sulfur cluster sensor-regulators. Curr Opin Chem Biol 16:35–44. doi: 10.1016/j.cbpa.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Nesbit AD, Giel JL, Rose JC, Kiley PJ. 2009. Sequence-specific binding to a subset of IscR-regulated promoters does not require IscR Fe-S cluster ligation. J Mol Biol 387:28–41. doi: 10.1016/j.jmb.2009.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mey AR, Wyckoff EE, Kanukurthy V, Fisher CR, Payne SM. 2005. Iron and Fur regulation in Vibrio cholerae and the role of Fur in virulence. Infect Immun 73:8167–8178. doi: 10.1128/IAI.73.12.8167-8178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palyada K, Threadgill D, Stintzi A. 2004. Iron acquisition and regulation in Campylobacter jejuni. J Bacteriol 186:4714–4729. doi: 10.1128/JB.186.14.4714-4729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim JG, Choi SH. 2014. IscR is a global regulator essential for the pathogenesis of Vibrio vulnificus and induced by host cells. Infect Immun 82:569–78. doi: 10.1128/IAI.01141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SH, Lee BY, Lau GW, Cho YH. 2009. IscR modulates catalase A (KatA) activity, peroxide resistance and full virulence of Pseudomonas aeruginosa PA14. J Microbiol Biotechnol 19:1520–1526. doi: 10.4014/jmb.0906.06028. [DOI] [PubMed] [Google Scholar]
- 24.Reference deleted.
- 25.Wu Y, Outten FW. 2009. IscR controls iron-dependent biofilm formation in Escherichia coli by regulating type I fimbria expression. J Bacteriol 191:1248–1257. doi: 10.1128/JB.01086-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans DG, Evans DJ Jr, Tjoa W. 1977. Hemagglutination of human group A erythrocytes by enterotoxigenic Escherichia coli isolated from adults with diarrhea: correlation with colonization factor. Infect Immun 18:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anantha RP, McVeigh AL, Lee LH, Agnew MK, Cassels FJ, Scott DA, Whittam TS, Savarino SJ. 2004. Evolutionary and functional relationships of colonization factor antigen I and other class 5 adhesive fimbriae of enterotoxigenic Escherichia coli. Infect Immun 72:7190–7201. doi: 10.1128/IAI.72.12.7190-7201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleckenstein JM, Lindler LE, Elsinghorst EA, Dale JB. 2000. Identification of a gene within a pathogenicity island of enterotoxigenic Escherichia coli H10407 required for maximal secretion of the heat-labile enterotoxin. Infect Immun 68:2766–2774. doi: 10.1128/IAI.68.5.2766-2774.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Svennerholm AM, Wiklund G. 1983. Rapid GM1-enzyme-linked immunosorbent assay with visual reading for identification of Escherichia coli heat-labile enterotoxin. J Clin Microbiol 17:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Froehlich B, Husmann L, Caron J, Scott JR. 1994. Regulation of rns, a positive regulatory factor for pili of enterotoxigenic Escherichia coli. J Bacteriol 176:5385–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller WG, Leveau JH, Lindow SE. 2000. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact 13:1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- 33.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299. [DOI] [PubMed] [Google Scholar]
- 37.Sjoling A, Wiklund G, Savarino SJ, Cohen DI, Svennerholm AM. 2007. Comparative analyses of phenotypic and genotypic methods for detection of enterotoxigenic Escherichia coli toxins and colonization factors. J Clin Microbiol 45:3295–3301. doi: 10.1128/JCM.00471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36. [PubMed] [Google Scholar]
- 39.Bailey TL, Gribskov M. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz CJ, Giel JL, Patschkowski T, Luther C, Ruzicka FJ, Beinert H, Kiley PJ. 2001. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc Natl Acad Sci U S A 98:14895–14900. doi: 10.1073/pnas.251550898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duprey A, Chansavang V, Fremion F, Gonthier C, Louis Y, Lejeune P, Springer F, Desjardin V, Rodrigue A, Dorel C. 2014. “NiCo Buster”: engineering E. coli for fast and efficient capture of cobalt and nickel. J Biol Eng 8:19. doi: 10.1186/1754-1611-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. 2006. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol 60:1058–1075. doi: 10.1111/j.1365-2958.2006.05160.x. [DOI] [PubMed] [Google Scholar]
- 43.Evans DJ Jr, Evans DG, Gorbach SL. 1973. Production of vascular permeability factor by enterotoxigenic Escherichia coli isolated from man. Infect Immun 8:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahl JW, Rasko DA. 2012. Analysis of global transcriptional profiles of enterotoxigenic Escherichia coli isolate E24377A. Infect Immun 80:1232–1242. doi: 10.1128/IAI.06138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McHugh JP, Rodriguez-Quinones F, Abdul-Tehrani H, Svistunenko DA, Poole RK, Cooper CE, Andrews SC. 2003. Global iron-dependent gene regulation in Escherichia coliA new mechanism for iron homeostasis. J Biol Chem 278:29478–29486. [DOI] [PubMed] [Google Scholar]
- 46.Hancock V, Ferrieres L, Klemm P. 2008. The ferric yersiniabactin uptake receptor FyuA is required for efficient biofilm formation by urinary tract infectious Escherichia coli in human urine. Microbiology 154:167–175. doi: 10.1099/mic.0.2007/011981-0. [DOI] [PubMed] [Google Scholar]
- 47.De Lorenzo V, Herrero M, Giovannini F, Neilands JB. 1988. Fur (ferric uptake regulation) protein and CAP (catabolite-activator protein) modulate transcription of fur gene in Escherichia coli. Eur J Biochem 173:537–546. doi: 10.1111/j.1432-1033.1988.tb14032.x. [DOI] [PubMed] [Google Scholar]
- 48.Lin X-M, Wu L-N, Li H, Wang S-Y, Peng X-X. 2008. Downregulation of Tsx and OmpW and upregulation of OmpX are required for iron homeostasis in Escherichia coli. J Proteome Res 7:1235–1243. doi: 10.1021/pr7005928. [DOI] [PubMed] [Google Scholar]
- 49.Pilonieta MC, Bodero MD, Munson GP. 2007. CfaD-dependent expression of a novel extracytoplasmic protein from enterotoxigenic Escherichia coli. J Bacteriol 189:5060–5067. doi: 10.1128/JB.00131-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishi J, Sheikh J, Mizuguchi K, Luisi B, Burland V, Boutin A, Rose DJ, Blattner FR, Nataro JP. 2003. The export of coat protein from enteroaggregative Escherichia coli by a specific ATP-binding cassette transporter system. J Biol Chem 278:45680–45689. doi: 10.1074/jbc.M306413200. [DOI] [PubMed] [Google Scholar]
- 51.Munson GP, Scott JR. 2000. Rns, a virulence regulator within the AraC family, requires binding sites upstream and downstream of its own promoter to function as an activator. Mol Microbiol 36:1391–1402. [DOI] [PubMed] [Google Scholar]
- 52.Fleckenstein JM, Kopecko DJ, Warren RL, Elsinghorst EA. 1996. Molecular characterization of the tia invasion locus from enterotoxigenic Escherichia coli. Infect Immun 64:2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michie KA, Boysen A, Low HH, Moller-Jensen J, Lowe J. 2014. LeoA, B and C from enterotoxigenic Escherichia coli (ETEC) are bacterial dynamins. PLoS One 9:e107211. doi: 10.1371/journal.pone.0107211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee DJ, Bingle LE, Heurlier K, Pallen MJ, Penn CW, Busby SJ, Hobman JL. 2009. Gene doctoring: a method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol 9:252. doi: 10.1186/1471-2180-9-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vinella D, Loiseau L, Ollagnier de Choudens S, Fontecave M, Barras F. 2013. In vivo [Fe-S] cluster acquisition by IscR and NsrR, two stress regulators in Escherichia coli. Mol Microbiol 87:493–508. doi: 10.1111/mmi.12135. [DOI] [PubMed] [Google Scholar]
- 57.Rajagopalan S, Teter SJ, Zwart PH, Brennan RG, Phillips KJ, Kiley PJ. 2013. Studies of IscR reveal a unique mechanism for metal-dependent regulation of DNA binding specificity. Nat Struct Mol Biol 20:740–747. doi: 10.1038/nsmb.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muir A, Hopfer U. 1985. Regional specificity of iron uptake by small intestinal brush-border membranes from normal and iron-deficient mice. Am J Physiol 248:G376–G379. [DOI] [PubMed] [Google Scholar]
- 59.Allen KP, Randolph MM, Fleckenstein JM. 2006. Importance of heat-labile enterotoxin in colonization of the adult mouse small intestine by human enterotoxigenic Escherichia coli strains. Infect Immun 74:869–875. doi: 10.1128/IAI.74.2.869-875.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheikh A, Luo Q, Roy K, Shabaan S, Kumar P, Qadri F, Fleckenstein JM. 2014. Contribution of the highly conserved EaeH surface protein to enterotoxigenic Escherichia coli pathogenesis. Infect Immun 82:3657–3666. doi: 10.1128/IAI.01890-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheikh J, Czeczulin JR, Harrington S, Hicks S, Henderson IR, Le Bouguenec C, Gounon P, Phillips A, Nataro JP. 2002. A novel dispersin protein in enteroaggregative Escherichia coli. J Clin Invest 110:1329–1337. doi: 10.1172/JCI16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crossman LC, Chaudhuri RR, Beatson SA, Wells TJ, Desvaux M, Cunningham AF, Petty NK, Mahon V, Brinkley C, Hobman JL, Savarino SJ, Turner SM, Pallen MJ, Penn CW, Parkhill J, Turner AK, Johnson TJ, Thomson NR, Smith SG, Henderson IR. 2010. A commensal gone bad: complete genome sequence of the prototypical enterotoxigenic Escherichia coli strain H10407. J Bacteriol 192:5822–5831. doi: 10.1128/JB.00710-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gonzales L, Sanchez S, Zambrana S, Iniguez V, Wiklund G, Svennerholm AM, Sjoling A. 2013. Molecular characterization of enterotoxigenic Escherichia coli isolates recovered from children with diarrhea during a 4-year period (2007 to 2010) in Bolivia. J Clin Microbiol 51:1219–1225. doi: 10.1128/JCM.02971-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McConnell MM, Chart H, Field AM, Hibberd M, Rowe B. 1989. Characterization of a putative colonization factor (PCFO166) of enterotoxigenic Escherichia coli of serogroup O166. J Gen Microbiol 135:1135–1144. [DOI] [PubMed] [Google Scholar]
- 65.Gilligan PH, Robertson DC. 1979. Nutritional requirements for synthesis of heat-labile enterotoxin by enterotoxigenic strains of Escherichia coli. Infect Immun 23:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yeo WS, Lee JH, Lee KC, Roe JH. 2006. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol Microbiol 61:206–218. doi: 10.1111/j.1365-2958.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- 67.Evans DG, Silver RP, Evans DJ Jr, Chase DG, Gorbach SL. 1975. Plasmid-controlled colonization factor associated with virulence in Ecsherichia coli enterotoxigenic for humans. Infect Immun 12:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.