

ABSTRACT
Vancomycin resistance in Gram-positive bacteria results from the replacement of the d-alanyl–d-alanine target of peptidoglycan precursors with d-alanyl–d-lactate or d-alanyl–d-serine (d-Ala-d-Ser), to which vancomycin has low binding affinity. VanT is one of the proteins required for the production of d-Ala-d-Ser-terminating precursors by converting l-Ser to d-Ser. VanT is composed of two domains, an N-terminal membrane-bound domain, likely involved in l-Ser uptake, and a C-terminal cytoplasmic catalytic domain which is related to bacterial alanine racemases. To gain insight into the molecular function of VanT, the crystal structure of the catalytic domain of VanTG from VanG-type resistant Enterococcus faecalis BM4518 was determined. The structure showed significant similarity to type III pyridoxal 5′-phosphate (PLP)-dependent alanine racemases, which are essential for peptidoglycan synthesis. Comparative structural analysis between VanTG and alanine racemases as well as site-directed mutagenesis identified three specific active site positions centered around Asn696 which are responsible for the l-amino acid specificity. This analysis also suggested that VanT racemases evolved from regular alanine racemases by acquiring additional selectivity toward serine while preserving that for alanine. The 4-fold-lower relative catalytic efficiency of VanTG against l-Ser versus l-Ala implied that this enzyme relies on its membrane-bound domain for l-Ser transport to increase the overall rate of d-Ser production. These findings illustrate how vancomycin pressure selected for molecular adaptation of a housekeeping enzyme to a bifunctional enzyme to allow for peptidoglycan remodeling, a strategy increasingly observed in antibiotic-resistant bacteria.
IMPORTANCE
Vancomycin is one of the drugs of last resort against Gram-positive antibiotic-resistant pathogens. However, bacteria have evolved a sophisticated mechanism which remodels the drug target, the d-alanine ending precursors in cell wall synthesis, into precursors terminating with d-lactate or d-serine, to which vancomycin has less affinity. d-Ser is synthesized by VanT serine racemase, which has two unusual characteristics: (i) it is one of the few serine racemases identified in bacteria and (ii) it contains a membrane-bound domain involved in l-Ser uptake. The structure of the catalytic domain of VanTG showed high similarity to alanine racemases, and we identified three specific active site substitutions responsible for l-Ser specificity. The data provide the molecular basis for VanT evolution to a bifunctional enzyme coordinating both transport and racemization. Our findings also illustrate the evolution of the essential alanine racemase into a vancomycin resistance enzyme in response to antibiotic pressure.
INTRODUCTION
Glycopeptides, such as vancomycin and teicoplanin, are key agents for the treatment of infections caused by antibiotic-resistant Gram-positive bacteria, but widespread clinical resistance to these drugs has become a major public health problem (1). Vancomycin blocks bacterial cell wall synthesis by binding the d-alanyl–d-alanine (d-Ala-d-Ala) C-terminal dipeptide of the muramyl pentapeptide moiety of peptidoglycan precursors, thereby interfering with the transglycosylase and transpeptidase activities of penicillin-binding proteins (PBPs) (2). Resistance arises from the ability of bacteria to produce modified peptidoglycan precursors ending in d-alanyl–d-lactate (d-Ala-d-Lac) or d-alanyl–d-serine (d-Ala-d-Ser), to which glycopeptides exhibit weak affinity (3). Nine vancomycin resistance (van) gene clusters have been described in enterococci and have been distinguished according to their specificities to incorporate either d-Ala-d-Lac (VanA, VanB, VanD, and VanM) or d-Ala-d-Ser (VanC, VanG, VanE, VanL, and VanN) in late precursors (3–6).
Synthesis of the d-Ala-d-Ser dipeptide requires two proteins: VanT, a membrane-bound racemase that converts l-Ser into d-Ser, and a ligase (VanC, VanG, VanE, VanL, and VanN) that links d-Ser to d-Ala (7). The pool of vancomycin-susceptible precursors is reduced by the hydrolysis of the d-Ala terminating dipeptide and pentapeptide species by the d,d-dipeptidase/d,d-pentapeptidase VanXY (8).
Serine racemases are mainly found in mammals, where d-Ser acts as a modulator of brain function (9, 10). In prokaryotes, this activity appears to be present primarily in glycopeptide-resistant Gram-positive bacteria (11). The VanT racemase was first characterized in a VanC-type-resistant Enterococcus gallinarum isolate (4, 11–13). The vanTC gene encodes a 698-amino-acid polypeptide containing an N-terminal (residues 1 to 322) membrane-bound domain that belongs to the acyltransferase superfamily (Pfam clan CL0316) whose role is attributed to l-Ser transport (4), and a C-terminal (residues 323 to 698) cytoplasmic domain where the catalytic activity is located (11, 13). This domain architecture is conserved among the VanTE, VanTG, and VanTN representatives (14–16) but differs in the vanL gene cluster (5), where two separate genes, vanTmL and vanTrL, encode, respectively, the membrane-bound and the cytosolic domains, implying that these two functionalities are required for resistance. The C-terminal domain of VanTC is 25% identical in sequence with Enterococcus faecalis alanine rasemase (EfAlr) (17) and conserves the pyridoxal 5′-phosphate (PLP) attachment motif present in the Alr protein. This domain functions as an Alr as it interconverts l-Ala and d-Ala, although this activity is 6-fold lower than that toward l-Ser (13). Alrs belong to the type III class of PLP-dependent enzymes (10) based on their structural arrangement (17–20). All characterized members of this enzyme family are homodimeric, with the two protomers associated in a head-to-tail fashion. Each subunit displays an N-terminal (α/β)8/TIM barrel domain and a C-terminal β-barrel fold domain. The active site is located in a cleft between the two domains at the dimer interface, where the PLP cofactor is tethered as an internal aldimine (Schiff base) with the ε-amino group of a catalytic lysine.
Alrs are highly specific for alanine racemization, and structure-function studies have led to a generally accepted reaction mechanism which involves a lysine and a tyrosine acting as acid-base catalysts for, respectively, donating and abstracting an α-hydrogen from the bound isomer of the substrate, d-Ala or l-Ala (20–22). In the l → d catalytic conversion, the l-Ala substrate first displaces the lysine and forms with PLP an l-external aldimine, which is then deprotonated by the tyrosine from the other subunit to produce an unstable carbanion. This carbanion is then reprotonated on the opposite face by the lysine to generate a d-external aldimine with PLP. The final step is the loss of the d-Ala product by transamination, and this recycles the internal lysine l-aldimine (20). These two key catalytic residues are conserved between Alrs and VanT enzymes (11), suggesting that the mechanism for racemization of serine is similar to that of alanine.
While this comparative analysis provided insights into the general reaction catalyzed by VanT enzymes, the molecular basis of their specificity toward l-Ser has not been investigated in detail. A study of the Alr from Bacillus stearothermophilus (BsAlr) suggested that the highly conserved Tyr354 is a key determinant for alanine specificity (23). Its replacement with an asparagine, which occupies the corresponding position in VanT enzymes, led to a 62-fold increase in serine racemase activity with no significant change in alanine racemization (23). However, no VanT enzyme has been structurally characterized, limiting our understanding of the molecular function of this unique serine racemase family and its role in vancomycin resistance.
In this study, we determined the crystal structure of the VanTG catalytic domain and studied the role of active site residues in catalysis and specificity by site-directed mutagenesis. Our data provide the molecular basis for the altered specificity of VanT toward l-Ser and provide clues as to the possible route of evolution of this resistance-related activity from that of common alanine racemases.
RESULTS
VanTG structure, active site, and comparison with Alrs.
In order to determine the structure of VanT enzymes, we expressed and purified the cytoplasmic domains of the VanT proteins from the vanC, vanG, vanL, and vanN vancomycin resistance operons. We successfully crystallized the VanTG domain encompassing residues 339 to 712 and determined its structure by the single anomalous dispersion (SAD) method to a resolution of 2.02 Å using a selenomethionine-derivatized protein crystal (Table 1).
TABLE 1 .
VanTG crystal X-ray diffraction data collection and refinement statistics
| Parameter | Data |
|---|---|
| Diffraction data | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 78.7, 82.3, 117.5 |
| β (°) | 90.1 |
| Resolution (Å) | 20.0–2.00 |
| Rsyma | 0.112 (0.547) |
| I/σ(I) | 16.8 (2.86) |
| Completeness (%) | 100 (100) |
| Redundancy | 7.7 (6.8) |
| Refinement statistics | |
| Resolution (Å) | 19.9–2.02 |
| No. of reflections (working, test) | 97,095, 1,871 |
| R factor/free R factorb | 18.2/23.3 (33.1/37.3) |
| No. of refined atoms | |
| Protein (no. of molecules) | 11,591 (4) |
| Solvent | 28 |
| Water | 700 |
| B factors | |
| Protein | 35.2 |
| Solvent | 28.9 |
| Water | 35.8 |
| RMSD of: | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.037 |
The crystal structure of VanTG revealed four protein chains arranged into two almost-identical homodimers which superimposed with a root mean square deviation (RMSD) of 0.6 Å across equivalent Cα atoms (their entire chains), and thus we focused our analysis on the “AB” dimer. The subunits for each VanTG dimer interacted in a “head-to-tail” fashion and displayed an N-terminal (α/β)8/TIM barrel and a C-terminal 9-stranded β-barrel. Both domains in the VanTG subunit contributed to the dimerization interface, which covered 2,615 Å2 and involved over 75 amino acids from each subunit (Fig. 1A). This extensive nature of the dimerization interface is consistent with the predominantly dimeric nature of VanTG in solution, as determined by size exclusion chromatography (data not shown). The structure showed significant similarity with that of EfAlr (17), BsAlr (20), and Vibrio cholerae Alr (VcAlr) (Fig. 1B). A structural homology search also revealed that VanTG is similar to other structurally characterized racemases, such as the broad-spectrum racemase BsrV and Bar from, respectively, V. cholerae (24) (Fig. 1B) and Pseudomonas putida (25), and to the lysine racemase Lyr from Proteus mirabilis (25). This analysis also confirmed that VanTG is a member of the type III PLP-dependent class of enzymes with significant overall structural similarity to specific and nonspecific amino acid racemases.
FIG 1 .
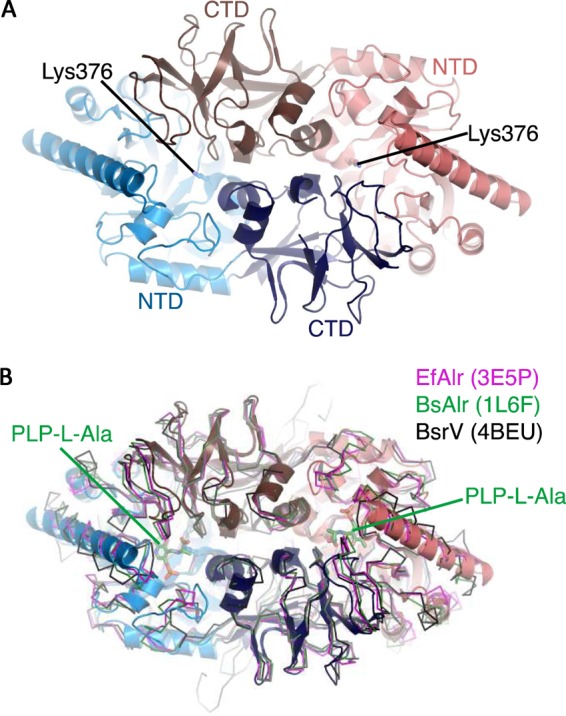
Structure of VanTG. (A) Homodimer of VanTG, showing the head-to-tail arrangement. One subunit (red and brown) contains the (α/β)8/TIM barrel domain at the N terminus (red) and the β-strands domain at the C terminus (brown). The PLP-binding site is indicated by the conserved Lys376 in the N-terminus. (B) Superposition of the VanTG homodimer colored as for panel A with various alanine racemase homologues shown in a ribbon diagram. The PLP cofactor bound to l-Ala in the BsAlr structure is shown as green sticks.
Multiple-sequence alignment of Alr, broad-spectrum, lysine-specific, and VanT racemases indicated that all these enzymes share active site residues (Fig. 2). However, a phylogenetic reconstruction differentiated three distinct groups according to amino acid specificity, with Lyr grouping with the broad-spectrum racemases (Fig. 2). This analysis indicated that while these enzymes share an overall structural architecture, they clearly contain distinct sequence signatures that correlate with their substrate specificity.
FIG 2 .

Sequence relationships of bacterial amino acid racemases. (Top) Multiple-sequence alignment of VanTs, Alrs, and broad-spectrum racemases. VanTG was aligned with VanT racemases (red) from van operons (VanTC, VanTE, VanTN, and VanTrL), broad-spectrum racemases (green) (V. cholerae BsrV, P. putida Bar, and P. mirabilis PmLyr), and Alrs homologues (blue) (E. faecalis Alr, C. difficile Alr, and B. stearothermophilus Alr). The numbering above the sequences corresponds to that of VanTG. Secondary elements from VanTG and BsAlr structures are shown, respectively, above and below the primary sequences. Conserved amino acids are shown in blue, those of the PLP-binding site are in black, and those responsible for VanT specificity are red. The key Lys/Tyr acid-base pair for the abstraction/donation of the α-hydrogen is indicated in green. The figure was generated with ESPript 3.0 (bottom). Phylogenetic reconstruction of the aligned racemases was performed using the maximum likelihood algorithm in the MEGA package and visualized with FigTree.
As with previously characterized racemase enzymes, the VanTG active site is located at the interface between the N-terminal TIM barrel and the C-terminal β-barrel domain at the bottom of a deep, positively charged pocket (see Fig. S1 in the supplemental material). We did not observe electron density consistent with the presence of PLP in any active site of the VanTG structure, based on a close analysis of the simulated annealing omit map (see Fig. S2 in the supplemental material). We experimented with modeling of PLP bound in an aldimine bond with each of the Lys376 residues; however, crystallographic refinement of this model resulted in negative density features surrounding multiple cofactor atoms, including the presumptive aldimine bond. Alternatively, refinement of the occupancy of the cofactor resulted in only insignificant occupancy values (no greater than 0.29, as in the chain C active site). Therefore, we were confident that our crystallographic data were best represented by one sulfate and multiple water molecules in each active site rather than the presence of the PLP cofactor. Crystallization of VanTG in the presence of excess PLP also did not result in determination of a PLP-bound VanTG structure. However, the superposition of the PLP-bound BsAlr (PDB 1L6F) and VanTG structures showed a high level of structural congruence between the PLP-coordinating and active site residues (Fig. 3). The superposition did not reveal any significant steric clashes between the PLP molecule and VanTG active site residues, suggesting that the position of PLP is conserved in these enzymes. This alignment indicated that the VanTG residues which are likely involved in PLP binding include Lys376, Tyr380, Arg470, His498, Gly559, Val560, and Tyr622′ (where ′ designates the residue from the other VanTG subunit). Notably, each of these residues is conserved between all Alr and VanT homologs, except for Val560, which is replaced by either an Ile or a Gly (Fig. 2). The hydrogen-bonding network in the VanTG active site involving the backbone amides of Tyr622′ and Met652′ and two water molecules was spatially identical to a similar network in BsAlr involving Tyr284′ and Met312′, a water molecule, and the carboxylate of the amino acid substrate (Fig. 3). The lysine/tyrosine acid-base pair (Lys39 and Tyr265) in BsAlr corresponded to VanTG residues Lys376 and Tyr603, and this pair is also conserved in other VanT enzymes (Fig. 2). However, the position of Tyr603 in VanTG was different from that of Tyr265 in BsAlr, likely due to the lack of PLP bound to VanTG, which led to the change in conformation of the loop carrying this catalytic residue (Fig. 3).
FIG 3 .
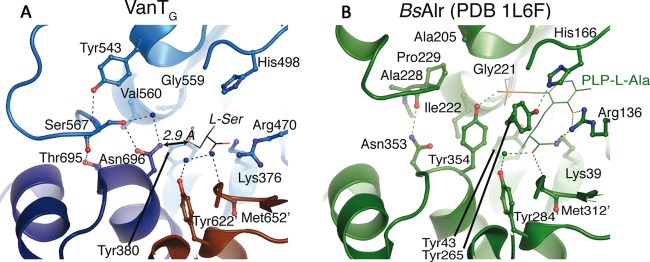
Comparison of VanTG (A) and BsAlr (B) active sites. Active site residues are shown by sticks. The PLP-binding site in VanTG is shown in blue, and the adjacent monomer is brown. l-Ser, shown as stick lines, was modeled by manual placement onto the coordinates of l-Ala from PLP-Ala in the BsAlr structure (green). Water molecules are indicated as spheres.
Taken together, these observations indicated significant structural similarities between VanT and Alr enzymes which extend into the catalytic site, and from these we expect that both enzyme groups rely on the same catalytic mechanism. The degree of similarity also implied that the distinct amino acid specificity of these enzyme groups could be structurally rationalized.
Role of critical residues in serine selectivity.
Kinetic analysis demonstrated that VanTG possesses racemase activity against both l-Ala and l-Ser substrates (Fig. 4A; see also Table S2 in the supplemental material) and is 4-fold more efficient in converting l-Ala than l-Ser, based on the kcat/Km ratios (Fig. 4B). The catalytic efficiency of VanTG against l-Ala was similar to that of VcAlr (24) but much less than that of BsAlr (23).
FIG 4 .

Comparison of alanine and serine racemase activities of VanTG mutants. (A) Catalytic efficiencies of alanine and serine racemase activities. Detailed kinetic parameters (mean values of at least three independent measurements) are indicated in Table S2 in the supplemental material. (B) l-Ala/ l-Ser catalytic efficiency ratios. Values are indicated on top of the bars.
To gain deeper insight into the structural elements responsible for VanTG activity against l-Ser, we analyzed the differences between active sites of VanTG and of the strict l-Ala racemase BsAlr. The VanTG active site contained 4 amino acids (Tyr543, Ser567, Thr695, and Asn696) that formed a hydrogen-bonded network not observed in the BsAlr enzyme which was due to a change in the conformation of the loop containing Ser567 (Fig. 3). Importantly, Asn696 was positioned within 3.5 Å of the methyl group of PLP–l-Ala bound to BsAlr. A model of l-Ser at this position showed its side chain hydroxyl within 2.9 Å of Asn696 (Fig. 3); therefore, this residue would be appropriately positioned to interact with the serine substrate. Furthermore, in VanTG, the Asn696 and Ser567 residues form hydrogen bonds with a water molecule that may assist in coordination of PLP, since it occupies the same position as BsAlr Tyr354, whose hydroxyl interacts with the PLP phosphate group. All four positions were conserved within, but not between, the VanT and Alr classes (i.e., Tyr-Ser-Thr-Asn in VanT enzymes and Ala-Pro-His/Asn-Tyr in Alr enzymes), suggesting that they play key but different roles in their respective enzyme classes (Fig. 2).
Based on this information, we probed the VanT-specific active site residues Tyr543, Ser567, Thr695, and Asn696 by site-directed mutagenesis to elucidate their contributions to substrate selectivity. We constructed 13 VanTG variants by replacing these residues with their equivalents in BsAlr and tested them for their ability to catalyze the conversion of l-Ala and l-Ser (see Table S2 in the supplemental material).
The kinetic parameters of the VanTG Tyr543Ala and Thr695Ser mutants were similar to those of the wild-type enzyme (Fig. 4A; see also Table S2 in the supplemental material). This indicated that a single alteration in the hydrogen bonds formed by these residues is not sufficient to affect VanTG racemase activity or substrate selectivity. In contrast, the Ser567Pro mutation led to a significant decrease in both racemase activities, affecting the Km and kcat values for l-Ala and l-Ser conversion (Fig. 4A; see also Table S2); the corresponding catalytic efficiencies dropped 12- and 8-fold, respectively. Since the serine/alanine racemase efficiency ratio for this VanTG variant did not change significantly, 2.7 versus 4.4 for the wild type (Fig. 4B), it is likely that the observed effect is due to destabilization of the active site. In support of this notion, the combination of Ser567Pro with the Tyr543Ala substitution had a less detrimental effect on the enzyme’s activity, while the Tyr543Ala/Ser567Pro double mutant retained significant activity against l-Ala and l-Ser (Fig. 4A; see also Table S2). The latter substitution would provide the space necessary to accommodate the larger proline side chain. These results indicated that, although Tyr543, Thr695, and Ser567 active site residues are conserved among VanT enzymes (Fig. 2), they have no direct effect on the substrate specificity of VanTG.
The remaining VanT-specific active site residue VanTG Asn696 corresponded to Tyr354 in BsAlr. The mutation Asn696Ala did not have a strong effect on the catalytic efficiency against either l-Ala or l-Ser. This substitution reduced the kcat values for l-Ala and l-Ser conversion by 3- and 4-fold, respectively, but this effect was offset by corresponding 3- and 4-fold drops in Km values (see Table S2 in the supplemental material). In sharp contrast, the Asn696Tyr substitution resulted in a 30-fold drop in l-Ser conversion without affecting the enzyme’s activity against l-Ala, leading to an increase of the alanine/serine racemase efficiency ratio up to 217 (Fig. 4B). Importantly, the Km value for the activity of the Asn696Tyr mutant against l-Ser increased 5-fold and that for l-Ala decreased 4-fold (see Table S2). These observations indicated that Asn696 is critical to confer VanTG substrate selectivity and that a tyrosine substitution at this position likely obstructs the binding of the larger side chain of the l-Ser substrate.
We thus postulated that the effect of the Asn696Tyr mutation on VanTG activity against l-Ala can be offset by additional mutations that would allow for full recovery of the activity against this substrate. This strategy could have been adopted in Alr, and thus additional mutations may effectively convert VanTG into an alanine racemase. To test this hypothesis, we generated the VanTG double mutants Tyr543Ala/Asn696Tyr and Ser567Pro/Asn696Tyr and the triple mutant Tyr543Ala/Ser567Pro/Asn696Tyr. Kinetic analysis of these variants showed that the two double mutants had higher alanine/serine efficiency ratios than the wild-type enzyme (65 and 128, respectively, versus 4.4 for the wild type). These values, however, were much lower than that of the Asn696Tyr mutant (217) (Fig. 4B). The fact that the Ser567Pro/Asn696Tyr double mutation increased serine racemase activity to an extent lower than Asn696Tyr could reflect the decrease in conformational flexibility necessary for accommodating the larger tyrosine residue caused by the Ser567Pro replacement. Strikingly, the triple Tyr543Ala/Ser567Pro/Asn696Tyr mutant was severely compromised in serine racemase activity, with a 7-fold increase in the Km and an 11-fold drop in the kcat value for l-Ser (see Table S2 in the supplemental material). In contrast, the activity of this mutant against l-Ala was less affected and showed a 4-fold change in both the Km and kcat values. The catalytic efficiency of the triple mutant for l-Ser conversion was severely reduced and 75-fold lower than that of the wild type. This VanTG variant demonstrated a catalytic efficiency for l-Ala conversion comparable to that of the wild type, and thus its alanine/serine racemase activity ratio was the highest observed, 345, in the variants (Fig. 4B). Combined, our data point to Asn696 as the key residue for VanTG serine racemase specificity and that substitution of its chemical environment by the BsAlr equivalents, as shown by analysis of the triple Tyr543Ala/Ser567Pro/Asn696Tyr mutant, resulted in nearly complete conversion of VanTG into a strict alanine racemase.
DISCUSSION
Bacteria use d-amino acids, commonly d-Ala and d-Glu, as building blocks for their peptidoglycan layer, and this isomer is thought to provide resistance to most proteases. Emerging evidence suggests that d-amino acids regulate cell wall remodeling and strength (26) and can affect biofilm formation and stability (27). Amino acid racemases, which convert the natural l-amino acids to their d-counterparts, play important roles in bacterial biology. They are classified as PLP-dependent and -independent enzymes which follow two distinct reaction mechanisms (10). Among the PLP-dependent group, the alanine racemases (Alrs) are ubiquitous and essential for growth of bacteria but are absent in mammals, which makes them attractive targets for antimicrobials. Consequently, Alrs have been extensively studied for their reaction chemistry and three-dimensional structure, providing a good understanding of their molecular mechanism (17, 19, 20). In contrast, PLP-dependent serine racemases are very rare in bacteria and, to the best of our knowledge, the only known role of these enzymes is for incorporation of d-Ser in the peptidoglycan of vancomycin-resistant bacteria (28). Broad-spectrum racemases, including BsrV from V. cholerae, produce d-Ser (24), but the biological role of this d-amino acid is unknown. The VanT vancomycin resistance serine racemases are similar to Alrs (11), suggesting that these proteins could be derived from the same ancestor. Yet, VanTs are distinct from Alr based on the presence of an atypical membrane-bound domain that is presumably involved in l-Ser transport (4).
As the role of d-Ser in vancomycin resistance has been clearly established, we were interested in elucidating the molecular basis and the evolution of VanT specificity. We thus determined the crystal structure of the VanTG C-terminal catalytic domain. The structure showed significant overall similarity with those of Alrs from B. stearothermophilus (20) and E. faecalis (17). We took advantage of this homology to probe the active site of VanTG by using site-directed mutagenesis.
Analysis of the VanTG and BsAlr structures supports the notion that VanT and Alr have a common ancestor (11). The evolution in substrate specificity was clearly dependent on substitution of residues at key positions of the active site surrounding the substrate amino acid side chain. Mutagenesis pointed to the crucial role of the conserved VanTG Asn696 in serine recognition. Replacement of this residue by a tyrosine, which would mimic the active site in Alrs, decreased the ability of VanTG for l-Ser conversion by 30-fold without affecting alanine racemase activity. This result is in agreement with the finding that the reverse mutation, Tyr354Asn, in BsAlr increases serine racemase activity by 62-fold (23). However, in contrast to this study, where BsAlr Tyr354Asn had a relatively minor effect on the Km for l-Ser, the Km value of the VanTG Asn696Tyr mutant for l-Ser was 5-fold higher than that of the wild type. Thus, Asn696Tyr in VanTG is likely to cause steric hindrance to the binding of l-Ser. Since the turnover of l-Ser by the Asn696Tyr mutant was decreased by 3-fold, asparagine at this position must play an indirect role in catalysis, perhaps by stabilizing the carbanion intermediate through a hydrogen bond to the serine hydroxyl side chain; this possibility is consistent with the modeling of l-Ser into the active site of VanTG structure and the analysis of the BsAlr Tyr354Asn mutant (23).
Mutagenesis also highlighted the role of VanTG active site residues proximal to Asn696. The steric hindrance imposed by Tyr696 was less pronounced in the double mutants (see Table S2 in the supplemental material); the additional mutations might provide space necessary to accommodate Tyr696. The VanTG triple mutant (Tyr543Ala/Ser567Pro/Asn696Tyr) demonstrated almost exclusive alanine racemase activity, consistent with the hypothesis that the hydrogen-bonding network of Tyr543, Ser567, Thr695, and Asn696 positions the Asn696 amide appropriately for interaction with the l-Ser hydroxyl group.
The importance of this area of the active site of racemases in determining amino acid specificity is supported by the recent discovery of broad-spectrum amino acid racemases (24). A comparison of BsrV, which is active against 10 amino acids, with VanTG indicated that the PLP-coordinating residues are conserved while the 4 amino acids that were subjected to mutagenesis in VanTG are not (Fig. 2); specifically, VanTG Asn696 was equivalent to Pro391 in BsrV. Such variation may lead to a more accessible active site in BsrV compared to Alr or VanTG, resulting in broader specificity. Similarly, the Bar and Lyr l-lysine-specific racemases had smaller amino acids (alanine and threonine, respectively) at the same position, enabling these enzymes to accommodate the longer lysine side chain. These observations are in further support of the relevance of this area of the racemase active sites for specificity.
Many antibiotic resistance enzymes can be traced back to primary “housekeeping” functions in bacterial biology, and the current understanding suggests that mutations, gene duplications, or overexpression of essential genes can provide new or extended specificities. This “proto-resistance” concept (29) is supported by the evolution of several antibiotic resistance enzymes, such as the β-lactamases (30) and RND efflux systems (31). We have shown that such a phenomenon occurred in the evolution of the VanXY vancomycin resistance d,d-dipeptidase/d,d-pentapeptidase. This enzyme is related in sequence and structure to members of the M15B peptidase family, which includes d-Ala-d-Ala-containing pentapeptide-specific hydrolases (8). VanY, a d-Ala-d-Ala-specific pentapeptidase, evolved into VanXY through acquisition of the “bisubstrate loop,” which added hydrolysis of d-Ala-d-Ala to that of a d-Ala-d-Ala-containing pentapeptide. Furthermore, this adaptive functionality is specific: VanXY is not active against the dipeptide and pentapeptide ending in d-Ser, provided by the molecular architecture of the active site. VanTG followed a similar evolutionary path: it retained activity against l-Ala but acquired additional specificity toward l-Ser due to the active site modifications centered around Asn696.
Replacement of a preexisting strict alanine racemase by a strict serine racemase would appear more efficient to confer resistance rather than evolution to an enzyme with both specificities. However, given that VanTG retains alanine racemase activity, the mutations acquired by a VanT-Alr progenitor were probably insufficient to evolve with a strict serine racemase. This could result from the fact that, even with the presence of Asn696, the active site of VanTG can still accommodate the smaller l-Ala. Thus, VanT enzymes have evolved by expanding, rather than switching, their substrate specificity compared to their VanT-Alr progenitors.
Given the dual specificity of VanTG and its relatively low activity toward l-Ser, it is likely that fusion of the racemase domain with a membrane-bound acyltransferase which imports l-Ser provides improved racemization and thus better adaptation for vancomycin resistance. Fusion of two genes with complementary functions is a strategy for antibiotic resistance evolution, which has been increasingly observed and results in extremely broad-spectrum resistance (32). It also has the advantage of gene coexpression, which reduces the biological cost associated with resistance (33). The most striking example is the bifunctional AAC(6′)/APH(2″) in Gram-positive cocci, which combines two aminoglycoside-modifying activities, acetylation and phosphorylation, with complementary substrate profiles as separate domains of a single protein (34). Of note, all the VanT racemases, except that of VanL-type resistance, resulted from gene fusion and have evolved as bifunctional enzymes (5). In VanL, the two functions are encoded by separate and adjacent genes; this likely represents an intermediate step in evolution. Apparently, this strategy was an efficient alternative to further specification of a racemase active site to an l-Ser substrate which, as already mentioned, might not be possible due to the smaller l-Ala side chain that could still be accommodated in the l-Ser-binding site.
Overall, this detailed structural and functional analysis of VanTG reinforces the notion that antibiotic resistance arises from preexisting housekeeping enzymes via acquisition of genetic changes that lead to expanded specificity or higher activity. The selective pressure of antibiotics can also select for the emergence and spread of bifunctional enzymes, and VanT racemases provide a prime example for a sophisticated evolution that has combined both strategies.
MATERIALS AND METHODS
Cloning and mutagenesis.
The C-terminal portion of vanTG (encoding amino acids 339 to 712) was amplified from E. faecalis BM4518 genomic DNA by PCR and cloned into p15Tv-LIC (35), which provided an N-terminal His6 tag fusion followed by a tobacco etch virus (TEV) protease cleavage site between the tag and residue 339 of VanTG. Mutations in vanTG were introduced by using the QuikChange mutagenesis kit (Stratagene) with oligonucleotide pairs (see Table S1 in the supplemental material).
Protein expression and purification.
The N-terminal His6-tagged wild-type and mutant VanTG proteins were produced in E. coli BL21-CodonPlus(DE3)-RIPL (Novagen) harboring the corresponding hybrid plasmid. Freshly transformed cells were grown in 0.5 liters of LB medium containing 25 µg/ml of chloramphenicol and 100 µg/ml of ampicillin. The culture was incubated at 37°C to an optical density at 600 nm of 0.8, at which point it was induced for 16 h at 16°C with 1 mM isopropyl-1-thio-β-d-galactopyranoside. For structural analysis, selenomethionine-substituted vanTG was expressed using the standard M9 high-yield growth procedure according to the manufacturer’s (Shanghai Medicilon) instructions and E. coli BL21(DE3)-CodonPlus. Cells were harvested by centrifugation and resuspended in 20 ml of buffer A (50 mM HEPES [pH 7.5], 300 mM NaCl, 10% [vol/vol] glycerol) supplemented with 10% (vol/vol) BugBuster 10× protein extraction reagent (Novagen), 25 units of Benzonase (Sigma-Aldrich), and 5 mM imidazole. The mixture was stirred for 10 min at room temperature and centrifuged at 20,000 × g for 45 min, and the supernatant was applied to a 1-ml HisTrap Fast Flow column (GE Healthcare, Uppsala, Sweden) equilibrated with buffer A containing 40 mM imidazole. The protein was eluted using buffer A with a gradient of 40 to 500 mM imidazole over 20 ml. For structural analysis, the His6 tag was removed by cleavage with TEV protease overnight at 4°C in dialysis buffer containing 0.3 M NaCl, 50 mM HEPES (pH 7.5), 5% glycerol, and 0.5 mM tris(2-carboxyethyl)phosphine, followed by binding to a HisTrap Fast Flow column and capture of the flowthrough. Fractions containing the VanTG proteins were identified by SDS-PAGE, pooled, and dialyzed overnight against 50 mM HEPES (pH 7.5), 300 mM NaCl, 5% (vol/vol) glycerol, and 1 mM Tris(2-carboxyethyl)phosphine, concentrated when needed with a Centriprep 30 concentrator, and stored at −80°C.
Kinetic analysis.
Determination of Ala and Ser racemase activities of VanTG proteins was based on the amino acid oxidase-lactate dehydrogenase (LDH)-coupled assay (36). The reaction was carried out in a 0.1-ml total volume containing 50 mM HEPES (pH 8.0), 0.2 mM NADH, 0.01 µg of LDH, 0.03 µg of d-amino acid oxidase (D-AAO), 100 units of catalase at 37°C, 15 µg of VanTG enzyme, and various concentrations of l-Ala or l-Ser. VanTG converts l-Ala and l-Ser into d-Ala and d-Ser, respectively. D-AAO converts d-Ala and d-Ser into, respectively, pyruvate and 3-hydroxypyruvate, which are reduced by LDH. The consumption of NADH, reflected in the formation of l-lactate or 2,3-dihydroxypropane, was followed at 340 nm. The steady-state kinetic parameters (mean values of at least three independent measurements) were obtained by fitting experimental data to the Hanes-Woolf equation (37) using the program EnzFitter (Biosoft, Cambridge, United Kingdom).
Crystallization and structure determination.
Selenomethionine-derivatized VanTG (residues 339 to 712) was crystallized at room temperature by the hanging drop method with 1 µl of a 23-mg/ml protein solution mixed with 1 µl of reservoir solution (0.2 M ammonium sulfate, 0.2 M sodium chloride, 0.1 M morpholineethanesulfonic acid [pH 6.5], and 30% [wt/vol] polyethylene glycol monomethyl ether 5000). This crystal was cryoprotected with paratone oil in a liquid nitrogen stream before data collection. Diffraction data at 100 K were collected at the Advanced Photon Source (APS), Argonne National Laboratory, Life Sciences Collaborative Access Team beamline 21-ID-G fitted with a MarMosaic 300 charge-coupled device at the selenium absorption peak (0.97856 Å). X-ray data were reduced with HKL-3000 (38). The VanTG structure was solved by SAD phasing using Phenix.autosol (39), which identified 27 of the 28 selenium sites in the asymmetric unit (four protein chains, each with seven selenomethionine residues). An initial model of the protein was built using Phenix.autobuild followed by rounds of manual model building and refinement with Coot (40) and Phenix.refine. Isotropic B-factors were refined with translation-libration-screw motion (TLS) parameterization (TLS groups were residues 340 to 582 and residues 583 to 711 for each protein chain). To obtain a bias-free model view of the VanTG active sites, we generated a simulated annealing omit map by deleting all active site residues, ions, and water molecules shown in Fig. 3 as well as all atoms within 5 Å of these groups, followed by simulated annealing using the Phenix.refine algorithm (default parameters). The resulting FO-FC map was inspected with Coot (40). The final VanTG model included residues 340 to 711 of four protein chains forming two homodimers. Average B-factor and bond angle/length RMSD values were calculated using Phenix. All geometry was verified using the Phenix and Coot validation tools plus the PDB Adit server. The VanTG structure had a good backbone with the following percentages of residues in the most favored, additional allowed, generously allowed, and disallowed regions, respectively, of the Ramachandran plot: 90.8%, 8.9%, 0.2%, and 0%.
Structural analysis.
Structure similarity searches were performed using the PDBeFold server (41, 42). Structure superpositions and analysis were performed with PyMOL (43). l-Ser was modeled in the active site of VanTG by manual placement onto the coordinates of l-Ala from PLP–l-Ala in B. stearothermophilus Alr (PDB 1L6F) (20). The side chain rotamer of l-Ser was the m rotamer (χ1 angle = −65°).
Sequence alignment and phylogenetic reconstruction.
Sequences of VanT, Alr, and broad-spectrum and lysine racemases were aligned using Clustal Omega (44) and visualized with the ESPript 3 server (http://espript.ibcp.fr) (45). Phylogenetic reconstruction was performed using the maximum likelihood algorithm in the MEGA package (46) and visualized with FigTree (http://tree.bio.ed.ac.uk/software/figtree).
Protein structure accession number.
The VanTG structure has been deposited in the Protein Databank under the accession code 4ECL.
SUPPLEMENTAL MATERIAL
Electrostatic surface representation of the VanTG dimer, indicating the positively charged active sites. Download
Electron density of the expected PLP-binding site in VanTG subunits A to D (top four pictures). The PLP-binding site is shown for BsAlr (bottom picture). Areas with green mesh show the simulated annealing omit Fo-Fc density at 2 σ after omitting all residues, ions, and waters in the active site according to Fig. 3 and all atoms within 5 Å of these groups. Download
Oligonucleotide primers for mutagenesis of vanTG
Kinetic parameters for racemization of l-Ala and l-Ser by VanTG and mutants.
ACKNOWLEDGMENTS
We thank R. Di Leo for technical assistance and Z. Wawrzak at the Life Sciences Collaborative Access Team/Advanced Photon Source for X-ray data collection.
This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services (contracts HHSN272200700058C and HHSN272201200026C) and by an unrestricted grant from Reckitt Benckiser.
Footnotes
Citation Meziane-Cherif D, Stogios PJ, Evdokimova E, Egorova O, Savchenko A, Courvalin P. 2015. Structural and functional adaptation of vancomycin resistance VanT serine racemases. mBio 6(4):e00806-15. doi:10.1128/mBio.00806-15.
Contributor Information
Gilles P. van Wezel, Leiden University.
Gerard D. Wright, McMaster University.
REFERENCES
- 1.Butler MS, Hansford KA, Blaskovich MA, Halai R, Cooper MA. 2014. Glycopeptide antibiotics: back to the future. J Antibiot (Tokyo) 67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- 2.Reynolds PE. 1989. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 8:943–950. doi: 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- 3.Courvalin P. 2006. Vancomycin resistance in gram-positive cocci. Clin Infect Dis 42(Suppl 1):S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 4.Arias CA, Peña J, Panesso D, Reynolds P. 2003. Role of the transmembrane domain of the VanT serine racemase in resistance to vancomycin in Enterococcus gallinarum BM4174. J Antimicrob Chemother 51:557–564. doi: 10.1093/jac/dkg128. [DOI] [PubMed] [Google Scholar]
- 5.Boyd DA, Willey BM, Fawcett D, Gillani N, Mulvey MR. 2008. Molecular characterization of Enterococcus faecalis N06-0364 with low-level vancomycin resistance harboring a novel d-Ala-d-Ser gene cluster, vanL. Antimicrob Agents Chemother 52:2667–2672. doi: 10.1128/AAC.01516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu D, Zhang L, Kong Y, Du J, Chen S, Chen J, Ding J, Jiang H, Shen X. 2008. Enzymatic characterization and crystal structure analysis of the d-alanine–d-alanine ligase from Helicobacter pylori. Proteins 72:1148–1160. doi: 10.1002/prot.22009. [DOI] [PubMed] [Google Scholar]
- 7.Meziane-Cherif D, Saul FA, Haouz A, Courvalin P. 2012. Structural and functional characterization of VanG D-Ala:D-Ser ligase associated with vancomycin resistance in Enterococcus faecalis. J Biol Chem 287:37583–37592. doi: 10.1074/jbc.M112.405522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meziane-Cherif D, Stogios PJ, Evdokimova E, Savchenko A, Courvalin P. 2014. Structural basis for the evolution of vancomycin resistance D,D-peptidases. Proc Natl Acad Sci U S A 111:5872–5877. doi: 10.1073/pnas.1402259111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshimura T, Goto M. 2008. d-Amino acids in the brain: structure and function of pyridoxal phosphate-dependent amino acid racemases. FEBS J 275:3527–3537. doi: 10.1111/j.1742-4658.2008.06516.x. [DOI] [PubMed] [Google Scholar]
- 10.Conti P, Tamborini L, Pinto A, Blondel A, Minoprio P, Mozzarelli A, De Micheli C. 2011. Drug discovery targeting amino acid racemases. Chem Rev 111:6919–6946. doi: 10.1021/cr2000702. [DOI] [PubMed] [Google Scholar]
- 11.Arias CA, Martín-Martinez M, Blundell TL, Arthur M, Courvalin P, Reynolds PE. 1999. Characterization and modelling of VanT: a novel, membrane-bound, serine racemase from vancomycin-resistant Enterococcus gallinarum BM4174. Mol Microbiol 31:1653–1664. doi: 10.1046/j.1365-2958.1999.01294.x. [DOI] [PubMed] [Google Scholar]
- 12.Arias CA, Courvalin P, Reynolds PE. 2000. vanC cluster of vancomycin-resistant Enterococcus gallinarum BM4174. Antimicrob Agents Chemother 44:1660–1666. doi: 10.1128/AAC.44.6.1660-1666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arias CA, Weisner J, Blackburn JM, Reynolds PE. 2000. Serine and alanine racemase activities of VanT: a protein necessary for vancomycin resistance in Enterococcus gallinarum BM4174. Microbiology 146:1727–1734. [DOI] [PubMed] [Google Scholar]
- 14.Abadía Patiño L, Courvalin P, Perichon B. 2002. vanE gene cluster of vancomycin-resistant Enterococcus faecalis BM4405. J Bacteriol 184:6457–6464. doi: 10.1128/JB.184.23.6457-6464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Depardieu F, Bonora MG, Reynolds PE, Courvalin P. 2003. The vanG glycopeptide resistance operon from Enterococcus faecalis revisited. Mol Microbiol 50:931–948. doi: 10.1046/j.1365-2958.2003.03737.x. [DOI] [PubMed] [Google Scholar]
- 16.Lebreton F, Depardieu F, Bourdon N, Fines-Guyon M, Berger P, Camiade S, Leclercq R, Courvalin P, Cattoir V. 2011. d-Ala-d-Ser VanN-type transferable vancomycin resistance in Enterococcus faecium. Antimicrob Agents Chemother 55:4606–4612. doi: 10.1128/AAC.00714-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priyadarshi A, Lee EH, Sung MW, Nam KH, Lee WH, Kim EE, Hwang KY. 2009. Structural insights into the alanine racemase from Enterococcus faecalis. Biochim Biophys Acta 1794:1030–1040. doi: 10.1016/j.bbapap.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Couñago RM, Davlieva M, Strych U, Hill RE, Krause KL. 2009. Biochemical and structural characterization of alanine racemase from Bacillus anthracis (Ames). BMC Struct Biol 9:53. doi: 10.1186/1472-6807-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asojo OA, Nelson SK, Mootien S, Lee Y, Rezende WC, Hyman DA, Matsumoto MM, Reiling S, Kelleher A, Ledizet M, Koski RA, Anthony KG. 2014. Structural and biochemical analyses of alanine racemase from the multidrug-resistant Clostridium difficile strain 630. Acta Crystallogr D Biol Crystallogr 70:1922–1933. doi: 10.1107/S1399004714009419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe A, Yoshimura T, Mikami B, Hayashi H, Kagamiyama H, Esaki N. 2002. Reaction mechanism of alanine racemase from Bacillus stearothermophilus: x-ray crystallographic studies of the enzyme bound with N-(5′-phosphopyridoxyl)alanine. J Biol Chem 277:19166–19172. doi: 10.1074/jbc.M201615200. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe A, Yoshimura T, Mikami B, Esaki N. 1999. Tyrosine 265 of alanine racemase serves as a base abstracting alpha-hydrogen from l-alanine: the counterpart residue to lysine 39 specific to d-alanine. J Biochem 126:781–786. doi: 10.1093/oxfordjournals.jbchem.a022517. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe A, Kurokawa Y, Yoshimura T, Kurihara T, Soda K, Esaki N, Watababe A. 1999. Role of lysine 39 of alanine racemase from Bacillus stearothermophilus that binds pyridoxal 5′-phosphate. Chemical rescue studies of Lys39 → Ala mutant. J Biol Chem 274:4189–4194. doi: 10.1074/jbc.274.7.4189. [DOI] [PubMed] [Google Scholar]
- 23.Patrick WM, Weisner J, Blackburn JM. 2002. Site-directed mutagenesis of Tyr354 in Geobacillus stearothermophilus alanine racemase identifies a role in controlling substrate specificity and a possible role in the evolution of antibiotic resistance. Chembiochem 3:789–792. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Espaillat A, Carrasco-López C, Bernardo-García N, Pietrosemoli N, Otero LH, Álvarez L, de Pedro MA, Pazos F, Davis BM, Waldor MK, Hermoso JA, Cava F. 2014. Structural basis for the broad specificity of a new family of amino-acid racemases. Acta Crystallogr D Biol Crystallogr 70:79–90. doi: 10.1107/S1399004713024838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu HM, Kuan YC, Chu CH, Hsu WH, Wang WC. 2012. Crystal structures of lysine-preferred racemases, the non-antibiotic selectable markers for transgenic plants. PLoS One 7:e48301. doi: 10.1371/journal.pone.0048301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cava F, Lam H, de Pedro MA, Waldor MK. 2011. Emerging knowledge of regulatory roles of d-amino acids in bacteria. Cell Mol Life Sci 68:817–831. doi: 10.1007/s00018-010-0571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R. 2010. d-Amino acids trigger biofilm disassembly. Science 328:627–629. doi: 10.1126/science.1188628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynolds PE, Courvalin P. 2005. Vancomycin resistance in enterococci due to synthesis of precursors terminating in d-alanyl-d-serine. Antimicrob Agents Chemother 49:21–25. doi: 10.1128/AAC.49.1.21-25.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morar M, Wright GD. 2010. The genomic enzymology of antibiotic resistance. Annu Rev Genet 44:25–51. doi: 10.1146/annurev-genet-102209-163517. [DOI] [PubMed] [Google Scholar]
- 30.Hall BG, Barlow M. 2004. Evolution of the serine beta-lactamases: past, present and future. Drug Resist Updat 7:111–123. doi: 10.1016/j.drup.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Yoon EJ, Courvalin P, Grillot-Courvalin C. 2013. RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: major role for AdeABC overexpression and AdeRS mutations. Antimicrob Agents Chemother 57:2989–2995. doi: 10.1128/AAC.02556-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Fisher JF, Mobashery S. 2009. The bifunctional enzymes of antibiotic resistance. Curr Opin Microbiol 12:505–511. doi: 10.1016/j.mib.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 33.Foucault ML, Depardieu F, Courvalin P, Grillot-Courvalin C. 2010. Inducible expression eliminates the fitness cost of vancomycin resistance in enterococci. Proc Natl Acad Sci U S A 107:16964–16969. doi: 10.1073/pnas.1006855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferretti JJ, Gilmore KS, Courvalin P. 1986. Nucleotide sequence analysis of the gene specifying the bifunctional 6′-aminoglycoside acetyltransferase 2″ aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J Bacteriol 167:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, Mark ID. 2009. A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol Biol 498:105–115. doi: 10.1007/978-1-59745-196-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badet B, Roise D, Walsh CT. 1984. Inactivation of the dadB Salmonella typhimurium alanine racemase by D and L isomers of beta-substituted alanines: kinetics, stoichiometry, active site peptide sequencing, and reaction mechanism. Biochemistry 23:5188–5194. doi: 10.1021/bi00317a016. [DOI] [PubMed] [Google Scholar]
- 37.Henderson PJF. 1992. Statistical analysis of enzyme kinetic data, p 277–313. In Eisenthal R, Danson MJ (ed), Enzyme assays, a practical approach. Oxford University Press, New York, NY. [Google Scholar]
- 38.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. 2006. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 42.Holm L, Rosenström P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA. [Google Scholar]
- 44.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Ω. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cox G, Stogios PJ, Savchenko A, Wright GD. 2014. Structural and molecular basis for resistance to aminoglycoside antibiotics by the adenylyltransferase ANT(2")-Ia. mBio 6(1):e02180–14. doi: 10.1128/mBio.02180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electrostatic surface representation of the VanTG dimer, indicating the positively charged active sites. Download
Electron density of the expected PLP-binding site in VanTG subunits A to D (top four pictures). The PLP-binding site is shown for BsAlr (bottom picture). Areas with green mesh show the simulated annealing omit Fo-Fc density at 2 σ after omitting all residues, ions, and waters in the active site according to Fig. 3 and all atoms within 5 Å of these groups. Download
Oligonucleotide primers for mutagenesis of vanTG
Kinetic parameters for racemization of l-Ala and l-Ser by VanTG and mutants.