

Abstract
The cytochrome P450 enzyme CYP-sb21 from Sebekia benihana is capable of catalyzing the site-specific hydroxylation of the immunosuppressant cyclosporine (CsA), leading to the single product γ-hydroxy-N-methyl-l-Leu4-CsA (CsA-4-OH). Unlike authentic CsA, this hydroxylated CsA shows significantly reduced immunosuppressive activity while it retains a side effect of CsA, the hair growth stimulation effect. Although CYP-sb21 was previously identified to be responsible for CsA-specific hydroxylation in vivo, the in vitro activity of CYP-sb21 has yet to be established for a deeper understanding of this P450 enzyme and further reaction optimization. In this study, we reconstituted the in vitro activity of CYP-sb21 by using surrogate redox partner proteins of bacterial and cyanobacterial origins. The highest CsA site-specific hydroxylation activity by CYP-sb21 was observed when it was partnered with the cyanobacterial redox system composed of seFdx and seFdR from Synechococcus elongatus PCC 7942. The best bioconversion yields were obtained in the presence of 10% methanol as a cosolvent and an NADPH regeneration system. A heterologous whole-cell biocatalyst using Escherichia coli was also constructed, and the permeability problem was solved by using N-cetyl-N,N,N-trimethylammonium bromide (CTAB). This work provides a useful example for reconstituting a hybrid P450 system and developing it into a promising biocatalyst for industrial application.
INTRODUCTION
Cyclosporine (CsA) (Fig. 1), a lipophilic cyclopeptide consisting of 11 amino acids, was first isolated from the soil fungus Tolypocladium inflatum (1). It is a well-known immunosuppressant drug that has been widely used in organ transplantation to prevent allograft rejection and in the treatment of autoimmune diseases (2, 3). Despite its immunosuppressant activity, CsA provokes several side effects, such as nephrotoxicity, hypertension, and hirsutism (4, 5). Among these side effects, the strong hair-growth-stimulating activity of CsA has attracted significant attention from both the academic community and the cosmetics industry due to its great potential to treat alopecia (6, 7). Mechanistically, it was revealed that CsA stimulates the growth of both murine hair epithelial cells and epidermal keratinocytes at an optimum concentration through inhibiting the expression and translocation of some protein kinase C isozymes that act as negative hair-growing factors (8).
FIG 1.

Bioconversion of CsA to CsA-4-OH by CYP-sb21 with heterologous redox systems.
Unfortunately, CsA was not practical for use as a hair-restoring agent due to its strong immunosuppressive activity. To decouple these two activities, a wide spectrum of CsA derivatives with alternative amino acid substitutions was prepared and evaluated (9), among which the derivative γ-hydroxy-N-methyl-l-Leu4-CsA (CsA-4-OH) (Fig. 1) was found to have lost immunosuppressive activity while retaining the considerable hair-growth-promoting effect (10). Regarding preparation, the highly regioselective hydroxylation of CsA by a chemical catalyst is extremely challenging. Thus, thousands of actinomycete strains were screened to select Sebekia benihana KCTC 9610 as the biotransformation strain, which is able to convert CsA into CsA-4-OH (Fig. 1) albeit at a low conversion rate. Later, the cytochrome P450 enzyme CYP-sb21 of this strain was identified to be responsible for the highly regiospecific hydroxylation at the fourth N-methyl leucine in CsA (11). Recently, CYP-sb21 was named CYP107Z14 based on the nomenclature system developed by David Nelson (12).
Cytochrome P450 enzymes (CYPs or P450s) are a superfamily of heme-containing proteins that mainly catalyze the oxidative reactions of a tremendous variety of compounds such as terpenes, steroids, polyketides, peptides, and other xenobiotic chemicals (13–15). Many P450 biocatalysts have been applied in pharmaceutical industries because they are capable of catalyzing some very specific regio- and stereoselective hydroxylation or epoxidation reactions of complex substrates to generate high-value-added products (16).
The sequential transfer of two reducing equivalents from NAD(P)H to the heme-iron reactive center of P450 enzymes via the delivery of a redox partner system is a key step in the P450 catalytic cycle. The efficiency of electron transfer directly affects the overall rate of P450-catalyzed reactions. To date, at least three major redox partner systems have been identified. The two-component class I redox system for most bacterial and mitochondrial CYPs comprises an iron-sulfur protein (ferredoxin [Fdx]) and a flavin adenine dinucleotide (FAD)-containing ferredoxin reductase (FdR) (17). The class II redox system, which mainly serves eukaryotic P450s, employs an FAD/flavin mononucleotide (FMN)-containing fused functional domain as the cytochrome P450 reductase (CPR) (18). The class III system is made up of an FMN/Fe2S2-containing reductase domain termed “RhFRED” that is naturally fused with a P450 enzyme from Rhodococcus sp. strain NCIMB 9784 (19). Novel P450s have been increasingly emerging as explosive genome sequencing data become available (20). More classification systems for P450 reductases have also been proposed (21). However, the optimal (either native or heterologous) redox partners often remain unknown or are difficult to express for many potential industrial P450s. Thus, it often becomes necessary to construct hybrid P450 reaction systems by using surrogate redox partner proteins in order to achieve desired catalytic processes (22).
For the P450 CsA hydroxylase CYP-sb21, seven putative fdx genes and four fdR genes were mined from the whole genome of S. benihana (11). Each of these 11 genes was individually inactivated by using the PCR-targeted gene disruption system. None of Fdxs or FdRs turned out to be CYP-sb21 specific. Although it was implied that Fdx-sb8 and FdR-sb3 might be the major redox partners for the regiospecific CsA hydroxylation by CYP-sb21 in vivo, this has not been validated, since an expression system for these S. benihana redox proteins has not yet been developed.
In this study, we confirmed that CYP-sb21 is the CsA-4-OH synthase in vitro by using four different surrogate redox partner systems, including spinach ferredoxin and spinach ferredoxin-NADP+ reductase (spFdx and spFdR, respectively), the ferredoxin (seFdx) (SynPcc7942_1499) and ferredoxin-NADP+ reductase (seFdR) (SynPcc7942_0978) from the cyanobacterium Synechococcus elongatus PCC 7942, RhFRED from Rhodococcus sp. NCIMB 9784, and a hybrid Rhodococcus-spinach reductase, RhFRED-Fdx (22). By using optimal cyanobacterial redox partners, a number of in vitro reaction factors was optimized to improve the overall rate of conversion of CsA to CsA-4-OH, such as the cosolvent and the NADPH regeneration system. Finally, a whole-cell biotransformation system for the cost-effective production of CsA-4-OH was developed. This system could be applied for the scaled-up production of this valuable hair-growth-stimulating agent in the future.
MATERIALS AND METHODS
Materials.
Antibiotics were obtained from SolarBio (Beijing, China). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). The codon-optimized cyp-sb21 gene was synthesized by Cosmo Genetech (Seoul, South Korea). Oligonucleotides were synthesized by Sangon Biotech (Shanghai, China), and their sequences are shown in Table 1. Pfu high-fidelity DNA polymerase, all restriction endonucleases, and T4 DNA ligase were bought from Fermentas (Vilnius, Lithuania) or TaKaRa (Dalian, China). The kits for plasmid extraction and DNA purification were purchased from Omega Bio-Tek (Jinan, China) and Promega (Madison, WI, USA), respectively. His-tagged protein purification was performed by using Ni-nitrilotriacetic acid (NTA) resin (Qiagen, Valencia, CA, USA), Amicon Ultra centrifugal filters (Millipore, Billerica, MA, USA), and PD-10 desalting columns (GE Healthcare, Piscataway, NJ, USA). CsA and CsA-4-OH were prepared as previously reported (11).
TABLE 1.
Primers used in this study
| Oligonucleotide | Primer sequence (5′–3′) | Function of underlined bases |
|---|---|---|
| CYP-sb21-F | GGAATTCCATATGGACACCGTTAATCTG | NdeI |
| CYP-sb21-R1 | CGGAATTCGCGCAGCAGAACCGGCAG | EcoRI |
| Fdx-F | GGAATTCCATATGATGGCAACCTACAAGG | NdeI |
| Fdx-R | CCGCTCGAGTTAGTAGAGGTCTTCTTC | XhoI |
| FdR-F | CGGGATCCATGTTGAATGCGAGTGTG | BamHI |
| FdR-R | CCCAAGCTTCTAGTAGGTTTCAACATGCC | HindIII |
| RhFRED-F | CAGATTCATATGGTGCTGCACCGCCATCAA | NdeI |
| RhFRED-R | CCCAAGCTTGAGTCGCAGGGCCAGCCG | HindIII |
| CYP-sb21-R2 | TTGATGCCGGTGCAGGCGCAGCAGAACC | cyp-sb21 overhang |
| RhFRED-Fdx-F | GGTTCTGCTGCGCCTGCACCGGCATCAAC | FMN domain overhang |
| RhFRED-Fdx-R | CCCAAGCTTTTATGCCGTCAGTTCTTC | HindIII |
| GDH-F | GGAATTCCATATGTATAAAGATTTAAAGG | NdeI |
| GDH-R | CGCGGATCCTTATCCGCGTCCTGCTTG | BamHI |
| CYP-sb21-Duet-F | GGAATTCCATATGGACACCGTTAATCTG | NdeI |
| CYP-sb21-Duet-R | CCGCTCGAGTCAGCGCAGCAGAACCGGCAG | XhoI |
Construction of recombinant plasmids.
Standard molecular cloning techniques were used to construct recombinant plasmids. Primers used for this study are shown in Table 1. The codon-optimized cyp-sb21 gene was inserted between the NdeI and EcoRI restriction sites of pET-28b to create pET-28b-cyp-sb21. To construct the cyp-sb21-RhFRED fusion gene, the cyp-sb21 fragment and the RhFRED gene fragment were PCR amplified by using pET-28b-cyp-sb21 and the previously constructed plasmid pET28b-pikC-RhFRED (23) as the templates and primer pairs CYP-sb21-F/CYP-sb21-R1 and RhFRED-F/RhFRED-R, respectively. Subsequently, the cyp-sb21 and RhFRED genes were double digested with NdeI/EcoRI and EcoRI/HindIII, respectively, and ligated into NdeI/HindIII-pretreated pET28b to create pET28b-cyp-sb21-RhFRED. The cyp-sb21-RhFRED-fdx hybrid gene was prepared by overlap extension PCR (24). The gene encoding CYP-sb21 was amplified from pET28b-cyp-sb21-RhFRED by using primer set CYP-sb21-F/CYP-sb21-R2. The RhFRED-fdx gene fragment was amplified from pET28b-RhFRED-fdx (22) by using primer pair RhFRED-Fdx-F/RhFRED-Fdx-R. The two PCR products with designed overlap sequences were mixed, annealed, extended, and finally amplified with primer pair CYP-sb21-F2/RhFRED-Fdx-R, resulting in the fused cyp-sb21-RhFRED-fdx gene, which was subcloned into pET-28b via the NdeI and HindIII restriction sites to obtain pET-28b-cyp-sb21-RhFRED-fdx. The vectors pET28b-sefdx and pET28b-sefdR for expression of Fdx and FdR of S. elongatus PCC 7942 were gifts from Xuefeng Lu, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences. For construction of the pCDFDuet-sefdR-sefdx coexpression vector, the sefdx and sefdR genes bearing the corresponding restriction sites were PCR amplified and sequentially inserted into the NdeI-XhoI and BamHI-HindIII sites of pCDFDuet-1 (25). Plasmids pET-28b-cyp-sb21 and pCDFDuet-sefdR-sefdx were cotransformed into Escherichia coli BL21(DE3) cells to obtain whole-cell transformation strain W1. pET-28b-gdh-cyp-sb21 was constructed as follows. First, the gdh gene from Bacillus subtilis was ligated into pET-28b, and cyp-sb21 was ligated into pCDFDuet-1. The latter plasmid was then digested with EcoRI/XhoI to obtain a fragment that includes the T7 promoter sequence. The resulting DNA fragment was ligated into pET28b-cyp-sb21 to obtain pET-28b-gdh-cyp-sb21. Plasmids pET-28b-gdh-cyp-sb21 and pCDFDuet-sefdR-sefdx were cotransformed into E. coli, resulting in strain W2. All of the above-described constructs (Table 2) were confirmed through DNA sequencing by Sangon Biotech (Shanghai, China).
TABLE 2.
Strains and plasmids used in this study
| E. coli strain or plasmid | Characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| DH5α | Cloning host | Life Technologies |
| BL21(DE3) | Expression host | Life Technologies |
| W1 | Recombinant E. coli strain coexpressing CYP-sb21, seFdx, and seFdR | This work |
| W2 | Recombinant E. coli strain coexpressing GDH, CYP-sb21, seFdx, and seFdR | This work |
| Plasmids | ||
| pET28b-cyp-sb21 | Vector for heterologous expression of CYP-sb21 | This work |
| pET28b-cyp-sb21-RhFRED | Vector for expression of the CYP-sb21-RhFRED fusion protein | This work |
| pET28b-cyp-sb21-RhFRED-fdx | Vector for expression of the CYP-sb21-RhFRED-Fdx fusion protein | This work |
| pCDFDuet-1-seFdx/seFdR | Vector for coexpression of seFdx and seFdR | This work |
| pET28b-gdh-cyp-sb21 | Vector for coexpression of GDH and CYP-sb21 | This work |
Protein purification.
The constructed plasmid was transformed into Escherichia coli BL21(DE3). A single colony of the transformant was inoculated into LB medium containing 50 mg/liter kanamycin. The seed culture grown overnight was used for 1:100 inoculation of 1 liter of LB medium containing 50 mg/liter kanamycin, 1 mM thiamine, 10% glycerol, and rare salt solution at 37°C (26). Protein expression was induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and 1 mM δ-aminolevulinic acid was added as the heme synthetic precursor until the optical density at 600 nm (OD600) reached 0.6 to 1.0. The cells were then incubated with shaking for 20 h at 18°C. The following protein purification procedure was carried out according to a previously developed procedure (27). All purified enzymes were verified to be >95% pure, as assessed by SDS-PAGE (Fig. 2A), and stored at −80°C. The UV-visible spectra were obtained on a DU 800 spectrophotometer (Beckman Coulter). The functional concentration of CYP-sb21 was calculated from the CO-bound reduced difference spectrum by using an extinction coefficient (ε450–490) of 91,000 M−1 · cm−1 (28). The concentrations of redox partner proteins were determined by a Bradford assay (29), using bovine serum albumin (BSA) as a standard.
FIG 2.

(A) SDS-PAGE analysis of purified proteins. (B) CO-bound reduced difference spectrum of purified CYP-sb21.
In vitro enzyme assays for CYP-sb21 activities.
The standard reaction mixture contained 10 μM CYP-sb21, 10 μM RhFRED (or 10 μM Fdx plus 10 μM FdR), 200 μM CsA (diluted from a 20 mM stock solution in methanol), and 1 mM NADPH in 100 μl of reaction buffer (50 mM NaH2PO4, 10% glycerol [pH 7.4]). Optionally, 5 U glucose dehydrogenase (GDH) and 20 mM glucose were added as an NADPH regeneration system (30). For self-sufficient versions of CYP-sb21 enzymes, 10 μM CYP-sb21-RhFRED or CYP-sb21-RhFRED-Fdx was used for replacement of the separated P450 and redox partner proteins in the above-described standard reaction mixture. P450 catalysis via the H2O2 shunt pathway uses H2O2 as the sole oxygen and electron donor, thus not requiring P450 reductase and NADPH (31). To test this possibility, 200 μM H2O2 was added to replace P450 reductase and NADPH in the CYP-sb21 reaction mixture. The reaction was stopped after incubation of the mixture at 30°C for 16 h by the addition of 100 μl methanol. The samples were centrifuged to remove precipitated proteins, and the supernatants were analyzed by high-performance liquid chromatography (HPLC) with an SB-C18 column (3.5 μm, 4.6 by 150 mm; Agilent) in a gradient system consisting of 25.0% methanol in deionized water (solvent A) and 100% acetonitrile (solvent B). One cycle of the buffer B gradient was programmed as follows: 40% buffer B for 4 min, 40 to 61% buffer B for 20 min, and 61 to 100% buffer B for 40 min, followed by 40% buffer B for 45 min. Detection was performed at 210 nm. The flow rate was 1.0 ml/min, and the injection volume was 60 μl.
NADPH coupling efficiency.
According to a previously established procedure (32), the NADPH coupling efficiency was determined as follows. NADPH consumption was monitored at 340 nm by using the Synergy HT plate reader (BioTek) and calculated with an extinction coefficient of 6.22 mM−1 · cm−1. The substrate consumption rate was measured by HPLC. All measurements were performed in duplicate. The coupling efficiency was calculated as the percentage of NADPH used for product formation over the total consumed NADPH.
LC-Q-TOF/MS analysis of products.
Liquid chromatography-quadrupole time of flight mass spectrometry (LC-Q-TOF/MS) analysis was carried out with a maXis ultrahigh-resolution TOF system (Bruker Daltonik, Germany), using a Thermo Scientific Hypersil Gold column (5 μm, 2.1 mm by 100 mm) at 30°C with H2O plus 25% methanol (MeOH) as solvent A and acetonitrile as solvent B. The gradient elution profile was as follows: 40% solvent B from 0 to 4 min, 40 to 61% solvent B from 4 to 15 min, 61 to 100% solvent B from 15 to 32 min, and 40% solvent B from 32 to 35 min. The flow rate was set at 0.2 ml/min, and the injection volume was 1 μl.
Optimization of the cosolvent system.
CsA is poorly soluble in water, with the maximum aqueous solubility being estimated to be ∼5 to 10 μM (http://www.lclabs.com/products/37-c-6000-cyclosporin-a/sds). To make CsA more water miscible, thereby improving the conversion rate, cosolvents, including ethanol, dimethyl sulfoxide (DMSO), and methanol, were added at 1%, 10%, or 20% (vol/vol) to the CYP-sb21 enzymatic assay mixtures. The impacts of the type and percentage of cosolvents on CsA hydroxylation were comparatively evaluated by HPLC.
Whole-cell biotransformation.
E. coli cells harboring pET28b-cyp-sb21/pCDFDuet-sefdR-sefdx (W1) or pET28b-gdh-cyp-sb21/pCDFDuet-sefdR-sefdx (W2) were cultured in LB medium containing 50 mg/liter kanamycin and streptomycin at 37°C. Protein expression was performed as described above. The cells were harvested by centrifugation and resuspended in 50 mM potassium phosphate buffer (pH 7.5) with 0.3% (wt/vol) N-cetyl-N,N,N-trimethylammonium bromide (CTAB), resulting in a suspension containing 200 mg (wet weight) of cells per ml. Next, CsA and NADPH were each added at a 100 μM final concentration and coincubated at 30°C in an orbital shaker at 150 rpm for 12 h. For biotransformation by W2, 100 mM glucose was additionally supplemented. Biotransformation was performed in a 50-ml shaking flask containing 5 ml of the reaction slurry. The reaction mixture was thoroughly extracted twice with an equal volume of ethyl acetate. The combined organic extracts were dried by nitrogen flow and resuspended in 100 μl MeOH as a testing sample. All samples were analyzed by HPLC.
Statistical analysis.
The t test was used to evaluate the significance of differences between groups. Each experimental value was expressed as the mean ± standard deviation (SD). A P value of <0.05 was taken to indicate statistical significance.
RESULTS
Expression of S. benihana CYP-sb21 and surrogate redox partners in E. coli.
Since CYP-sb21 was expressed at a low level in E. coli initially (data not shown), the cyp-sb21 sequence was optimized according to E. coli codon preferences. Codon-optimized cyp-sb21 and the corresponding fusion genes were successfully expressed and purified to homogeneity (Fig. 2A). The CO-bound reduced difference spectra of different versions of CYP-sb21 proteins showed the characteristic peak at 450 nm, confirming the expression of functional P450 enzymes (Fig. 2B). Also, surrogate redox partner proteins, such as seFdx, seFdR, and RhFRED, were expressed and purified by SDS-PAGE (Fig. 2A).
In vitro conversion of CsA to CsA-4-OH by different versions of CYP-sb21 enzymes.
Reconstitution of the in vitro activity of CYP-sb21 was initiated by using the commercially available spinach redox partner proteins spFdx and spFdR. Unfortunately, this commonly used redox system failed to support CYP-sb21 activity (Fig. 3A). Since a previous in vivo study (11) provided solid evidence that CYP-sb21 should be responsible for the conversion of CsA to CsA-4-OH, this unsuccessful result is likely due to the mismatch of the P450 enzyme and redox partners.
FIG 3.
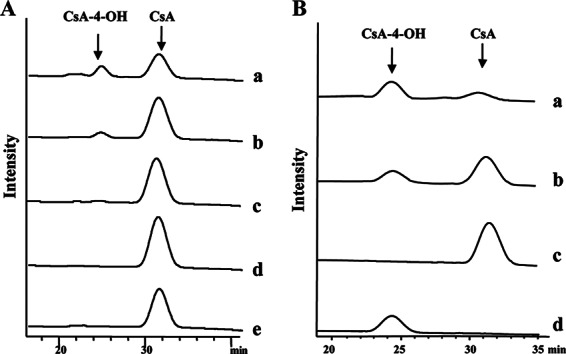
HPLC analysis of different CYP-sb21 reaction mixtures. (A) CsA hydroxylation by CYP-sb21 using different surrogate electron transfer systems. (a) seFdx and seFdR from S. elongatus; (b) Rhodococcus sp. RhFRED; (c) CYP-sb21 fused to RhFRED; (d) CYP-sb21 fused to RhFRED-Fdx; (e) spFdx and spFdR from spinach. (B) The NADPH regeneration system increases the biotransformation of CsA to CsA-4-OH. (a) GDH and glucose were added; (b) control without GDH and glucose; (c) CsA standard; (d) CsA-4-OH standard. In all cases, the reactions were carried out at 30°C for 16 h.
Thus, we sought to test two other redox systems, including the cyanobacterial system seFdx/seFdR and RhFRED from Rhodococcus sp. As a result, both seFdx/seFdR and RhFRED (Fig. 3A) were able to support CYP-sb21 for the generation of the same more polar product. High-resolution mass spectrometric analysis showed an m/z value of 1,240.8254 (corresponding to the molecular formula C62H111N11O13Na [calculated m/z 1,240.8261]) for this product, which is 16 Da higher than the molecular mass of [CsA + Na]+ (observed m/z 1,224.8305; calculated m/z 1,224.8311). The same retention time and coelution with the authentic CsA-4-OH standard confirmed that hydroxylation occurred at the γ position of N-methyl-l-Leu4 of CsA. Quantitatively, the conversion rates were 25.0% and 17.3% for the CYP-sb21 reaction using seFdx/seFdR and RhFRED, respectively.
We also used the self-sufficient P450 enzymes CYP-sb21-RhFRED and CYP-sb21-RhFRED-Fdx to hydroxylate CsA. CYP-sb21-RhFRED converted 9.4% of CsA into CsA-4-OH, while CYP-sb21-RhFRED-Fdx only marginally catalyzed the same reaction (Fig. 3A).
Optimization of the CYP-sb21/seFdx/seFdR reaction system in vitro.
CsA is poorly soluble in water. It was reported previously that a potential biocompatible organic solvent could improve the biocatalytic activity against hydrophobic compounds (33). Thus, different cosolvents, including ethanol, DMSO, and methanol, were tested in order to make CsA more water miscible, thereby improving the conversion rate. Among the tested organic cosolvent systems, 10% methanol gave the highest bioconversion rate, 53.0% (Table 3). Under these conditions, however, the NADPH coupling efficiency was only 7.9%. Notably, the amount of CsA-4-OH decreased when the concentrations of all cosolvents were >10%. This is likely due to enzyme denaturation by high concentrations of organic solvents.
TABLE 3.
Improvement of bioconversion efficiency by using organic cosolventsa
| Cosolvent | Concn (%) | Conversion rate (%) |
|---|---|---|
| Ethanol | 1 | 19.2 |
| 10 | 32.3 | |
| 20 | 26.8 | |
| DMSO | 1 | 20.1 |
| 10 | 39.6 | |
| 20 | 29.0 | |
| Methanol | 1 | 25.0 |
| 10 | 53.0 | |
| 20 | 44.9 |
Reaction conditions were 30°C for 16 h for the CYP-sb21/seFdx/seFdR reaction system. All experiments were performed in duplicate, and all standard errors were <10%.
NADPH is a cofactor required for P450 catalysis. It is well known that an NADPH regeneration system can significantly increase the P450 catalytic efficiency (34–36). In some cases, such a system is even necessary for the practical synthesis of important chemicals (37). Thus, we further optimized the CYP-sb21 reaction by using the GDH/glucose NADPH regeneration system, and 83.5% conversion of CsA was achieved (Fig. 3B).
Whole-cell biotransformation.
The whole-cell system has become increasingly important in industry. It avoids expensive procedures for enzyme purification and protects the enzyme from inactivation (38). Recombinant E. coli strain W1 coexpressing CYP-sb21, seFdx, seFdR was constructed, grown, and harvested as a whole-cell catalyst. We attempted to improve substrate solubility and uptake by adding a cosolvent, including 10% methanol, 10% DMSO, or 10% ethanol, as described above, during development of the in vitro enzymatic assay. Unfortunately, there was no product detected. This might be due to the impermeability of the bacterial outer membrane to CsA.
To solve this problem, a commonly used surfactant, CTAB, was added to the whole-cell biotransformation system, consisting of 200 mg (wet weight) of cells/ml, 100 μM NADPH, and 100 μM CsA. Upon optimization of the CTAB concentration (data not shown), the conversion rate was increased to 45.4% ± 4.2% when the cells were treated with 0.3% (wt/vol) CTAB (Fig. 4). Moreover, recombinant E. coli strain W2 coexpressing GDH further increased the rate of biotransformation of CsA to 53.5% ± 5.2%. The percentage of transformation by W2 is statistically different from that for W1 (P < 0.05).
FIG 4.
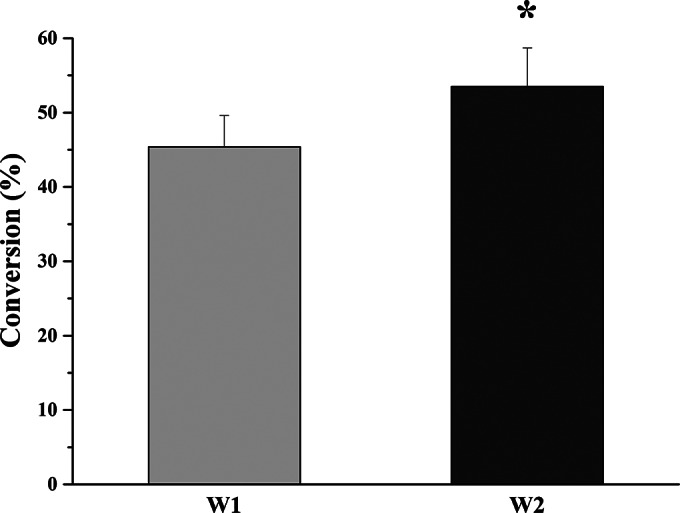
Whole-cell biotransformation of CsA to CsA-4-OH by W1 (E. coli cells harboring pET-28b-cyp-sb21 and pCDFDuet-sefdR-sefdx) and W2 (E. coli cells harboring pET-28b-gdh-cyp-sb21 and pCDFDuet-sefdR-sefdx). Assays were independently repeated three times. The asterisk indicates statistical significance (P < 0.05).
DISCUSSION
The processes of biotransformation of CsA to CsA-4-OH were previously investigated in vivo. Through screening of >100 soil actinomycetes, the rare actinomycete S. benihana was identified to hold the unique ability to regiospecifically hydroxylate the clinical immunosuppressant CsA at the γ position of the fourth N-methyl leucine, leading to CsA-4-OH with a significant loss of immunosuppressive activity, while maintaining the hair-growth-stimulating effect (39). Maximum bioconversion (54%) of 50 mg/liter (≈42 μM) CsA was achieved in a 5-day culture of S. benihana when molybdenum salts were added to the optimized medium (39).
Later, whole-genome sequencing and analysis of S. benihana and targeted gene disruptions enabled the identification of the cyclosporine-specific P450 hydroxylase gene cyp-sb21 (11). To improve CsA hydroxylation, an extra copy of the CYP-sb21 gene was integrated into the S. benihana chromosome. Alternatively, CYP-sb21 was heterologously expressed in a number of engineered Streptomyces coelicolor strains. Unfortunately, only low rates of conversion of CsA to CsA-4-OH were achieved with these approaches (11). The poor cross-membrane transfer of the substrate and product might be the major reason for the low conversion rates. In addition, suboptimal redox partners could be another limiting factor for the activity of CYP-sb21 in S. coelicolor.
In this study, we were able to overexpress the codon-optimized cyp-sb21 gene in E. coli and purify this P450 enzyme in its active form. By comparatively testing a group of surrogate electron transfer partners, including plant spFdx/spFdR, cyanobacterial seFdx/seFdR, Rhodococcus RhFRED, and hybrid Rhodococcus-spinach RhFRED-Fdx systems, the in vitro activity of CYP-sb21 was reconstituted. The CYP-sb21/seFdx/seFdR catalytic system showed the highest level of production of CsA-4-OH (Fig. 3A), while the commercially available spFdx/spFdR system did not support CYP-sb21 activity at all (Fig. 3A). These results highlight the significance of the selection of redox partners when reconstituting the activity of a P450 enzyme (22). We also examined the catalytic activity of CYP-sb21 using 200 μM H2O2 as the sole electron and oxygen donor. However, CYP-sb21 was unable to take advantage of the H2O2 shunt pathway (data not shown).
During the in vitro CYP-sb21 reactions, inhomogeneous aggregates formed immediately after CsA was added, indicating a significant solubility problem of the substrate. Thus, three different cosolvents were tested in the enzyme reaction system. It was found that 10% methanol improved the conversion rate from 25.0% to 53.0% (Table 3). However, this solubility problem was not completely solved, since insoluble matter still existed (although at a greatly reduced level) in the reaction solution. Further enhancement of the cosolvent concentration to 20% was unsuccessful, likely due to the denaturation of enzymes by high concentrations of organic solvents. Thus, how to increase the organic solvent tolerance of CYP-sb21 is an important question to be addressed in our future investigations.
Notably, the very low solubility of CsA prevented us from determining the kinetic parameters of CYP-sb21. Nonetheless, we surmise that the Km value for the unnatural substrate CsA is likely to be high (i.e., low binding affinity). The poor solubility of CsA may partially explain the very low rate of turnover (only 11.2 h−1 and 0.7 h−1 for 0.5-h and 16-h reactions, respectively) of CYP-sb21. The instability of CYP-sb21 resulting from the addition of 10% methanol and the radicals generated from significant uncoupled reactions could be another important reason.
NADPH is the required electron resource for P450 monooxygenase that affects the rate of reaction. This importance has been demonstrated by a previous study (11) showing that knockout of the SCO5426 gene (39) in S. coelicolor improved CsA hydroxylation by CYP-sb21 due to the increased supply of NADPH (11). GDH has been successfully applied for cofactor regeneration in many industrial processes (40), since it oxidizes glucose in the presence of the cofactor NAD(P)+ to form glucono-δ-lactone and NADPH. Our data also showed that the GDH/glucose-based NADPH regeneration system is an essential component for the efficient conversion of CsA to CsA-4-OH (Fig. 3B and 4).
In vitro enzymatic conversion is apparently more straightforward and environmentally friendly. However, it is not preferred for industrial applications due to the high cost of enzyme purification, low operational stability, and sensitivity of enzymes to process conditions (38). Practically, it is more attractive to use whole-cell catalysts. Since CYP-sb21 plus seFdx/seFdR turned out to be the best combination in vitro, a whole-cell catalyst system composed of E. coli cells coexpressing CYP-sb21 along with seFdx and seFdR was constructed. Upon using 0.3% (wt/vol) CTAB as an effective permeabilizer that can disintegrate cell membranes by removing Mg2+ and Ca2+ and increase the transport of substrates and products (41, 42), 45.4% conversion was achieved over 12 h with 100 μM CsA. When GDH was coexpressed, this conversion rate was further increased to 53.5%, which corresponds to an ∼20-times-higher conversion efficiency than that previously reported for bioconversion from CsA to 4-OH-CsA using S. benihana (39). Moreover, the E. coli whole-cell catalyst system has many other advantages, such as easy manipulation, high yield, rapid growth cycle, and easy product recovery.
It is well known that CTAB can make E. coli more porous, hence improving the performance of whole-cell transformation. For example, Bagherinejad et al. reported that CTAB-treated cells increased the biotransformation of penicillin G by 9% in a penicillin G acylase-containing recombinant E. coli strain (38). The nitrilase activity of Alcaligenes faecalis ECU0401 was increased 4.5-fold when cells were permeabilized by using 0.3% (wt/vol) CTAB (43). A recombinant lipase was expressed in E. coli by using olive oil as the substrate, which gave a conversion rate of almost 100% when cells were treated with 0.3% (wt/vol) CTAB for 20 h, while this rate decreased to <50% when untreated cells were used (44). Our results also support the usefulness of CTAB for the whole-cell transformation of CsA.
The established whole-cell transformation system could be developed into a very useful approach for cost-effective synthesis of the hair stimulator CsA-4-OH. However, more protein engineering and process engineering work should be performed in the future. On one hand, random mutagenesis/directed evolution or rational design/site-directed mutagenesis could be employed to increase the catalytic performance of CYP-sb21 through improving the electron transfer efficiency by altering P450-redox partner interactions and to enhance the organic cosolvent resistance of CYP-sb21. On the other hand, process factors during whole-cell biotransformation, such as temperature, pH, dissolved oxygen concentrations, and mass transfer, need to be systematically optimized.
The identification of suitable redox partners for the P450 cyclosporine hydroxylase CYP-sb21 of S. benihana enabled us to reconstitute the in vitro activity of CYP-sb21. With this knowledge on surrogate redox partners, we expect that a fungal CsA-4-OH-producing strain of Tolypocladium inflatum (the native CsA producer) could be engineered by introducing the codon-optimized genes encoding CYP-sb21 and seFdx/seFdR into the fungal genome. Since CsA-4-OH would be synthesized intracellularly from CsA, the problem of cross-membrane transfer could be solved provided that CsA-4-OH can be exported by the CsA efflux pump. This might be the best option for the cost-effective production of CsA-4-OH if this engineering work could be done by using an industrial high-CsA producer.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China under grant number NSFC 31422002, funding from the Recruitment Program of Global Experts (S.L.), and the National Research Foundation (NRF) of Korea (no. NRF-2014R1A2A1A11052236) (E.-S.K.).
We thank Jianjun Li and Xuefeng Lu at the Qingdao Institute of Bioenergy and Biotechnology, Chinese Academy of Sciences, for providing us with the plasmids for expression of seFdx and seFdR. We also thank David Nelson at the University of Tennessee Health Science Center for naming CYP-sb21.
REFERENCES
- 1.Borel JF, Feurer C, Gubler HU, Stahelin H. 1976. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions 6:468–475. doi: 10.1007/BF01973261. [DOI] [PubMed] [Google Scholar]
- 2.Flechner SM. 1983. Cyclosporine: a new and promising immunosuppressive agent. Urol Clin North Am 10:263–275. [PubMed] [Google Scholar]
- 3.Tedesco D, Haragsim L. 2012. Cyclosporine: a review. J Transplant 2012:230386. doi: 10.1155/2012/230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harper JI, Kendra JR, Desai S, Staughton RC, Barrett AJ, Hobbs JR. 1984. Dermatological aspects of the use of cyclosporin A for prophylaxis of graft-versus-host disease. Br J Dermatol 110:469–474. doi: 10.1111/j.1365-2133.1984.tb04662.x. [DOI] [PubMed] [Google Scholar]
- 5.Kahan BD, Flechner SM, Lorber MI, Golden D, Conley S, Van Buren CT. 1987. Complications of cyclosporine-prednisone immunosuppression in 402 renal allograft recipients exclusively followed at a single center for from one to five years. Transplantation 43:197–204. doi: 10.1097/00007890-198702000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Paus R, Stenn KS, Link RE. 1989. The induction of anagen hair growth in telogen mouse skin by cyclosporine A administration. Lab Invest 60:365–369. [PubMed] [Google Scholar]
- 7.Kim CD, Lee MH, Sohn KC, Kim JM, Li SJ, Rang MJ, Roh SS, Oh YS, Yoon TJ, Im M, Seo YJ, Lee JH, Park JK. 2008. Induction of synapse associated protein 102 expression in cyclosporin A-stimulated hair growth. Exp Dermatol 17:693–699. doi: 10.1111/j.1600-0625.2007.00694.x. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T, Kamimura A. 2001. Cyclosporin A promotes hair epithelial cell proliferation and modulates protein kinase C expression and translocation in hair epithelial cells. J Investig Dermatol 117:605–611. doi: 10.1046/j.0022-202x.2001.01452.x. [DOI] [PubMed] [Google Scholar]
- 9.Kim SN, Ahn HJ, Lee CW, Lee MH, Kim JH, Kim JI, Kim SJ, Cho HS, Lee HS, Kim HJ. July 2004. Use of 3-position cyclosporine derivatives for hair growth. US patent 6,762,164.
- 10.Kim SN, Ahn HJ, Lee CW, Lee MH, Kim JH, Kim JI, Kim SJ, Cho HS, Lee HS, Kim HJ. August 2004. The use of nonimmunosuppressive [γ-hydroxy-N-methyl-l-leucine4]cyclosporin derivatives for treating hair loss. US patent 20040161399 A1.
- 11.Lee MJ, Kim HB, Yoon YJ, Han K, Kim ES. 2013. Identification of a cyclosporine-specific P450 hydroxylase gene through targeted cytochrome P450 complement (CYPome) disruption in Sebekia benihana. Appl Environ Microbiol 79:2253–2262. doi: 10.1128/AEM.03722-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson DR. 2009. The cytochrome P450 homepage. Hum Genomics 4:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guengerich FP. 2001. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 14.Coon MJ. 2005. Cytochrome P450: nature's most versatile biological catalyst. Annu Rev Pharmacol Toxicol 45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- 15.Pikuleva IA, Waterman MR. 2013. Cytochromes P450: roles in diseases. J Biol Chem 288:17091–17098. doi: 10.1074/jbc.R112.431916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernhardt R. 2006. Cytochromes P450 as versatile biocatalysts. J Biotechnol 124:128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 17.Munro AW, Girvan HM, McLean KJ. 2007. Variations on a (t)heme—novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat Prod Rep 24:585–609. doi: 10.1039/b604190f. [DOI] [PubMed] [Google Scholar]
- 18.Munro AW, Girvan HM, McLean KJ. 2007. Cytochrome P450—redox partner fusion enzymes. Biochim Biophys Acta 1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Roberts GA, Grogan G, Greter A, Flitsch SL, Turner NJ. 2002. Identification of a new class of cytochrome P450 from a Rhodococcus sp. J Bacteriol 184:3898–3908. doi: 10.1128/JB.184.14.3898-3908.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannemann F, Bichet A, Ewen KM, Bernhardt R. 2007. Cytochrome P450 systems—biological variations of electron transport chains. Biochim Biophys Acta 1770:330–344. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 21.Kelly SL, Kelly DE. 2013. Microbial cytochromes P450: biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us? Philos Trans R Soc Lond B Biol Sci 368:20120476. doi: 10.1098/rstb.2012.0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Liu Y, Yan J, Cao S, Bai F, Yang Y, Huang S, Yao L, Anzai Y, Kato F, Podust LM, Sherman DH, Li S. 2014. New reactions and products resulting from alternative interactions between the P450 enzyme and redox partners. J Am Chem Soc 136:3640–3646. doi: 10.1021/ja4130302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Podust LM, Sherman DH. 2007. Engineering and analysis of a self-sufficient biosynthetic cytochrome P450 PikC fused to the RhFRED reductase domain. J Am Chem Soc 129:12940–12941. doi: 10.1021/ja075842d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heckman KL, Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Lu X, Li JJ. 2013. Conversion of fatty aldehydes into alk(a/e)nes by in vitro reconstituted cyanobacterial aldehyde-deformylating oxygenase with the cognate electron transfer system. Biotechnol Biofuels 6:86. doi: 10.1186/1754-6834-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anzai Y, Li S, Chaulagain MR, Kinoshita K, Kato F, Montgomery J, Sherman DH. 2008. Functional analysis of MycCI and MycG, cytochrome P450 enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem Biol 15:950–959. doi: 10.1016/j.chembiol.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Y, Wilson D, Zhao L, Liu H, Sherman DH. 1998. Hydroxylation of macrolactones YC-17 and narbomycin is mediated by the pikC-encoded cytochrome P450 in Streptomyces venezuelae. Chem Biol 5:661–667. doi: 10.1016/S1074-5521(98)90293-9. [DOI] [PubMed] [Google Scholar]
- 28.Omura T, Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem 239:2379–2385. [PubMed] [Google Scholar]
- 29.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez-Martin A, von Buhler CJ, Tieves F, Fernandez S, Ferrero M, Urlacher VB. 2014. Whole-cell biotransformation with recombinant cytochrome P450 for the selective oxidation of Grundmann's ketone. Bioorg Med Chem 22:5586–5592. doi: 10.1016/j.bmc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Hrycay EG, Bandiera SM. 2012. The monooxygenase, peroxidase, and peroxygenase properties of cytochrome P450. Arch Biochem Biophys 522:71–89. doi: 10.1016/j.abb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Ba L, Li P, Zhang H, Duan Y, Lin Z. 2013. Semi-rational engineering of cytochrome P450sca-2 in a hybrid system for enhanced catalytic activity: insights into the important role of electron transfer. Biotechnol Bioeng 110:2815–2825. doi: 10.1002/bit.24960. [DOI] [PubMed] [Google Scholar]
- 33.Ni Y, Su Y, Li H, Zhou J, Sun Z. 2013. Scalable biocatalytic synthesis of optically pure ethyl (R)-2-hydroxy-4-phenylbutyrate using a recombinant E. coli with high catalyst yield. J Biotechnol 168:493–498. doi: 10.1016/j.jbiotec.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 34.Schewe H, Kaup BA, Schrader J. 2008. Improvement of P450BM-3 whole-cell biocatalysis by integrating heterologous cofactor regeneration combining glucose facilitator and dehydrogenase in E. coli. Appl Microbiol Biotechnol 78:55–65. doi: 10.1007/s00253-007-1277-1. [DOI] [PubMed] [Google Scholar]
- 35.Schewe H, Holtmann D, Schrader J. 2009. P450BM-3-catalyzed whole-cell biotransformation of alpha-pinene with recombinant Escherichia coli in an aqueous-organic two-phase system. Appl Microbiol Biotechnol 83:849–857. doi: 10.1007/s00253-009-1917-8. [DOI] [PubMed] [Google Scholar]
- 36.Mouri T, Shimizu T, Kamiya N, Goto M, Ichinose H. 2009. Design of a cytochrome P450BM3 reaction system linked by two-step cofactor regeneration catalyzed by a soluble transhydrogenase and glycerol dehydrogenase. Biotechnol Prog 25:1372–1378. doi: 10.1002/btpr.231. [DOI] [PubMed] [Google Scholar]
- 37.Di Nardo G, Gilardi G. 2012. Optimization of the bacterial cytochrome P450 BM3 system for the production of human drug metabolites. Int J Mol Sci 13:15901–15924. doi: 10.3390/ijms131215901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bagherinejad MR, Korbekandi H, Tavakoli N, Abedi D. 2012. Immobilization of penicillin G acylase using permeabilized Escherichia coli whole cells within chitosan beads. Res Pharm Sci 7:79–85. [PMC free article] [PubMed] [Google Scholar]
- 39.Park NS, Myeong JS, Park HJ, Han K, Kim SN, Kim ES. 2005. Characterization and culture optimization of regiospecific cyclosporin hydroxylation in rare actinomycetes species. J Microbiol Biotechnol 15:188–191. [Google Scholar]
- 40.Xu Z, Jing K, Liu Y, Cen P. 2007. High-level expression of recombinant glucose dehydrogenase and its application in NADPH regeneration. J Ind Microbiol Biotechnol 34:83–90. [DOI] [PubMed] [Google Scholar]
- 41.Vaara M. 1992. Agents that increase the permeability of the outer membrane. Microbiol Rev 56:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng S, Wei D, Song Q, Zhao X. 2006. Immobilization of permeabilized whole cell penicillin G acylase from Alcaligenes faecalis using pore matrix crosslinked with glutaraldehyde. Biotechnol Lett 28:1129–1133. doi: 10.1007/s10529-006-9067-x. [DOI] [PubMed] [Google Scholar]
- 43.He YC, Zhang ZJ, Xu JH, Liu YY. 2010. Biocatalytic synthesis of (R)-(−)-mandelic acid from racemic mandelonitrile by cetyltrimethylammonium bromide-permeabilized cells of Alcaligenes faecalis ECU0401. J Ind Microbiol Biotechnol 37:741–750. doi: 10.1007/s10295-010-0720-y. [DOI] [PubMed] [Google Scholar]
- 44.Gao B, Su E, Lin J, Jiang Z, Ma Y, Wei D. 2009. Development of recombinant Escherichia coli whole-cell biocatalyst expressing a novel alkaline lipase-coding gene from Proteus sp. for biodiesel production. J Biotechnol 139:169–175. doi: 10.1016/j.jbiotec.2008.10.004. [DOI] [PubMed] [Google Scholar]