

Abstract
Hydroxy acid dehydrogenases, including l- and d-lactate dehydrogenases (L-LDH and D-LDH), are responsible for the stereospecific conversion of 2-keto acids to 2-hydroxyacids and extensively used in a wide range of biotechnological applications. A common feature of LDHs is their high specificity for NAD+ as a cofactor. An LDH that could effectively use NADPH as a coenzyme could be an alternative enzymatic system for regeneration of the oxidized, phosphorylated cofactor. In this study, a d-lactate dehydrogenase from a Sporolactobacillus inulinus strain was found to use both NADH and NADPH with high efficiencies and with a preference for NADPH as its coenzyme, which is different from the coenzyme utilization of all previously reported LDHs. The biochemical properties of the D-LDH enzyme were determined by X-ray crystal structural characterization and in vivo and in vitro enzymatic activity analyses. The residue Asn174 was demonstrated to be critical for NADPH utilization. Characterization of the biochemical properties of this enzyme will contribute to understanding of the catalytic mechanism and provide referential information for shifting the coenzyme utilization specificity of 2-hydroxyacid dehydrogenases.
INTRODUCTION
Lactic acid, existing as d- and l-isomers, is an important organic acid that can be widely used in food, cosmetic, chemical, and many other industries (1, 2). The most important application of lactic acid is the production of biodegradable polymer poly(lactic acid) (3). Since the thermoresistance of pure poly(l-lactic acid) is rather low, development of a stereocomplex of poly(l-lactic acid) and poly(d-lactic acid) with a melting point as high as that of petroleum-based polymers is considered an advance in poly(lactic acid) modification (4, 5).
l-Lactate dehydrogenase (L-LDH) and d-lactate dehydrogenase (D-LDH) are responsible for the synthesis of l- and d-lactic acids, respectively (2, 6). Given the potential use of chiral hydroxyl acids as valuable synthons and precursors for chiral products in the biotechnological industry (7), it is of significant interest to gain in-depth insights into the substrate specificities, catalytic kinetics, and molecular mechanisms of 2-hydroxyacid dehydrogenase enzymes. The l-isomer-specific 2-hydroxyacid dehydrogenase is a cytoplasmic enzyme present in essentially all major organ systems. This enzyme has been extensively characterized and used as an indicator of pathological conditions (8). However, the d-isomer-specific 2-hydroxyacid dehydrogenase has gained much less attention. Sequence alignments have shown that D-LDH and L-LDH display significant differences in their amino acid sequences and belong to two distinct families: the d- and l-2-hydroxyacid dehydrogenases, respectively (9).
Hydroxy acid dehydrogenases are responsible for the stereospecific conversion of 2-keto acids to 2-hydroxyacids, having wide biotechnological applications, like synthesis of antibiotics, flavor development in dairy products, and production of valuable synthons (10). Due to their highly specific activity in the reduction of pyruvate and the high stability of the enzyme and substrate, LDHs serve as an efficient system for cofactor regeneration (11). A unifying feature of all previously reported lactate dehydrogenases is their strict specificity for NAD+ as a cofactor (6). However, suitable systems for the regeneration of NADP+ are rare. Richter et al. (12) described a structure-guided alteration in the cofactor specificity of the L-LDH from Bacillus subtilis for regeneration of NADP+. To date, NADP+-dependent LDHs have not yet been found in nature (6). Therefore, a D-LDH that can effectively use NADPH as coenzyme would expand the alternative enzymatic systems for the regeneration of NADPH.
Here, we biochemically characterized a D-LDH from Sporolactobacillus inulinus CASD, which has been found to be a d-lactic acid producer with an optical purity above 99.3% (13). Based on the annotated genome sequence (14), the gene product was designated d-lactate dehydrogenase 744 (DLDH744). Interestingly, we found that DLDH744 preferentially uses NADPH as a coenzyme, by contrasting the apparent Km values for NADH and NADPH.
MATERIALS AND METHODS
Bacterial strains and plasmids.
S. inulinus CASD was cultivated in 40 g/liter glucose and 10 g/liter yeast extract. Calcium carbonate (30 g/liter) was added to neutralize d-lactic acid during cultivation. The seed culture was prepared as follows: a loop of cells from a slant culture was inoculated into 20 ml of the above sterile medium in 50-ml conical flasks and incubated for 48 h at 42°C without shaking.
Escherichia coli strains were grown aerobically in Luria-Bertani (LB) medium and set on a rotary shaker. E. coli DH5α (Tiangen, Beijing, China) was used for general cloning, while E. coli BL21(DE3) (Tiangen) was used for protein expression. Plasmids pMD 19-T (TaKaRa, Dalian, China) and pET-28a(+) (Novagen, Lausanne, Switzerland) were used as vectors.
Gene cloning and construction of protein-overexpressing recombinant strains.
The draft genome sequence of S. inulinus CASD has been deposited in GenBank under accession number AFVQ00000000 (14). According to the nucleotide sequence of the annotated gene (NODE_744_orf00003, 1,008 bp including the stop codon), a forward primer (5′-GGTCATGAAAATCATTATGTTCAG-3′) and a reverse primer (5′-GCAAGCTTGTTTTCATCAGCTACTTTGTT-3′) were designed to introduce BspHI and HindIII restriction sites on either side of the gene. The amplified DNA fragments were ligated into the pMD 19-T vector for sequencing. After validating the insert by sequencing, the recombinant plasmid was digested with BspHI and HindIII endonucleases (TaKaRa, Dalian, China). The digested DNA fragments were then inserted into the pET-28a(+) plasmid that had been digested with NcoI and HindIII endonucleases (TaKaRa). The reconstructed pET-28a-D-LDH plasmid was transformed into E. coli BL21(DE3) for protein expression.
Expression and purification of d-lactic acid dehydrogenase DLDH744.
For protein expression, E. coli BL21(DE3) harboring the recombinant plasmid was incubated in LB medium containing 30 μg/ml kanamycin at 37°C with shaking. When the culture reached an optical density at 600 nm of 0.6 to 0.8, 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce gene expression. After induction at 25°C for an additional 8 h, cells were harvested by centrifugation. The harvested cells were resuspended in binding buffer (20 mM Tris-HCl [pH 7.8], 500 mM sodium chloride, and 10 mM imidazole) and disrupted by sonication. The suspension was centrifuged at 12,000 × g for 20 min to remove cell debris. DLDH744 was purified by nickel affinity chromatography according to the manufacturer's protocol (Tiandz, Beijing, China). The enzyme was eluted with elution buffer (20 mM Tris-HCl [pH 7.8], 500 mM sodium chloride, and 300 mM imidazole) (15).
The resultant effluent was filtered and loaded onto a HisTrap HP 5-ml column for desalination (GE Healthcare, Shanghai, China). The enzyme was eluted with phosphate-buffered saline (PBS; pH 7.0) at a flow rate of 5 ml/min. The fractions containing the enzyme were detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The native molecular mass of DLDH744 was estimated by native PAGE using bovine serum albumin (68.0 kDa) as a reference. The protein concentration was determined by the Bradford method using bovine serum albumin as a standard.
Enzymatic activity assay.
After purification, DLDH744 activity was determined with pyruvate as a substrate. The initial rate of oxidation of NAD(P)H was calculated by measuring the absorbance at 340 nm in 400 μl of the following mixture: 100 mM PBS (pH 5.5), 20 mM sodium pyruvate, 0.2 mM NAD(P)H, and 10 μl of enzyme (16). The reaction was initiated by the addition of enzyme. One unit was defined as the amount of enzyme converting 1 μM NAD(P)H per min. Specific activity was expressed as units per milligram of protein.
Measurement of the kinetic parameters of DLDH744.
The kinetic constants of purified enzyme were calculated using 10 μg DLDH744 enzyme in 100 mM PBS (pH 5.5) at 30°C. The Km values for NADH and NADPH were measured using 20 mM pyruvate as the substrate, with cofactor concentrations between 0.01 mM and 0.3 mM. The data were extracted using ChemSW software. The resulting kinetic parameters were calculated from multiple measurements.
Crystallization, X-ray data collection, processing, and structure determination.
The initial crystallization screening was performed in 96-well plates at 289 K using the hanging-drop vapor-diffusion method with Hampton Research commercial crystallization kits (Crystal Screen, Crystal Screen 2, Index, and PEG/Ion kits). The protein concentrations for crystallization screening were 5 and 10 mg/ml in 20 mM Tris-HCl (pH 7.5) and 5% glycerol. For X-ray diffraction experiments, the crystals were quick-soaked in reservoir solution containing 20% (vol/vol) glycol as a cryoprotectant for 15 to 20 s. The soaked crystals were mounted in nylon loops and flash-cooled in liquid nitrogen at 100 K for data collection (17, 18). An X-ray diffraction data set was collected at 100 K using an in-house X-ray source (Rigaku MicroMax-007HF desktop rotating-anode X-ray generator with a Cu target operated at 40 kV and 30 mA) and Saturn 944+ detector with a 70-mm crystal-to-detector distance at a wavelength of 1.5418 Å. The 360 diffraction frames were collected with 0.5° oscillation per image at 100 K. The crystal diffracted to 3.15 Å resolution. The set of diffraction data were indexed, integrated, and subsequently scaled using HKL-2000 (19). The initial structure solution of DLDH744 was obtained by molecular replacement using the program PHASER (20) with the crystal structure of d-isomer-specific 2-hydroxyacid dehydrogenase from Lactobacillus delbrueckii subsp. bulgaricus (previously known as Lactobacillus bulgaricus) (PDB ID 2YQ4) as a search model. Manual model building and refinement were performed with COOT (21) following rigid body refinement, energy minimization, and individual B-factor refinement. The quality of the final refined model was verified using the program Procheck (22). Final refinement statistics are summarized in Table S1 in the supplemental material (23).
Site-directed mutagenesis.
The plasmid pET-28a-D-LDH was extracted from the recombinant strain using the Plasmid minikit (Omega Bio-Tek, Norcross, GA, USA). The mutant strains used in this study were constructed by site-directed PCR mutagenesis using Pyrobest DNA polymerase (TaKaRa, Dalian, China) (24). The primers used for the introduction of the mutations are shown in Table 1. The PCR was performed according to standard procedures using plasmid pET-28a-D-LDH as the template. After the PCR, the remaining template was digested with the restriction enzyme DpnI (TaKaRa, Dalian, China) at 37°C for 2 h. The DpnI-digested PCR product was then purified and transformed into E. coli BL21(DE3). The cells were spread onto LB agar plates containing 30 μg/ml kanamycin. Single clones were selected for identification using gene sequencing.
TABLE 1.
Primers used for site-directed mutagenesis of DLDH744a

+, sense sequence; −, antisense sequence. Underlined codons indicate the mutation site.
Determination of in vivo cofactor concentration in strains harboring the wild-type (WT) or mutant enzyme.
As genetic manipulation of the parent strain S. inulinus CASD is currently not feasible, E. coli BW25113 was chosen as the alternative host strain to investigate the coenzyme preference in vivo. To eliminate the influence of the native lactate dehydrogenases in E. coli, the genes of l-lactate dehydrogenase (lldD) and d-lactate dehydrogenase (ldhA) of BW25113 were first knocked out. The gene fragment of DLDH744 or mutant N174Y was inserted into pGEX-KG vector (GE Healthcare), which drives expression by the Ptac promoter between the HindIII and EcoRI restriction sites. The resultant recombinant plasmids PGEX-DLDH744 and PGEX-N174Y were then transformed into E. coli BW25113ΔlldD ΔldhA. The engineered strains harboring the recombinant plasmid (E. coli 744WT and E. coli N174Y) were incubated in LB medium overnight and then transferred into fermentation medium (20 g/liter glucose, 10 g/liter yeast extract, 15 g/liter tryptone, 15 g/liter NaCl, 3 g/liter calcium carbonate). The cells were collected after 12 h of cultivation without shaking, and the in vivo concentrations of lactic acid, NAD+, NADH, NADP+, and NADPH were determined.
Analytical methods.
The concentrations of NADP+ were measured using the EnzyFluo NADP+/NADPH assay kit (BioAssay Systems, Hayward, CA, USA), and the concentrations of NADH were measured using the EnzyChrom NAD+/NADH assay kit (BioAssay Systems) as described in the manufacturers' protocols. Briefly, the cells were washed with cold PBS and pelleted for each sample. Then, the samples (∼105 cells) were homogenized with either 100 μl NAD(P)+ extraction buffer for NAD(P)+ determination or 100 μl NAD(P)H extraction buffer for NAD(P)H determination. Each solution was first heated at 60°C for 5 min, and then 20 μl assay buffer and 100 μl of the opposite extraction buffer were added to neutralize the extracts. The supernatants were used for cofactor concentration assay.
Nicotinamide nucleotide transhydrogenase catalyzes the reversible transfer of hydrogen between NAD and NADP according to the reaction NADH + NADP+ ↔ NAD + NADPH (25). The transhydrogenase activity of DLDH744 was determined by measuring the concentration of NADH in 800 μl of the following mixture: 100 mM PBS (pH 5.5), 0.2 mM NAD+, 0.2 mM NADPH, and 10 μl of enzyme. NADH kinases were reported to directly phosphorylate NADH to produce NADPH (26). The possible reverse NADH kinase activity of DLDH744 was determined by measuring the concentration of phosphate in 800 μl of the following mixture: 20 mM sodium pyruvate, 0.2 mM NADPH, and 10 μl of enzyme in Milli-Q water (Millipore, Bedford, MA, USA). The phosphate released from NADP+ was measured using the malachite green phosphate assay kit (BioAssay Systems) according to the manufacturer's protocol.
d-Lactic acid was measured using a high-performance liquid chromatography (HPLC) system (Agilent 1260 series) equipped with a chiral column (MCI Gel CRS10 W; Japan) and detection at 254 nm. The mobile phase was 2 mM CuSO4 at a flow rate of 0.5 ml/min (25°C).
Protein structure accession number.
The complete tertiary structure of DLDH744 in complex with NAD+ determined in this study has been submitted to PDB under PDB ID 4XKJ.
RESULTS
Enzymatic properties of DLDH744.
DLDH744 is a 335-amino-acid enzyme with a calculated molecular mass of 36.7 kDa. The molecular mass of the expressed protein was approximately 37.0 kDa as determined by SDS-PAGE analysis, indicating that the recombinant protein had been expressed successfully in E. coli. Native PAGE analysis showed a molecular mass of approximately 70.0 kDa, suggesting that DLDH744 exists as a homodimer. The purified enzyme, here referred as DLDH744, had clear activities on pyruvate and d-lactic acid but not on l-lactic acid with NAD(H) as coenzyme. The optimal reaction pH for DLDH744 was 5.5 (see Fig. S1 in the supplemental material). Additionally, Cu2+ and Zn2+ strongly inhibited DLDH744 enzyme activities (see Fig. S2 in the supplemental material). The enzyme was stable at temperatures below 32°C and inactivated rapidly above 38°C (see Fig. S3 in the supplemental material).
NADH and NADPH as coenzymes for DLDH744.
As mentioned above, all lactate dehydrogenases reported to date have high specificity for NAD+ as a cofactor (6). The Km value for the NADH of DLDH744, when catalyzing the reduction of pyruvate to lactate, was 0.114 ± 0.0056 mM. Interestingly, NADPH was also found to be an effective cofactor for pyruvate reduction to lactate. Notably, the Km value for NADPH in the same reaction was 0.036 ± 0.0004 mM, which was much lower than that of NADH. These results indicated that the enzyme is NAD(P)+ dependent, with a preference for NADPH. To confirm these results, products from the reaction using NADH or NADPH as a coenzyme were measured by HPLC equipped with a chiral column. As shown in Fig. 1, pyruvate was reduced exclusively to d-lactic acid by DLDH744 using either NADH or NADPH as the sole coenzyme. The reaction mixtures with NADH and NADPH produced the same concentrations of d-lactate during the same reaction times. Additionally, the absence of l-lactic acid in the reaction system indicated that DLDH744 is a strict d-lactate dehydrogenase. No product was detected in the absence of either coenzyme or substrate.
FIG 1.

HPLC analyses of products of pyruvate reduction by purified DLDH744 with NADPH or NADH as cofactors. (a) Reaction with NADPH as coenzyme; (b) reaction with NADH as coenzyme; (c) control without a coenzyme added; (d) l-lactic acid standard; (e) d-lactic acid standard.
Overall crystal structure of DLDH744.
For further investigation of this enzyme, the complete tertiary structure of DLDH744 in complex with NAD+ was determined by X-ray crystallography. The structure of DLDH744 was determined by molecular replacement method and refined as shown in Table S1 in the supplemental material. Like typical NAD+-dependent dehydrogenases, a homodimer of DLDH744 was found in one asymmetric unit. Two identical monomers (with Cα atoms' root mean square [RMS] difference of 1.339 Å) associate at the cofactor-binding domain to form the homodimer. Each monomer covers full-length DLDH744 (Met1-Ala332), which is composed of two separate domains, the substrate-binding domain (Met1-Tyr101) and the cofactor-binding domain (Ser102-Ala332). These two domains are separated by a deep cleft (Fig. 2). Each monomer contains one cofactor NAD+ in the cofactor binding domain. The cofactor-binding domain has a central β-sheet containing six parallel strands that are flanked on both sides by α-helices. This topology is a variant of the conventional dinucleotide-binding Rossmann fold that is widely conserved among various members of the NAD+-dependent dehydrogenase family. The smaller substrate-binding domain is also characterized by a modified Rossmann fold topology composed of a five-stranded parallel β-sheet flanked by five α-helices (Fig. 2).
FIG 2.
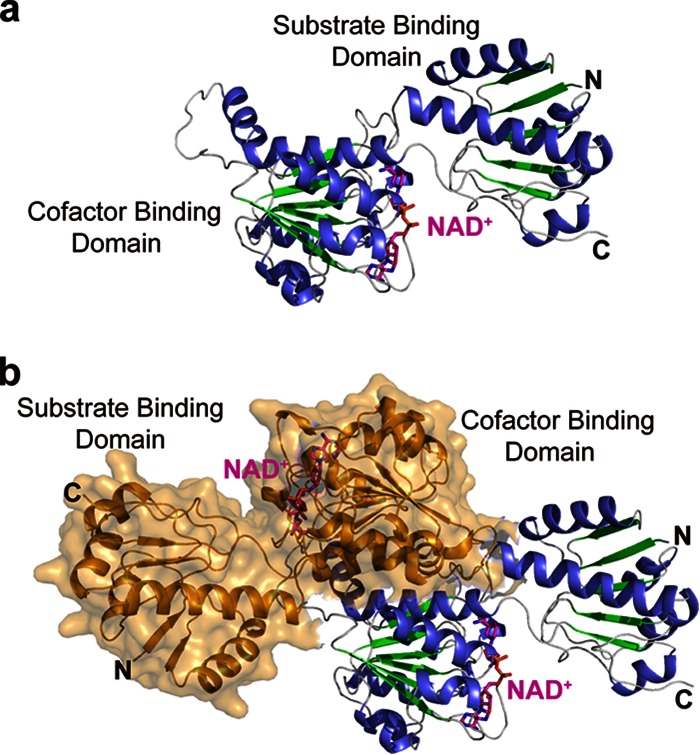
Overall crystal structure of DLDH744. (a) Crystal structure of one subunit in complex with cofactor NAD+. The secondary structures are colored in blue for α helix and green for β strand; NAD+ is colored in magenta and shown in sticks. (b) Dimeric structure of DLDH744. The other subunit in the dimer is represented by surface and colored in orange.
Exploration of the key sites critical for NADPH utilization.
As DLDH744 was found to have d-2-hydroxyacid dehydrogenase activity with the unusual ability to use both NADH and NADPH as coenzymes, it was of interest to investigate the sites that discriminate between NADH and NADPH. Structural studies of DLDH744 have identified some critical residues that contact the coenzyme (Fig. 3). The ribose ring (adjacent to the adenine) forms two hydrogen bonds with the carboxyl side chain of Asp175 (2.4 Å and 2.9 Å). The pyrophosphate atoms form weak hydrogen bonds with the main-chain nitrogen atoms of Arg155 (3.3 Å) and Ile156 (3.0 Å). Residues in this region are part of the NAD+-binding signature motif GXGXXGX, which is present from Gly152 to Gly158 in DLDH744. The nicotinamide ribose ring is stabilized by interactions with the main-chain oxygen atom of Val205 (2.6 Å) and the amine group of Arg234 (3.2 Å). The amine group of the nicotinamide ring is stabilized by Ala232 (3.2 Å), and the carbonyl group forms a hydrogen bond with main-chain nitrogen atom of Gly297 (3.0 Å).
FIG 3.
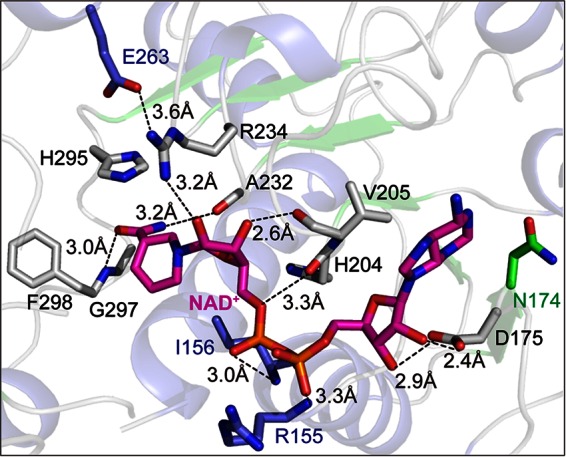
Structural analysis for identification of cofactor NAD+-binding site of DLDH744. Key amino acids necessary for cofactor NAD+ binding are shown in sticks, and the hydrogen bonds are shown as dashed lines.
Amino acid sequence alignment between DLDH744 and other functionally verified NAD+-dependent D-LDHs was performed using ClustalX2 and rendered using ESPript (27). The alignment indicated that most of the cofactor-binding sites are highly conserved among the NAD+-dependent D-LDHs (Fig. 4). Only Gly297 is different from the other D-LDHs, but the mutation G297A did not change the cofactor usage (data not shown). Ala232 is not a conserved residue in D-LDHs, and the corresponding residue in some other NAD+-dependent D-LDHs is also Ala. Therefore, the cofactor-binding residues are not the sites that affect NADPH utilization of DLDH744.
FIG 4.

Sequence alignment of DLDH744 with homologous enzymes. The secondary structure of D-LDH from L. bulgaricus (PDB ID 1J4A) is presented (top). The indicated numbers of the residues refer to the D-LDH from L. bulgaricus. Helices are marked with spirals, beta strands with arrows, and turns with the letter T. Identical residues and conserved substitutions are shaded in red and boxed in by blue rectangles, respectively. The cofactor-binding sites are identified by a black square. The nucleotide-binding signature domain GXGXXG(17X)D is identified by black triangles. Asn174 is a unique residue in DLDH744 that differs from residues in the same position in the other d-2-hydroxyacid dehydrogenases. L. pentosus, Lactobacillus pentosus; S. aureus, Staphylococcus aureus.
NADPH differs from NADH only by the presence of an additional phosphate group on the adenine ribose of the coenzyme. Therefore, we modeled NADP in the active site to predict which residues were involved in cofactor specificity. The residues beneath the adenine ribose of the coenzyme should play a role in using NADPH as a coenzyme (Fig. 5a). In the structure, the ribose ring forms two hydrogen bonds with the carboxyl side chain of Asp175. Asp175 was reported to be responsible for the NADH preference in some D-LDHs by hindering binding of the negatively charged and bulky phosphate group (28, 29). The D-LDH activity of the D175S mutant was decreased when using either NADH or NADPH as the coenzyme (Fig. 6), indicating that Asp175 was a key binding site for cofactor but not the residue responsible for discriminating between NADH and NADPH in DLDH744. Interestingly, the residues surrounding Asp175 are rich in diversity, as shown in the amino acid sequence alignment result (Fig. 4). Therefore, the adjacent amino acid residues beneath the adenine ribose of the coenzyme (position 174 to 177) were mutated to the corresponding amino acids of other NAD+-dependent LDHs to explore the cofactor specificity. The d-lactate dehydrogenase activity of these mutants was measured using NADH or NADPH as coenzyme. Among these mutants, N174Y exhibited sharply decreased D-LDH activity with NADPH, while its activity with NADH was not significantly affected compared with wild-type DLDH744 (Fig. 6).
FIG 5.

Comparison between the structures of DLDH744 modeled with NADP+ and D-LDH from Aquifex aeolicus in complex with NAD+. (a) Crystal structure of DLDH744 modeled with NADP+ in the cofactor binding site. The secondary structures are colored in blue for α-helix and green for β-strand; NAD+ is colored in magenta and shown in sticks. Asn174 and other amino acids near the adenine ribose are shown in sticks. (b) The crystal structure of D-LDH from A. aeolicus bound with NAD+ was shown in same color scheme as DLDH744. Tyr170 and corresponding amino acids in D-LDH from A. aeolicus are shown in sticks.
FIG 6.

d-Lactate dehydrogenase activity with the cofactors NADH and NADPH after site-directed mutagenesis. Lactate dehydrogenase activity of DLDH744 mutant enzymes with NADH (a) or with NADPH (b) as coenzyme. The error bars in the figure indicate the standard deviations from three parallel replicates.
To further investigate the role of Asn174 in the utilization of NADPH as a coenzyme, the corresponding amino acid (Tyr) of a d-lactate dehydrogenase from Lactobacillus delbrueckii subsp. bulgaricus DSM 20081, which has already been experimentally verified as an NAD-dependent D-LDH (30), was mutated to Asn. Although a very low activity with NADPH as coenzyme was detected by the wild-type enzyme (0.96 ± 0.01 U/mg) in our experiments, it might be due to the impurity of the reagent NAPDH since the purity of NADPH purchased from Sigma-Aldrich (lot no. SLBJ6612V) is about 97%. The specific enzyme activity with the mutant using NADPH as the coenzyme was determined to be 7.35 ± 0.40 U/mg, whereas the specific activity using NADH did not change drastically (from 81.8 ± 3.2 U/mg to 88.8 ± 2.6 U/mg). In addition, the amino acid sequences of 26 uncharacterized lactate dehydrogenases from Lactobacillus genomes were also aligned with DLDH744, and all contained a Tyr at the corresponding amino acid residue, consistent with D-LDH from Lactobacillus delbrueckii subsp. bulgaricus DSM 20081 (see Fig. S4 in the supplemental material). These results demonstrated again the importance of Asn174 in the utilization of NADPH as a coenzyme in DLDH744.
The kinetic parameters of the N174Y mutant in reactions using NADH and NADPH were determined using pyruvate as a substrate. The kinetic parameters using NADH by the N174Y mutant were not significantly different from those of the wild-type enzyme. After mutation, the Km value for NADH as a coenzyme was reduced, whereas that for NADPH was increased. The Vmax of the N174Y mutant using NADPH as a coenzyme was remarkably reduced to 0.91 U/mg. As a result, the turnover number (kcat) and overall catalytic efficiency (kcat/Km) of the mutant were significantly decreased (Table 2). Therefore, Asn174 was functionally verified as the key residue that is critical for NADPH utilization.
TABLE 2.
Kinetic constants of the wild-type (WT) and mutant (N174Y) d-lactate dehydrogenase DLDH744 for NADH and NADPH
| Enzyme | Means ± SD of kinetic constants for cofactor: |
|||||||
|---|---|---|---|---|---|---|---|---|
| NADH |
NADPH |
|||||||
| Km (mM) | Vmax (U/mg) | kcat (min−1) | kcat/Km (mM−1 s−1) | Km (mM) | Vmax (U/mg) | kcat (min−1) | kcat/Km (mM−1 s−1) | |
| WT | 0.114 ± 0.0056 | 7.50 ± 0.168 | 525.20 ± 11.80 | 77.02 ± 1.73 | 0.036 ± 0.0004 | 4.66 ± 0.014 | 326.53 ± 0.98 | 149.31 ± 0.45 |
| N174Y | 0.080 ± 0.0003 | 6.93 ± 0.001 | 485.28 ± 0.14 | 100.72 ± 0.03 | 0.056 ± 0.0002 | 0.91 ± 0.001 | 63.56 ± 0.04 | 18.77 ± 0.01 |
Determination of in vivo cofactor concentration.
To determine the coenzyme preference in vivo, the strain that has only DLDH744 (or the N174Y mutant enzyme) for lactic acid production was engineered. No l-lactate was detected in either strain, indicating that only the introduced exogenous d-lactate dehydrogenases (DLDH744 or its mutant) contributed to lactate formation. The concentrations of lactic acid, NAD+, NADH, NADP+, and NADPH were determined after fermentation. The lactic acid production by the E. coli strain with the wild-type enzyme (E. coli 744WT, 2.33 ± 0.11 g/liter) was consistent with that of the E. coli strain with the mutant enzyme (E. coli N174Y, 2.77 ± 0.20 g/liter). However, the concentration of NADPH in E. coli with the wild-type enzyme was one-half of that in E. coli with the mutant enzyme. Furthermore, the NADH concentration was lower and the NAD+ concentration was higher in the E. coli strain with the mutant enzyme than in that with the wild-type enzyme (Table 3). These results were also consistent with the finding that the mutant exhibited decreased D-LDH activity using NADPH, while its activity using NADH was not significantly different (Fig. 6). Thus, it is clear that DLDH744 has preferential specificity for NADPH in vivo.
TABLE 3.
In vivo concentrations of coenzymes in the recombinant E. coli hosts harboring wild-type or mutant DLDH744
| E. coli strain | Means ± SD of in vivo concn of coenzyme: |
|||
|---|---|---|---|---|
| NADH (μM) | NAD+ (μM) | NADPH (μM) | NADP+ (μM) | |
| 744WT | 0.616 ± 0.008 | 1.997 ± 0.097 | 0.117 ± 0.002 | 0.089 ± 0.001 |
| N174Y | 0.544 ± 0.009 | 3.074 ± 0.059 | 0.224 ± 0.008 | 0.078 ± 0.003 |
Transhydrogenase and reverse NADH kinase activities of DLDH744.
The energy-independent, soluble transhydrogenase UdhA of E. coli has been reported to convert NADPH and NAD+ to NADP+ and NADH, respectively, under high concentrations of NADPH (31). It is of interest to determine whether DLDH744 had a similar transhydrogenase activity and therefore used regenerated NADH as a coenzyme. No NADH was detected when purified DLDH744 was incubated with NADPH, even upon addition of NAD+ (data not shown). After the enzymatic reaction with pyruvate as a substrate and NADPH as a coenzyme, only NADP+ was identified as a product. The amount of NADP+ produced (0.199 mM) was consistent with the quantity of NADPH added (0.200 mM). Therefore, no hydrogen was transferred from NADPH to NAD+ to produce NADH. Additionally, NADH kinases were reported to directly phosphorylate NADH to produce NADPH, which subsequently enhanced the productivity of the NADPH-dependent biotransformation processes (31). To exclude the possibility that NADH was being produced from NADPH by reverse NADH kinase activity, the quantity of released phosphate was measured using Milli-Q water in the enzymatic reaction mixture. As expected, there was no liberation of phosphate from NADPH when DLDH744 and NAPDH were incubated with pyruvate. These results provided clear evidence that DLDH744 was capable of utilizing NADPH as a coenzyme.
DISCUSSION
Hydroxy acid dehydrogenases, which are key players in carbohydrate metabolism, catalyze the reversible conversion of keto acids into chiral hydroxyl acids concomitant with the oxidation of NAD(P)H to NAD(P)+ (7). Besides the NAD(P)+-dependent hydroxy acid dehydrogenases, the NAD(P)+-independent lactate dehydrogenases have also been found in some bacteria (6). We found no activity of DLDH744 toward pyruvate without the cofactors NAD+ or NADP+, indicating that it is an NAD(P)+-dependent lactate dehydrogenase.
D-LDH and glutamate dehydrogenase (GDH) belong to two distinct families: d-2-hydroxyacid dehydrogenases and Glu/Leu/Phe/Val dehydrogenases, respectively. Unlike d-LDHs, which are all reported to be NAD+ dependent to date, three types of GDHs have been identified: NAD+ dependent, NADP+ dependent, and dual-nucleotide specific (32). As DLDH744 could use both NADH and NADPH with high efficiencies and even with a preference for NADPH as the coenzyme both in vitro and in vivo, protein structures were first compared for DLDH744 and GDHs. However, the similarities between DLDH744 and GDH were quite low at the amino acid sequence level, making the direct comparisons of DLDH744 to GDH unreliable. Consequentially, the focus of the study was shifted to investigating the difference between DLDH744 and other reported NAD-linked LDHs with experimentally verified functionalities.
It was reported that primary sequence alone is not a reliable guide for predicting coenzyme specificity (32). Cofactor specificity appears to be a function of the arrangement of secondary structural elements. Therefore, protein structural analysis was also performed while investigating the specific NADP+-binding site of DLDH744. Bernard et al. (29) reported that Asp175 is responsible for the NADH preference of D-LDHs. This is not true for DLDH744, since the residue in this position is also aspartate but DLDH744 was able to use both NADH and NADPH. We found that the N174Y mutation of DLDH744 specifically affected the use of NADPH as a coenzyme. In the structure, Asp175 forms hydrogen bonds with the phosphate groups of the adenosine ribose, and the side chain of Asn174 is directed toward the surface of the cofactor-binding domain. The corresponding amino acid residue in other functionally verified NAD+-dependent D-LDHs was tyrosine, which contains a large and hydrophobic side chain, in contrast to the functional Asn174 in DLDH744. The contrast between the crystal structure for DLDH744 and the NAD+-dependent D-LDH from Aquifex aeolicus validates the prediction, as shown in Fig. 5b (33). The neutral, small side chain of asparagine adjacent to the crucial residue Asp175 has an advantage in reducing steric hindrance for binding of the negatively charged and bulky phosphate group of NADPH. Thus, mutation of Asn174 to Tyr increased steric hindrance and the mutant reduced the LDH activity with NADPH. Additionally, the Km value for NADH was reduced whereas the Km value for NADPH was increased after mutation, indicating that Asn174 plays an important role in cofactor specificity. It should be noticed that the Km value for NADPH was still lower than that for NADH by the mutation N174Y (Table 2). There might be other features of the enzyme that participate in giving it a higher affinity for NADPH.
To further investigate the importance of Asn174 in the utilization of NADPH as a coenzyme, the corresponding amino acid (Tyr) of the NAD-dependent d-lactate dehydrogenase from Lactobacillus delbrueckii subsp. bulgaricus DSM 20081 was mutated to Asn. Although the activity of d-lactate dehydrogenase from L. bulgaricus with NADPH after site mutation was not as high as the activity using NADH, the gain-of-function of this specific enzyme activity using NADPH as a coenzyme still demonstrated the importance of Asn174 in the utilization of NADPH as a coenzyme. Furthermore, structural analysis has shown that NADPH cannot enter the interdomain active-site cleft of the D-LDH from L. bulgaricus (28). DLDH744 and the D-LDH from L. bulgaricus share only 41% identities at the amino acid level. Unlike the NAD+-dependent D-LDH from L. bulgaricus, DLDH744 might interact with NADPH as a cofactor directly, perhaps in a different conformation from that in which it uses NADH as a cofactor. This scenario would be in accordance with a recent report describing the NADP+-dependent glutamate dehydrogenase from E. coli, in which the conformation was changed when using NADPH as a coenzyme from its NADH utilization pattern (32).
The l-lactate dehydrogenase from Bacillus subtilis (BsLDH) was reported to be engineered for regeneration of NADPH. The maximal specific activity of engineered BsLDH with NADPH was 90.8 U/mg with a Km value of 0.06 mM (12). Until now, no NADP+-dependent d-lactate dehydrogenase was found in nature (6). The affinity for NADPH of wild-type DLDH744 (Km value of 0.036 mM) is higher than that of the engineered BsLDH, although the activity of wild-type DLDH744 with NADPH was lower. Therefore, the NADP-preferring d-lactate dehydrogenase found in this study would expand the enzymatic systems for the regeneration of NADPH, especially for some stereospecific conversion reactions in which l-lactate dehydrogenase should not be applied.
In summary, to our knowledge, this study is the first to report a wild-type lactate dehydrogenase that uses both NADH and NADPH with high efficiency and shows preference for NADPH as a coenzyme. The characterization of the active residues of this enzyme contributes to the mechanistic understanding of its activities.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Basic Research Program of China (2011CBA00800), the Key Deployment Projects of Chinese Academy of Sciences (KGZD-EW-606), the National Natural Science Foundation of China (31300601), and the PUMC Youth Fund (33320140186).
We thank Ping Xu (Shanghai Jiao Tong University, Shanghai, China) for helpful discussions and Zhiyong Lou (Tsinghua University, Beijing, China) for help in revising the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01871-15.
REFERENCES
- 1.Peng LL, Wang LM, Che CC, Yang G, Yu B, Ma YH. 2013. Bacillus sp. strain P38: an efficient producer of L-lactate from cellulosic hydrolysate, with high tolerance for 2-furfural. Bioresour Technol 149:169–176. doi: 10.1016/j.biortech.2013.09.047. [DOI] [PubMed] [Google Scholar]
- 2.Wang LM, Cai YM, Zhu LF, Guo HL, Yu B. 2014. Major role of NAD-dependent lactate dehydrogenases in high optically pure L-lactic acid production by thermophilic Bacillus coagulans. Appl Environ Microbiol 80:7134–7141. doi: 10.1128/AEM.01864-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang X, Xue YF, Wang AY, Wang LM, Zhang GM, Zeng QT, Yu B, Ma YH. 2013. Efficient production of polymer-grade L-lactate by an alkaliphilic Exiguobacterium sp. strain under nonsterile open fermentation conditions. Bioresour Technol 143:665–668. doi: 10.1016/j.biortech.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 4.Ikada Y, Jamshidi K, Tsuji H, Hyon SH. 1987. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 20:904–906. doi: 10.1021/ma00170a034. [DOI] [Google Scholar]
- 5.Li Y, Wang LM, Ju JS, Yu B, Ma YH. 2013. Efficient production of polymer-grade D-lactate by Sporolactobacillus laevolacticus DSM442 with agricultural waste cottonseed as the sole nitrogen source. Bioresour Technol 142:186–191. doi: 10.1016/j.biortech.2013.04.124. [DOI] [PubMed] [Google Scholar]
- 6.Garvie EI. 1980. Bacterial lactate dehydrogenases. Microbiol Rev 44:106–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hummel W. 1999. Large-scale applications of NAD(P)-dependent oxidoreductases: recent developments. Trends Biotechnol 17:487–492. doi: 10.1016/S0167-7799(98)01207-4. [DOI] [PubMed] [Google Scholar]
- 8.Drent M, Cobben NAM, Henderson RF, Wouters EFM, van Dieijen-Visser M. 1996. Usefulness of lactate dehydrogenase and its isoenzymes as indicators of lung damage or inflammation. Eur Respir J 9:1736–1742. doi: 10.1183/09031936.96.09081736. [DOI] [PubMed] [Google Scholar]
- 9.Taguchi H, Ohta T. 1991. D-Lactate dehydrogenase is a member of the D-isomer-specific 2-hydroxyacid dehydrogenase family. Cloning, sequencing, and expression in Escherichia coli of the D-lactate dehydrogenase gene of Lactobacillus plantarum. J Biol Chem 266:12588–12594. [PubMed] [Google Scholar]
- 10.Holton SJ, Anandhakrishnan M, Geerlof A, Wilmanns M. 2013. Structural characterization of a D-isomer specific 2-hydroxyacid dehydrogenase from Lactobacillus delbrueckii subsp. bulgaricus. J Struct Biol 181:179–184. doi: 10.1016/j.jsb.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Weckbecker A, Groger H, Hummel W. 2010. Regeneration of nicotinamide coenzymes: principles and applications for the synthesis of chiral compounds. Adv Biochem Eng Biotechnol 120:195–242. doi: 10.1007/10_2009_55. [DOI] [PubMed] [Google Scholar]
- 12.Richter N, Zienert A, Hummel W. 2011. A single-point mutation enables lactate dehydrogenase from Bacillus subtilis to utilize NAD+ and NADP+ as cofactor. Eng Life Sci 11:26–36. doi: 10.1002/elsc.201000151. [DOI] [Google Scholar]
- 13.Wang LM, Zhao B, Li FS, Xu K, Ma CQ, Tao F, Li QG, Xu P. 2011. Highly efficient production of D-lactate by Sporolactobacillus sp. CASD with simultaneous enzymatic hydrolysis of peanut meal. Appl Microbiol Biotechnol 89:1009–1017. doi: 10.1007/s00253-010-2904-9. [DOI] [PubMed] [Google Scholar]
- 14.Yu B, Su F, Wang LM, Xu K, Zhao B, Xu P. 2011. Draft genome sequence of Sporolactobacillus inulinus strain CASD, an efficient D-lactic acid-producing bacterium with high-concentration lactate tolerance capability. J Bacteriol 193:5864–5865. doi: 10.1128/JB.05934-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q, Shao WL. 2010. Molecular genetic characterization of the thermostable L-lactate dehydrogenase gene (ldhL) of Thermoanaerobacter ethanolicus JW200 and biochemical characterization of the enzyme. Biochemistry (Mosc) 75:526–530. doi: 10.1134/S0006297910040188. [DOI] [PubMed] [Google Scholar]
- 16.Mu W, Yu S, Jiang B, Li X. 2012. Characterization of D-lactate dehydrogenase from Pediococcus acidilactici that converts phenylpyruvic acid into phenyllactic acid. Biotechnol Lett 34:907–911. doi: 10.1007/s10529-012-0847-1. [DOI] [PubMed] [Google Scholar]
- 17.Li B, Wang Q, Pan X, Fernández de Castro I, Sun Y, Guo Y, Tao X, Risco C, Sui SF, Lou Z. 2013. Bunyamwera virus possesses a distinct nucleocapsid protein to facilitate genome encapsidation. Proc Natl Acad Sci U S A 110:9048–9053. doi: 10.1073/pnas.1222552110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Sun Y, Guo Y, Lou Z. 2013. Structural perspective on the formation of ribonucleoprotein complex in negative-sense single-stranded RNA viruses. Trends Microbiol 21:475–484. doi: 10.1016/j.tim.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 22.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.DeLano WL. 2002. The PyMOL molecular graphics system. http://www.pymol.org. [Google Scholar]
- 24.Urban A, Neukirchen S, Jaeger KE. 1997. A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR. Nucleic Acids Res 25:2227–2228. doi: 10.1093/nar/25.11.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoek JB, Rydstrom J. 1988. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J 254:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bieganowski P, Seidle HF, Wojcik M, Brenner C. 2006. Synthetic lethal and biochemical analyses of NAD and NADH kinases in Saccharomyces cerevisiae establish separation of cellular functions. J Biol Chem 281:22439–22445. doi: 10.1074/jbc.M513919200. [DOI] [PubMed] [Google Scholar]
- 27.Gouet P, Robert X, Courcelle E. 2003. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Razeto A, Kochhar S, Hottinger H, Dauter M, Wilson KS, Lamzin VS. 2002. Domain closure, substrate specificity and catalysis of D-lactate dehydrogenase from Lactobacillus bulgaricus. J Mol Biol 318:109–119. doi: 10.1016/S0022-2836(02)00086-4. [DOI] [PubMed] [Google Scholar]
- 29.Bernard N, Johnsen K, Holbrook JJ, Delcour J. 1995. D175 discriminates between NADH and NADPH in the coenzyme binding site of Lactobacillus delbrueckii subsp. bulgaricus D-lactate dehydrogenase. Biochem Biophys Res Commun 208:895–900. doi: 10.1006/bbrc.1995.1419. [DOI] [PubMed] [Google Scholar]
- 30.Zheng Z, Sheng B, Ma C, Zhang H, Gao C, Su F, Xu P. 2012. Relative catalytic efficiency of ldhL-and ldhD-encoded products is crucial for optical purity of lactic acid produced by Lactobacillus strains. Appl Environ Microbiol 78:3480–3483. doi: 10.1128/AEM.00058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee WH, Kim MD, Jin YS, Seo JH. 2013. Engineering of NADPH regenerators in Escherichia coli for enhanced biotransformation. Appl Microbiol Biotechnol 97:2761–2772. doi: 10.1007/s00253-013-4750-z. [DOI] [PubMed] [Google Scholar]
- 32.Sharkey MA, Oliveira TF, Engel PC, Khan AR. 2013. Structure of NADP(+)-dependent glutamate dehydrogenase from Escherichia coli-reflections on the basis of coenzyme specificity in the family of glutamate dehydrogenases. FEBS J 280:4681–4692. doi: 10.1111/febs.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antonyuk SV, Strange RW, Ellis MJ, Bessho Y, Kuramitsu S, Inoue Y, Yokoyama S, Hasnain SS. 2009. Structure of D-lactate dehydrogenase from Aquifex aeolicus complexed with NAD+ and lactic acid (or pyruvate). Acta Crystallogr F Struct Biol Cryst Commun 65:1209–1213. doi: 10.1107/S1744309109044935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.