

Abstract
Lager brewing strains of Saccharomyces pastorianus are natural interspecific hybrids originating from the spontaneous hybridization of Saccharomyces cerevisiae and Saccharomyces eubayanus. Over the past 500 years, S. pastorianus has been domesticated to become one of the most important industrial microorganisms. Production of lager-type beers requires a set of essential phenotypes, including the ability to ferment maltose and maltotriose at low temperature, the production of flavors and aromas, and the ability to flocculate. Understanding of the molecular basis of complex brewing-related phenotypic traits is a prerequisite for rational strain improvement. While genome sequences have been reported, the variability and dynamics of S. pastorianus genomes have not been investigated in detail. Here, using deep sequencing and chromosome copy number analysis, we showed that S. pastorianus strain CBS1483 exhibited extensive aneuploidy. This was confirmed by quantitative PCR and by flow cytometry. As a direct consequence of this aneuploidy, a massive number of sequence variants was identified, leading to at least 1,800 additional protein variants in S. pastorianus CBS1483. Analysis of eight additional S. pastorianus strains revealed that the previously defined group I strains showed comparable karyotypes, while group II strains showed large interstrain karyotypic variability. Comparison of three strains with nearly identical genome sequences revealed substantial chromosome copy number variation, which may contribute to strain-specific phenotypic traits. The observed variability of lager yeast genomes demonstrates that systematic linking of genotype to phenotype requires a three-dimensional genome analysis encompassing physical chromosomal structures, the copy number of individual chromosomes or chromosomal regions, and the allelic variation of copies of individual genes.
INTRODUCTION
Lager brewing yeasts have been inadvertently selected and evolved over many hundreds of years by brewers to survive under the stressful conditions that they encounter during wort fermentation. These stressful conditions include high osmotic and hydrostatic pressures, low temperatures, and high CO2 and high ethanol concentrations (1). More recently, strain selection has focused on specific flavor profiles, which can be used as bar codes for specific beverages and for the yeast strain used to ferment it (2). However, understanding of the genetic and molecular basis for brewing yeast performance is very incomplete. A deeper knowledge and a deeper understanding of the genome content and structure of S. pastorianus strains are essential for further knowledge-based strain improvement programs.
Beer brewing yeasts include ale yeasts, which are taxonomically classified as Saccharomyces cerevisiae (3), and lager yeasts. The latter were long known to be S. carlsbergensis but were recently renamed S. pastorianus (4–6). Over the past few decades, it has become generally accepted that S. pastorianus strains are natural, interspecific hybrids of S. cerevisiae and a non-cerevisiae Saccharomyces species (4, 7–11). Publication of the first complete genome sequence of S. pastorianus Weihenstephan 34/70 (WS34/70) (12) and the subsequent release of three additional S. pastorianus sequences (13, 14) confirmed that S. pastorianus is a hybrid of S. cerevisiae and a species closely related to S. bayanus. This result was further refined by the recent discovery and genome analysis of S. eubayanus, whose genome sequence shows a 99.5% identity with that of the non-cerevisiae part of the S. pastorianus genome (15). Originally isolated from oak trees in Nothofagus (Southern beech) forests and stromata of Cyttaria hariotii (an ascomycetous parasite of Nothofagus spp.) in Patagonia, S. eubayanus has also recently been isolated in North America (16) and Asia (17). Phylogenetic analysis demonstrated that the Tibetan population of the S. eubayanus lineage was more closely related to S. pastorianus than to the type strain CBS12357 (PYCC 6148) from Patagonia (17).
The distribution of S. eubayanus is consistent with an earlier hypothesis that lager yeast strains may have originated from multiple, separate locations (18) and, thus, from distinct hybridization events. Indeed, lager strains of S. pastorianus can be divided into two populations on the basis of their DNA content, as estimated by array-based comparative genome hybridization (array-CGH) (18, 19). The hybrid ancestor of group I, which includes Saaz-type bottom-fermenting yeast, was proposed to be an allodiploid strain originating from the fusion of a haploid ale S. cerevisiae yeast cell with a haploid S. eubayanus cell. This initial model was, however, shown not to be completely correct, as deep sequencing analysis revealed that S. carlsbergensis was basically triploid with a diploid S. eubayanus and a haploid S. cerevisiae genome content (13). Group II strains, which include Frohberg-type bottom-fermenting yeasts, were proposed to have arisen from the fusion of a homozygous diploid S. cerevisiae strain with a haploid S. eubayanus strain (18).
The hybrid genomes of brewing yeast strains are not simple juxtapositions of the two subgenomes. Both group I and II strain genomes harbor numerous specific features, including the partial or complete loss of chromosomes, interchromosomal translocations between S. cerevisiae and/or S. eubayanus chromosomes, and introgression of sequences from one subgenome into the other. Additionally, chromosomal rearrangements, some of which result in copy number changes, are likely to have contributed to the series of events that shaped brewing yeast genomes in response to the selective pressures to which they were exposed in man-made brewing environments (5, 20, 21).
The availability of full genome sequences for both group I (S. carlsbergensis CBS1513) (13, 14) and group II (S. pastorianus WS34/70) (12) strains enabled a closer examination of the difference between these two types. Despite their supposedly different origins, group I and II strains showed breakpoint reuse in two genes (HSP82 and KEM1) (14), suggesting that these recurrent rearrangements in independent lineages may have provided advantageous evolutionary innovations in brewing environments (22). While it was established that group I and II strains harbored different numbers of structurally distinct chromosome types (29 and 36, respectively), recent reports did not unequivocally quantify chromosome counts and aneuploidy. This is illustrated by the fact that two studies identified different chromosome copy numbers and distributions for the same S. pastorianus (syn., S. carlsbergensis) strain, CBS1513 (31 versus 47 chromosomes) (13, 14).
The goal of the present study was to quantitatively analyze the diversity of S. pastorianus genomes by (i) determining and establishing the chromosome copy number in different lager strains of S. pastorianus, (ii) analyzing the chromosome copy number variation in different brewing strains, and (iii) examining the relation between the variation in chromosome copy number and strain phenotype. To this end, we sequenced and assembled de novo the genome of S. pastorianus strain CBS1483, which was then used as a model for setting up the methodology for determining chromosome copy number. In a second step, the genome sequence of CBS1483 was compared to the genome sequences of five newly sequenced lager strain genomes and to those of three already published ones. Finally, we compared two brewing-related performance traits (the diacetyl production profiles and flocculation characteristics) of three karyotypic variants of strain WS34/70, which exhibited differences in chromosome copy numbers.
MATERIALS AND METHODS
Strains.
The S. pastorianus strains used in this study are listed in Table 1. Stock cultures were grown at 20°C in shake flasks on complex YP (1% yeast extract, 2% peptone) medium supplemented with maltose (20 g liter−1). When stationary phase was reached, sterile glycerol was added to 30% (vol/vol), and 2-ml aliquots were stored in sterile vials at −80°C.
TABLE 1.
Strains used in this study
| Strain | Description | Reference or source |
|---|---|---|
| S. pastorianus CBS1483 | Group II brewer's yeast, Heineken's bottom yeast, July 1927 | 18 |
| S. pastorianus A1 | Group II, WS34/70 isolate | 42 |
| S. pastorianus A1+B11 | Group II, WS34/70 isolate | 42 |
| S. pastorianus A2 | Group II, WS34/70 isolate | 42 |
| S. pastorianus Spy1 | Group II, industrial strain descendant of S. pastorianus CBS1483 | |
| S. pastorianus Spy2 | Group II, industrial strain Mexican bottom-fermenting yeast | |
| S. pastorianus CBS1260 | Group II, Frohberg-type bottom-fermenting yeast, March 1937 | 13, 14 |
| S. pastorianus var. monacensis CBS1503 | Group I, S. monacensis type strain, original culture, 1908 | 13, 14 |
| S. pastorianus var. carlsbergensis CBS1513 | Group I, S. carlsbergensis type strain, October 1947 | 13, 14 |
| S. cerevisiae CEN.PK113-7D | MATa SUC2 MAL2-8c | 78, 79 |
| S. cerevisiae CEN.PK112 | MATa/MATα | Euroscarfa |
| S. cerevisiae FRY153 | 3n |
Cultivation and media.
S. pastorianus Weihenstephan 34/70 (Hefezentrum Weihenstephan TU, Munich, Germany, 2005) isolates A1, A1+B11, and A2 and the control strain, S. cerevisiae CEN.PK113-7D, were cultured in defined medium (WMM medium) containing 10 g · liter−1 (NH4)2SO4, 6 g · liter−1 KH2PO4, 0.25 g · liter−1 MgSO4·7H2O. Vitamins, trace elements (with the CaCl2·2H2O concentration reduced to 0.04 mg · liter−1), and the anaerobic growth factors ergosterol (10 mg · liter−1) and Tween 80 (420 mg · liter−1) were added (23). The WMM medium was supplemented with a complex mixture of sugars from corn syrup containing 9.5 g · liter−1 maltotriose, 27 g · liter−1 maltose, 4.5 g · liter−1 glucose, and 0.75 g · liter−1 fructose as consumable sugars.
From the stock cultures, the strains were cultivated in 60-ml septum flasks with a gas outlet containing 50 ml defined medium. The main cultures were performed in 500-ml septum bottle flasks with a gas outlet containing either 100 ml (for the flocculation assay) or 200 ml (for vicinal diketone measurement) of defined medium. Cultures were performed in Infors Multitron standard bioreactors (Infors HT, Bottmingen, Switzerland) set at 160 rpm with a 50-mm throw at 20°C.
Genome sequencing, assembly, and analysis.
Genome sequencing was performed using an Illumina HiSeq2000 sequencer (Illumina, San Diego, CA) at BaseClear (Leiden, The Netherlands). Genomic DNA of the CBS1483 strain was used to obtain four paired libraries with different insert sizes. Two 100-cycle paired-end libraries with insert sizes of 500 and 180 bp were sequenced. For the latter, the pairs of overlapping reads were merged into single longer pseudoreads. Another two 50-cycle mate pair libraries with 3- and 8-kb insert sizes were sequenced. The combined libraries comprised more than 10+8 reads, which represented ∼7 Gb, resulting in coverage of ∼270 times (see Table S1 in the supplemental material).
The genome assembly was performed on the pseudoread library using the gsAssembler (version 2.6) software package, also known as the Newbler software package (454 Life Sciences, Branford, CT), using the default settings. Further, scaffolding was performed on the assembled contigs using the SSPACE (version 2.0) program (24). SSPACE maps the paired 500-bp-insert-size library reads on the assembled contigs using the Bowtie aligner (version 0.12.5) (25) as the mapping tool. On the basis of the paired link information between the different contigs, the orientation and distance between consecutive contigs were determined and merged into scaffolds. In an iterative process, the 3-kb mate pair library was first mapped to the scaffolds formed in the previous round and then the 8-kb mate pair library was mapped to the scaffolds formed in the previous round using SSPACE. During the scaffolding process, gaps were introduced to preserve the distance between two placed contigs. In the next step, the GapFiller tool (version 1) (26) was applied and closed 549 out of 812 gaps, solving almost 11,000 unknown bases.
The scaffolds (see Table S4 in the supplemental material) were annotated by use of the MAKER2 pipeline (27) (see Table S5 in the supplemental material).
The chromosomal copy number variation was estimated by applying the Magnolya program (28). When there was no prior knowledge of the ploidy, the gamma setting was set to none. Magnolya uses a Poisson mixture model to estimate the integer copy number of an assembled contig with a minimum length of 500 bp.
To predict chromosome copy number, Magnolya can use two assembler types, Newbler (454 Life Sciences), which is suitable for long reads (≥100 bp), and ABySS (29), which is suitable for short reads. Both of these assemblers accept only fasta or fastq files in base space format. To be able to process the publically available data for strains CBS1503, CBS1513, and CBS1260 (http://www.ebi.ac.uk/ena/data/view/PRJEB4654), the original Solid4 color space data were converted to fastq. The color space data were first mapped to the CBS1483 assembled scaffolds using Bowtie (version 0.12.8) (25). The mapped reads were extracted from the sequence alignment map and converted into a base space fastq format using SamToFastq.jar from Picard tools (version 1.113; http://picard.sourceforge.net). In a final step, sequence reads were trimmed and filtered on the basis of the quality score obtained using the Trimmomatic trimmer (30). The converted data were suitable for chromosomal copy number determination with Magnolya using ABySS (version 1.3.7) as the assembler with a k-mer of 29.
Determination of heterozygous positions between alleles was performed using the genome analysis toolkit (GATK) Unified Genotyper (31). The pseudoreads and paired-end library were mapped to the assembled contigs using the Burrows-Wheeler algorithm (BWA) (32). The alignment result file (in.bam format) was sorted into categories (from n = 1 to n = 5) on the basis of the contig copy number previously predicted by Magnolya using an in-house script. The five ploidy groups generated were individually analyzed for single nucleotide variations by setting the ploidy and max_alternate_alleles parameters to the corresponding copy number.
Copy number variation analysis. (i) PCR.
To determine the copy number of specific chromosomes or parts of chromosomes in strain CBS1483, quantitative PCR (qPCR) of genomic DNA was employed, using pUG6 DNA (33) as an internal standard. The genomic DNA of each lager brewing strain and the DNA corresponding to plasmid pUG6 previously cut with restriction enzyme HindIII were mixed in a 1:1 molar ratio. The primers (see Table S6 in the supplemental material) were designed to exhibit a similar melting temperature (Tm) ranging from 59.5 to 61.5°C. The efficiency (E) of the qPCR assays for the target genes and the control amplicon was calculated using the formula 10(−1/m) − 1, where m is the slope of the function derived from the threshold cycle (CT)-versus-log dilution plot (0.02 to 200 ng input DNA) of the DNA template. Only primers exhibiting E values ranging from 1.92 to 1.94 were used for the quantification reactions (34). The PCRs were performed using a SYBR green PCR kit (Qiagen GmbH) on a Rotor GeneQ instrument (Qiagen GmbH). The quantification of each gene was based on a minimum of eight independent replicates. The raw data analysis was performed using LinRegPCR (version 11.0) software (35). The relative copy number (CNR) of the target gene was calculated using the formula (Etarget1)ΔCT(target1 − kanMX)/(Etarget2)ΔCT(target2 − kanMX), where kanMX denotes the PCR product of the control ble gene harbored by pUG6 and target1 and target2 denote the PCR products of chromosomal gene targets 1 and 2, respectively, used to determine the chromosome copy number ratio.
(ii) DNA content determination by flow cytometric analysis.
Samples of culture broth (equivalent to circa 107 cells) were taken from mid-exponential-phase shake flask cultures on WMM medium and centrifuged (5 min, 4,700 × g). The pellet was washed once with cold demineralized water (36), vortexed briefly, centrifuged again (5 min, 4,700 × g), and suspended in 800 μl 70% ethanol while vortexing. After addition of another 800 μl 70% ethanol, fixed cells were stored at 4°C until further staining and analysis. Staining of the cells with SYTOX green nucleic acid stain was performed as described in reference 37. Samples were analyzed on a BD Accuri C6 flow cytometer (BD Biosciences, Breda, The Netherlands) equipped with a 488-nm laser. The fluorescence intensity (DNA content) was represented using BD Accuri C6 software (BD Biosciences).
Analysis.
The optical density at 600 nm (OD660) was measured using a Biochrom Libra S60 spectrophotometer (Biochrom, Cambridge, United Kingdom). The sugars and ethanol in the culture supernatants were analyzed using high-pressure liquid chromatography on a chromatograph containing an Aminex HPX-87H ion exchange column (Bio-Rad, Veenendaal, The Netherlands) operating at 60°C with 5 mM H2SO4 as the mobile phase at a flow rate of 0.6 ml · min−1. Vicinal diketones were analyzed using static headspace gas chromatography (GC). Five milliliters of culture supernatant was heated to 65°C for 30 min prior to injection using a CTC Combi Pal headspace autoinjector (CTC Analytics AG, Zwingen, Switzerland). Samples were analyzed using a 7890A Agilent GC (Agilent, Amstelveen, The Netherlands) with an electron capture detector on a CP-Sil 8 CB capillary column (50 m by 530 μm by 1 μm; Agilent, Amstelveen, The Netherlands). The split ratio was 1:1 with a split flow of 8 ml nitrogen per minute. The injector temperature was set at 120°C and an oven temperature profile of 35°C for 3 min followed by an increase of 10°C · min−1 to 95°C was used. The electron capture detector temperature was set at 150°C with a makeup flow of 10 ml nitrogen per minute.
Flocculation assay.
Flocculation ability was analyzed using a modified Helm's test (38, 39). Culture samples were spun down and washed with 2 ml of 50 mM EDTA, followed by washing with 30 ml of cold distilled H2O (dH2O). After washing, the cells were resuspended in at least 25 ml flocculation buffer (50 mM sodium acetate buffer, pH 4.5) to obtain an OD660 of 2.5. Each strain and a water control (1.9 ml each) were distributed in 12 wells of a 2-ml 96-well deep-well plate (Greiner Bio-One, Alphen a/d Rijn, The Netherlands), of which 6 wells contained 100 μl of dH2O and 6 wells contained 100 μl of a 20% CaCl2 stock solution. After mixing, the cells were incubated for 5 min, followed by 45 s of vortexing and 5 min of sedimentation. One hundred microliters was drawn from 1 mm below the meniscus and pipetted in a 96-well microtiter plate (Greiner Bio-One, Alphen a/d Rijn, The Netherlands) containing 100 μl of 50 mM EDTA. The OD660 was measured in an Infinite M200 Pro microplate reader (Tecan Group Ltd., Männedorf, Switzerland) after 10 s of vortexing. The flocculation coefficient (percent) was calculated on the blank-corrected average for the six replicates using the following equation: [1 − (B/A)] × 100, where A is the average OD660 for the cells with CaCl2 and B is the average OD660 for the cells with dH2O.
Nucleotide sequence accession numbers.
The sequence data generated in this study are searchable at NCBI Entrez (http://www.ncbi.nlm.nih.gov/) under BioProject number PRJNA266750 (accession number SRP049726). Table 2 describes the structure of the searchable data.
TABLE 2.
Accession numbers of sequencing data generated and used in this studya
| Accession no. | ||
|---|---|---|
| Biosample | Expt | Run |
| SRS743281 | SRX758142 | SRR1649181 |
| SRX758143 | SRR1649182 | |
| SRX758140 | SRR1649179 | |
| SRX758141 | SRR1649180 | |
| SRS743282 | SRX758144 | SRR1649183 |
| SRX758145 | SRR1649190 | |
| SRX758149 | SRR1649191 | |
| SRS743429 | SRX758305 | SRR1658893 |
| SRS743428 | SRX758304 | SRR1658892 |
| SRS743430 | SRX758306 | SRR1658889 |
The data are searchable at NCBI Entrez (http://www.ncbi.nlm.nih.gov/) under BioProject number PRJNA266750 (accession number SRP049726), and the whole-genome shotgun accession number is JTFI00000000.
RESULTS
Assembly and annotation of Saccharomyces pastorianus CBS1483.
To enable and facilitate the de novo assembly of the genome sequence of S. pastorianus group II strain CBS1483, four different DNA libraries were prepared and sequenced using Illumina technology (see Table S1 in the supplemental material). The first two libraries were 100-cycle paired-end libraries with insert sizes of 180 and 500 bp. The overlapping read pair data for the 150-bp library were merged into single longer pseudoreads of ca. 140 bp. Subsequently, the pseudoreads were used to perform the primary assembly, which yielded 908 contigs of 500 bp or longer (Table 3). In a second assembly step, the 500-bp-insert-size library and the two 50-cycle mate pair libraries with 3- and 8-kb insert sizes, respectively, were iteratively used to further assemble the CBS1483 strain genome sequence. Scaffolding using the SSPACE (version 2.0) program (24) and GapFiller tool (26) resulted in a reduction from 908 contigs to 59 scaffolds (Table 3). The cumulative size of the 41st longest scaffold covered 22.1 Mb (which also included 196 kb of nucleotides introduced during the gap-filling step). The mitochondrial DNA (scaffold size, 41 to 73 kb) was also identified. The remaining 17 scaffolds had an average length of 4.7 kb and an aggregate size of 80 kb. Finally, the sequences of the contigs and scaffolds assembled de novo were aligned to the genome sequences of S. cerevisiae S288C (www.yeastgenome.org/) and S. eubayanus CBS12358 (15).
TABLE 3.
Assembly statistics for the S. pastorianus CBS1483 sequence
| Characteristic | Value |
|---|---|
| Assembly | |
| No. of contigs | 908 |
| Avg contig size (kbp) | 24.2 |
| N50a (kbp) | 52.5 |
| Size of largest contig (kbp) | 315.2 |
| Total sequence length (Mbp) | 21.97 |
| Scaffolding and gap filling | |
| No. of scaffolds of ≥500 bp | 59 |
| Avg scaffold size (kbp) | 378.8 |
| N50 (kbp) | 750.1 |
| Size of largest scaffold (kbp) | 1,464.6 |
| Total sequence length (Mbp) | 22.3b |
| No. of ORFs in the annotation | 10,279 |
Given a set of sequences of varying lengths, the N50 length is defined as the length N for which 50% of all bases in the sequences are included.
Of which 196,391 nucleotides were in 263 gaps.
Since lager brewing strains have been reported to be highly aneuploid (10, 18, 19), chromosome copy number is an essential feature of the overall genome architecture. To quantify the chromosome copy number, we used the Magnolya algorithm, which applies a Poisson mixture model for estimation of the copy number of contigs assembled from sequencing data without mapping the reads to a reference genome (28). Subsequently, by combining the results of the de novo assembly and the chromosome copy number analysis, a genome map that comprised 35 different chromosomal structures and a total count of 68 chromosomes was established (Fig. 1).
FIG 1.

Chromosomal structure and chromosome copy number of the lager brewing strain S. pastorianus CBS1483. The graph represents the ploidy prediction, generated with the Magnolya algorithm (28), of contigs that were de novo assembled by Newbler (454 Life Sciences) and aligned to the reference S. cerevisiae S288C (43) and S. eubayanus CBS12357 (15) genome sequences using NUCMER (MUMmer, version 3.21; www.mummer.sourceforge.net). Blue and red blocks, contributions of the S. cerevisiae (Sc) and S. eubayanus (Seub) subgenomes, respectively, to the S. pastorianus CBS1483 genome organization. The representation of the genomic organization of CBS1483 takes into account the individual chromosome ploidy and the 35 chromosomal structures identified in this study.
Consistent with three previously sequenced S. pastorianus genomes (12–14), a large majority of the translocation events identified in CBS1483 have already been described (see Table S2 in the supplemental material). However, the genome of strain CBS1483 revealed three specific chromosomal structures that could be identified only by integrating assembled scaffolds and chromosome copy number information. First, in contrast to other sequenced brewing strains (12–14), CBS1483 retained neither a full S. cerevisiae-type chromosome III (ScCHRIII) nor a full S. eubayanus-type CHRIII (SeubCHRIII). Instead, it harbored only a chimeric chromosome consisting of the left part of S. eubayanus CHRIII (including the left arm, centromere, and a major part of the right arm) and the distal part of the right arm of S. cerevisiae CHRIII. Such a chimeric CHRIII was also found in S. pastorianus WS34/70, but in that strain it occurred together with a full S. cerevisiae CHRIII (12). Second, the CBS1483 strain harbored a new, nonreciprocal introgression on CHRXII: a ca. 6-kb insertion of S. eubayanus DNA into S. cerevisiae CHRXII (Fig. 1; see also Fig. S1 in the supplemental material). Third, an S. eubayanus CHRI intrachromosomal region of 9.03 kb (including BUD14, ADE1, KIN3, and CDC15) (see Fig. S2 in the supplemental material) was found to be present at a higher copy number than the copy number for the rest of the chromosome, which could indicate another introgression of S. eubayanus DNA into S. cerevisiae CHRI. However, local reassembly of the reads did not confirm such an introgression; instead, the extra copy was found to be flanked by sequences showing high similarity with Ty-delta sites (see Fig. S2 in the supplemental material), which precluded a precise localization of the duplicated region.
Strikingly, the chromosome copy number differed dramatically for the 35 S. pastorianus CBS1483 chromosomal structures. Copy numbers were quantified and ranged from 1 copy, e.g., for S. cerevisiae CHRVI and CHRVII, to a maximum of 5 copies for S. cerevisiae CHRVIII (Table 4). The chromosomal copy number analysis results were consistent with the assembly results. For instance, two types of chromosome VII were identified in the assembly: a full S. cerevisiae CHRVII and a chimeric S. cerevisiae CHRVII-S. eubayanus CHRVII. This was further supported by the Magnolya analysis, which predicted a copy number integer for the central part of S. cerevisiae CHRVII (1 copy) different from that for the right and left ends of the same chromosome (4 copies). Moreover, it predicted a copy number of 3 for S. eubayanus CHRVII. These data establish that S. pastorianus CBS1483 carries 1 copy of a full S. cerevisiae CHRVII and 3 copies of the chimeric version (Fig. 1).
TABLE 4.
Chromosome copy number in nine different S. pastorianus strainsa
| Species and chromosomeb | Size (kb) | Chromosome copy no. |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group II |
Group I |
|||||||||
| CBS1483 (47.5)b | WS34/70 karyotype isolates |
Industrial lager strains |
||||||||
| A1 (50.5) | A1+B11 (56.0) | A2 (53.5) | Spy2 (39.1) | Spy1 (56.9) | CBS1260 (35.7) | CBS1513 (33.6) | CBS1503 (34.1) | |||
| S. cerevisiae | ||||||||||
| CHRI | 179 | 3 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 |
| CHRII | 780 | 3 | 3 | 3 | 3 | 2 | 3 | 2 | 1 | 1 |
| CHRIII | 300 | 0 | 1.5 | 1 | 2 | 0 | 2 | 0 | 0 | 0 |
| CHRIV | 1,464 | 3 | 2 | 3 | 3 | 2 | 3 | 2 | 1 | 1 |
| CHRV | 572 | 3 | 2 | 3 | 2 | 1 | 3 | 0 | 1 | 0 |
| CHRVI | 260 | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 0 | 0 |
| CHRVII | 1,050 | 1 | 3 | 2 | 3 | 1 | 2.75 | 2 | 1 | 0 |
| CHRVIII | 504 | 5 | 3 | 3 | 4 | 2 | 3 | 2 | 1 | 1 |
| CHRIX | 398 | 3 | 4 | 5 | 4 | 3 | 3 | 0 | 1 | 2 |
| CHRX | 750 | 2 | 3 | 3 | 3 | 2 | 1 | 2 | 1 | 1 |
| CHRXI | 639 | 2 | 3 | 3 | 3 | 1 | 2 | 2 | 0 | 1 |
| CHRXII | 983 | 2 | 2 | 3 | 2 | 2 | 3 | 1.5 | 0 | 0 |
| CHRXIII | 918 | 2 | 2 | 3 | 3 | 1 | 3 | 1 | 1 | 1 |
| CHRXIV | 758 | 2.25 | 3 | 3 | 3 | 2 | 2.25 | 2 | 1 | 0 |
| CHRX | 1,032 | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 1 |
| CHRXIV | 914 | 2 | 2 | 3 | 2 | 3 | 3 | 1 | 1 | 0 |
| S. eubayanus and hybridc | ||||||||||
| SeubCHRI | 149 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 1 |
| SeubCHRII to SeubCHRIV | 1,265 | 1 | 2 | 2 | 2 | 1.75 | 2 | 1 | 2 | 2 |
| SeubCHRIII-ScCHRIII | 295 | 4 | 3 | 4 | 3 | 4 | 3 | 2 | 1 | 1 |
| SeubCHRIV and SeubCHRII | 984 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 |
| SeubCHRV | 546 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 3 |
| SeubCHRVI | 253 | 3 | 3 | 2 | 3 | 1 | 3 | 2 | 3 | 3 |
| SeubCHRVII and ScCHRVII | 1,044 | 3 | 2 | 2 | 1 | 2 | 2 | 1 | 2 | 3 |
| SeubCHRVIII to SeubCHRXV | 804 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 2 |
| SeubCHRXI | 393 | 2 | 1 | 1 | 1 | 1 | 2 | 3 | 2 | 1 |
| SeubCHRX | 650 | 1 | 1 | 3 | 1 | 1.25 | 4 | 1 | 2 | 2 |
| SeubCHRXI | 652 | 2 | 2 | 2 | 2 | 2 | 3 | 2.75 | 3 | 2.5 |
| SeubCHRXII | 1,036 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 | 3 |
| SeubCHRXIII | 925 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| SeubCHRXIV | 757 | 2 | 2 | 2 | 2 | 2 | 3 | 1 | 2 | 3 |
| SeubCHRXV and SeubCHRVIII | 748 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 2 | 2 |
| SeubCHRVI | 910 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 3 |
| Total | 68.25 | 70.5 | 77 | 75 | 49.25 | 79 | 55 | 45 | 46.5 | |
The chromosome copy number was quantified by Magnolya.
The values in parentheses are the total genome size (in megabases).
Seub, S. eubayanus; Sc, S. cerevisiae.
Overall, the genome of S. pastorianus CBS1483 was composed of 56% S. cerevisiae chromosomal DNA, 34% S. eubayanus chromosomal DNA, and 10% chimeric S. cerevisiae-S. eubayanus chromosomal DNA distributed over 68 chromosomes. While arithmetically the genome size was nearly tetraploid (4n + 4), the distribution did not match any of the already formulated assumptions regarding lager yeast genome ploidy, which often refers to these yeasts as allopolyploid or as hybrids of a haploid genome and a diploid genome. Our data showed a much more complex chromosomal distribution picturing a composite genome organization (Fig. 1 and Table 4).
Confirmation of chromosome copy numbers in CBS1483 by qPCR and flow cytometry.
To check if the extraordinary, high chromosome copy numbers predicted by the Poisson mixture model were correct, these predictions were experimentally tested. Quantitative PCR was used as an independent method to analyze the representation of chromosomal sequences that were predicted to have a different copy number. To verify the model-based predictions of the CHRVII structures, PCR primers were designed for three different sites on CHRVII: in S. cerevisiae, EMP24 and MEP1 (ScEMP24 and ScMEP1, respectively), and in S. eubayanus, SHY1 (SeubSHY1). These genes were predicted to be present in 4, 1, and 3 copies, respectively (Fig. 2). For an unbiased copy number estimation, genomic DNA of CBS1483 was spiked with plasmid DNA (pUG6 DNA [33]). The relative, pUG6-normalized abundances of ScEMP24 and ScMEP1 (4.05 ± 0.1) and of SeubSHY1 and ScMEP1 (3.2 ± 0.05) (Fig. 2) perfectly matched the model-based chromosome copy numbers. Four additional experimental verifications of predicted copy numbers targeting sequences on CHRI (S. cerevisiae and S. eubayanus ADE1), CHRIII (S. eubayanus-type NFS1 and S. cerevisiae-type ABP1), CHRVIII (S. cerevisiae and S. eubayanus DUR3), and CHRXV (S. cerevisiae and S. eubayanus MDM20) were performed. The copy number ratio of the pUG6-normalized abundances of the ScADE1 and SeubADE1 alleles (4.3 ± 0.6) was consistent with the model prediction. Similarly, the ratio calculated for SeubNFS1 and ScABP1 (1.3 ± 0.5) was compatible with the presence of a unique, chimeric S. cerevisiae CHRIII-S. eubayanus CHRIII structure. The ratio between the DUR3 species alleles (5.6 ± 0.8) corroborated the predicted large (5-fold) copy number variation between S. cerevisiae CHRVIII and S. eubayanus CHRVIII. Finally, the ratio measured for the pUG6-normalized abundances of ScMDM20 and SeubMDM20 (3.4 ± 0.5) was in agreement with the predicted 3:1 distribution in S. cerevisiae and S. eubayanus (Fig. 2).
FIG 2.

Chromosome copy number determination by real-time quantitative PCR. (A) Representation of chromosomes I, III, VII, VIII, and XV and their copy numbers. Blue and red blocks denote the contributions of the S. cerevisiae and S. eubayanus subgenomes, respectively; colored triangles (red and blue) denote the positions of the marker genes used in the analysis. (B) Quantification of the relative copy number ratio of gene pairs located on chromosomes I, III, VII, VIII, and XV. The bar graph represents the normalized copy number ratio of a gene pair. Each gene copy number was normalized relative to that of control DNA spiked in genomic DNA of strain CBS1483. Data are presented as the average and standard deviation for at least eight independent reactions.
Two previous studies lend further support to the reliability of the model-based copy number estimations for strain CBS1483. Using a similar quantitative PCR approach, the CBS1483 ScARO10 and SeubARO10 alleles were shown to have a copy number ratio of 3 (40), matching the model-based prediction of S. cerevisiae CHRIV and S. eubayanus CHRII to CHRIV (Table 4). Furthermore, the observation that a single transformation was sufficient to completely delete the ScHXK1 allele in S. pastorianus CBS1483 (41) is consistent with the model-based prediction that this strain contains only a single copy of S. cerevisiae CHRVI.
In a second approach, the total DNA content of S. pastorianus strain CBS1483 was estimated by comparison with the total DNA content of strains of known ploidy (n for CEN.PK113-7D, 2n for CEN.PK122, 3n for FRY153) using flow cytometry. The fluorescence intensity of the peaks corresponding to the original ploidy of control strains and to its doubling showed a nearly perfect linear correlation (R2 = 0.998; Fig. 3A). On the basis of this correlation, the overall ploidy values of S. pastorianus CBS1483, based on n and 2n peaks characteristic of the G1-M phase of the cell cycle, were 3.8 and 7.5, respectively (Fig. 3B). If a haploid genome size of 12 Mb is assumed, the size of the CBS1483 genome should then be 46 Mb. These estimates were in full agreement with the chromosomal count predicted by the Magnolya algorithm, which estimated this size to be 47.5 (Table 4). Collectively, these data confirmed the reliability of the prediction model and therefore enabled us to extend the use of this method to multiple strains.
FIG 3.
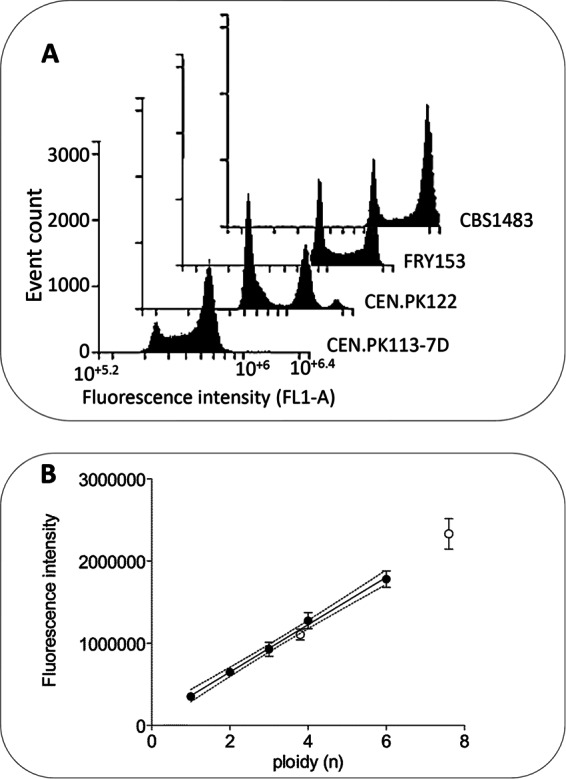
DNA content determination by flow cytometry. (A) DNA content was measured by flow cytometry analysis of Saccharomyces cerevisiae CEN.PK113-7D (n), CEN.PK122 (2n), and FRY153 (3n) and of S. pastorianus CBS1483. The cells were harvested in mid-log phase and stained with SYTOX green nucleic acid stain. The histogram represents the distribution of ungated FL1-A fluorescence. (B) Determination of the ploidy of S. pastorianus CBS1483. To establish a correlation between fluorescence intensity and ploidy, control strains (closed circles) CEN.PK113-7D (n/2n), CEN.PK122 (2n/4n), and FRY153 (3n/6n) were used. The data plotted are the fluorescence intensities of the flow cytometry histogram peaks with the highest cell count. The fluorescence intensity of the CBS1483 histogram peaks (open circles) was compared to a standard linear correlation line based on data for the control strains. Dashed lines, 95% confidence interval of the regression analysis. Fluorescence data indicate the average and mean deviation from duplicate experiments.
Chromosome copy number variation among S. pastorianus strains.
The extraordinary differences in the copy numbers of individual chromosomes in strain CBS1483 raised the question of the extent to which this genetic feature differs among S. pastorianus lager brewing strains. To address this question, five additional S. pastorianus strains were newly sequenced, thereby enabling us to extend our analysis beyond the already sequenced strains S. carlsbergensis (syn., S. pastorianus) CBS1513, S. monacensis CBS1503 (13, 14), and S. pastorianus Weihenstephan 34/70 (12) and CBS1260 (14). Two of the additionally sequenced strains (Spy1 and Spy2) are industrial brewing strains, and the other three are clonal variants of the widely used nonproprietary brewing lager yeast S. pastorianus WS34/70 (www.hefebank-weihenstephan.de/). Karyotyping of WS34/70 cultures indicated that supposedly pure commercial cultures of this strain are, in fact, composed of several karyotypic variants (42). Three Weihenstephan 34/70 isolates with reported stable karyotype differences (A1, A1+B11, and A2) (42) were sequenced and analyzed for chromosome copy number variation, bringing the total number of S. pastorianus strains included in the analysis to nine (seven group II and two group I strains; Table 4).
Two previous studies on S. pastorianus (S. carlsbergensis) strain CBS1513 (DBVPG 6033) reported different total chromosome copy numbers of 31 (14) and 47 (13). To validate and benchmark the use of the Magnolya algorithm for copy number estimations, we used the S. carlsbergensis CBS1513 sequence data generated by Hewitt and coworkers (14). This analysis predicted a total of 45 chromosomes (Table 4). Of 29 structurally different chromosomes identified by Walther and coauthors (13), 28 showed an identical chromosome copy number in our analysis. The main difference between our copy number assessment and that of Walther et al. (13) concerned CHRIII, for which only 1 copy was estimated by Magnolya, while Walther and coworkers reported 3 (13). The resulting copy number difference (45 versus 47 copies) (Table 4) may result from the different methodologies used for chromosome quantification. Alternatively, the difference may be due to the use of an independent genome sequencing data set (14) and thus reflects a chromosome copy number difference between clonal lines derived from the same strain.
A large difference in the total chromosome complement of strains CBS1483 (68 copies) and CBS1513 (45 to 47 copies) was observed. Analysis of seven additional lager brewing strains revealed even more extreme differences. The group I (Saaz) strains CBS1513 and CBS1503 showed similar chromosome copy numbers (45 and 46.5 copies, respectively). Conversely, the group II (Frohberg) strains revealed extremely different chromosome complements, ranging from 49 copies for strain CBS1260 (DBVPG 6257) to 79 copies for industrial strain Spy1. For the latter strain, this would correspond to a total genome size of 57 Mb (Table 4), which is almost 5-fold higher than the genome size of haploid S. cerevisiae strains (43).
Single-cell isolates of WS34/70 strains A1, A1+B11, and A2 showed strikingly different chromosome complements of 70, 77, and 75 copies, respectively. These results are consistent with those of an earlier karyotyping study (42) and indicate that chromosomal copy number can be highly dynamic, even in supposedly pure cultures.
Exploring allelic variation in S. pastorianus CBS1483.
The results presented in the previous paragraphs underline the suggestion that the assembled and already published genome sequences of S. pastorianus strains do not reflect the full variation of the genomic landscape of lager brewing strains. In addition to copy number variation per se, the multiple alleles carried by sister chromosomes may exhibit sequence variations. The extent of heterozygosity in S. pastorianus CBS1483 was estimated by using the GATK Unified Genotyper algorithm (www.broadinstitute.org/gatk) and by taking into account the calculated ploidy of individual chromosomes. This analysis revealed that 13,839 nucleotide positions were covered by more than one base, thus confirming the presence of heterozygous loci (see Table S3 in the supplemental material). These variations affected nucleotides that were located upstream or downstream of open reading frames (ORFs) (43.5%) as well as nucleotides in protein-encoding sequences (56.5%). Of the 7,815 mutations located in ORFs, only 4,024, located in 1,829 ORFs, resulted in an amino acid change (3,872 missense mutations) or in the introduction of a premature stop codon (152 nonsense mutations) (Fig. 4A). At 27 positions, three different nucleotides which could lead to the occurrence of three protein variants were encountered in the sequence data.
FIG 4.

Allelic variation in S. pastorianus CBS1483. (A) Pie chart representing the distribution of the chromosomal positions covered by more than one nucleotide, as identified by GATK (www.broadinstitute.org/gatk). The numbers in parentheses denote the number of positions affected by allelic variation. (B) Bar charts representing the nucleotide frequency at heterozygous positions in the genes ScOAF1 (3n), SeubFLO1 (4n), and ScHPF1 (5n). The position coverage (in percent) was calculated from the ratio of the number of reads covering the position containing one of the nucleotides over the total number of reads covering the evaluated position. The positions tested for allelic variation were covered by a minimum of 110 sequencing reads. Dashed lines, the theoretical distribution for the position coverage for a 2:1 distribution for 3n ploidy (ScOAF1), for a 2:1:1 distribution for a 4n ploidy (SeubFLO1), and for 3:1:1 and 2:2:1 distributions for 5n ploidy (ScHPF1).
To further investigate allelic variation, three open reading frames (ScOAF1, SeubFLO1, and ScHPF1) were more closely inspected. Three alleles of ScOAF1, a gene encoding a regulator involved in the transcriptional control of β-oxidation, were located on the 3 copies of S. cerevisiae CHRI in S. pastorianus CBS1483. These three alleles exhibited 13 different variable positions, 1 of which was situated in the promoter region and 12 of which occurred in the open reading frame. Of the latter 12 single-nucleotide variations (SNVs), 5 led to an amino acid change which could lead to proteins with different properties. However, the available sequence data did not allow the complete reconstruction of the two or three potentially separate alleles (Fig. 4B). A more precise reconstruction of discrete alleles was possible for SeubFLO1 and ScHPF1. The 4 copies of SeubFLO1, which encodes a lectin-like protein involved in flocculation, harbored only two positions with variations, and a change at one of these positions yielded an alteration of the protein sequence. Interestingly, three variants were found for this location and encoded predicted protein sequences with either a threonine, a serine, or an alanine at position 164. These alleles occurred in a 2:1:1 ratio (Fig. 4B). At 5n ploidy, only a single gene, ScHPF1, which encodes a haze-protective mannoprotein involved in an aggregated protein particle size reduction, showed a position within the open reading frame with multiple nucleotide coverage. Out of two SNVs identified in the ScHPF1 gene, only one resulted in an amino acid change, giving rise to two predicted protein sequences represented by a 4:1 allelic ratio (Fig. 4B). These examples demonstrate that the theoretical proteome of strain CBS1483 cannot be simply derived from the 10,172 protein sequences (Table 3; see also Table S4 in the supplemental material) predicted by genome annotation of the consensus sequence. Instead, the in silico CBS1483 proteome should include at least 1,829 additional protein variants.
Linking chromosome copy number variation to phenotype.
To test whether the differences in chromosome copy number correlated with the phenotype, we analyzed the three WS34/70 isolates (A1, A2+B11, A2) in more detail. In addition to the chromosomal rearrangements that enabled their identification, this revealed differences from the published Weihenstephan 34/70 sequence (12), indicating that these strains are not entirely isogenic. While strains A1 and A2 exhibited less than 200 nonsense and missense mutation differences, they carried a common set of 361 SNVs relative to the WS34/70 reference sequence. Out of the three isolates, the sequence of A1+B11 was the closest to the reference sequence, but it still exhibited over 500 nonsense and missense differences from the sequences of the A1 and A2 strains (see Fig. S3 in the supplemental material). Notwithstanding these differences, the sequences of these lager yeast genomes were highly similar and therefore provided an excellent baseline from which to study the impact of chromosome copy number on fermentation performance. The three strains were cultivated in WMM medium, a chemically defined medium that contains a mixture of maltose, maltotriose, glucose, and fructose in a 64:23:11:2 mass ratio. The three strains showed identical specific growth rates (0.15 h−1) and sugar consumption profiles at 20°C (see Fig. S4 in the supplemental material).
Diacetyl, a vicinal diketone, is a butter-tasting off flavor that is produced during fermentation as a by-product of valine metabolism. During lager production, diacetyl concentrations need to be reduced by the yeast cells to concentrations below the sensory threshold. The diacetyl concentration produced by a brewing strain is, therefore, a highly relevant industrial characteristic of S. pastorianus strains. The diacetyl production profiles of the three WS34/70 isolates were biphasic, with a production phase during the first 25 to 30 h of cultivation being followed by a diacetyl consumption phase. Strain A1 showed the highest diacetyl production peak (351 ± 10 μg/liter), a value that was 1.7- and 2.9-fold higher than the values found with strains A2 and A1+B11, respectively (Fig. 5). Sequence analysis did not reveal any differences in the valine biosynthesis genes (ILV2, ILV6, ILV5, ILV3, BAT1, and BAT2) of the three strains. However, the chromosomes harboring these genes did show copy number variation. Strain A1+B11, which presented the lowest diacetyl peak, showed the largest differences. The copy numbers of S. cerevisiae CHRXIV and S. eubayanus CHRXIV, which carry ILV2/YMR108W, encoding the catalytic subunit of α-acetolactate synthase, the enzyme that catalyzes the first step in valine biosynthesis, were identical in the three strains (3 and 2, respectively). However, chromosomes XII and X showed 1 and 2 additional copies, respectively, in strain A1+B11 relative to the numbers in strains A1 and A2. These chromosomes carry ILV5/YMR108W and ILV3/YJR016C, which encode the enzymes that catalyze the second and third steps in the valine biosynthesis pathway, respectively (Fig. 5).
FIG 5.

Correlation between diacetyl production profile and chromosome copy number of valine biosynthesis genes. (A) S. pastorianus strains A1 (closed square), A1+B11 (open circle), and A2 (open square) were grown at 20°C in WMM medium supplemented with a mixture of complex sugars from corn syrup containing 9.5 g · liter−1 maltotriose, 27 g · liter−1 maltose, 4.5 g · liter−1 glucose, and 0.75 g · liter−1 fructose. Diacetyl concentrations were measured using static headspace gas chromatography. Data are presented as the average and mean deviation from duplicate independent replicates. (B) Valine and diacetyl biosynthetic pathways and chromosome copy number of the genes involved in the metabolic routes in strains A1, A1+B11, and A2. The chromosome copy number was predicted using Magnolya (28).
Several strategies to control diacetyl in brewing yeasts are based on increasing the metabolic flux through the pathway from α-acetolactate to valine. Both the incremental reduction of the ILV6 copy number (44) and the increase in the copy number and overexpression of ILV5 in lager yeast (45–47) have previously been shown to lead to reduced diacetyl formation. The chromosome copy number analysis of the three variants would then suggest that in strain A1+B11, the extra copies of ILV5 (from 1 additional copy of CHRXII) and ILV3 (from 2 additional copies of CHRX), genes encoding enzymes that catalyze reactions downstream of the diacetyl precursor α-acetolactate, were sufficient to generate a low-diacetyl-concentration phenotype.
The ability to flocculate is another highly relevant trait of brewing strains (48). After 38 h of cultivation, when sugars were already completely consumed (see Fig. S4 in the supplemental material), strains A1, A1+B11, and A2 were sampled, washed, and analyzed for flocculence in the presence and absence of Ca2+. The three strains exhibited extremely different phenotypes in this assay. The A1 strain expressed a phenotype comparable to that of the nonflocculent laboratory strain CEN.PK113-7D, while strains A2 and A1+B11 showed clear flocculation phenotypes, with A1+B11 being more flocculent than A2 (Fig. 6). The flocculin-encoding genes LgFLO, FLO1, FLO5, FLO9, and FLO10 are responsible for a large part of the flocculation characteristics of lager yeasts (49, 50). In a first step, we examined sequence variations in the flocculin genes of the three WS34/70 variant strains. Whereas no variation was observed in LgFLO, FLO1, FLO5, or FLO10, one difference was detected in the FLO9 gene in the A1 strain that led to the replacement of a histidine residue by a tryptophan residue at position 215. In a second step, we compared the copy number of the various FLO genes in the three strains. Of the three SeubFLO genes (FLO1, FLO5, and FLO10), only FLO1 did not exhibit a copy number matching the chromosomal copy number in all strain variants, as it was not identified in the A1 strain. Similarly, the LgFLO gene from the A1 strain also showed a copy number comparable to the CHRI copy number. In contrast, the LgFLO gene in A1+B11 and A2 revealed 4 and 2 additional copies, respectively, relative to the copy number of CHRI. Although the mutation found in FLO9 and the absence of SeubFLO1 might affect the flocculation properties of the A1 strain, the largest difference between the three strains lay in the LgFLO gene, whose copy number exhibited a strong positive correlation (R2 = 0.972) with the flocculence of the WS34/70 strain isolates (Fig. 6), implying that the LgFLO gene might be the essential determinant of the flocculation phenotype.
FIG 6.

Flocculation and FLO gene copy number in S. pastorianus A1, A1+B11, and A2. (Top) The copy number of the annotated FLO genes was predicted using Magnolya (28). The copy number of the contig harboring the FLO gene was compared to the copy number of the contigs comprising the rest of the chromosome. *, the gene copy number matches the chromosome value. The sequence analysis of the FLO1 and FLO9 genes did not enable an unequivocal assignment of an S. cerevisiae or S. eubayanus allele type; these genes are therefore labeled “unknown.” (Bottom left) Flocculation properties of the A1, A1+B11, and A2 strains. The strains were grown in WMM medium at 20°C for 38 h. Cells were sampled by centrifugation, and flocculence was analyzed with the modified Helm's test (38, 39). The flocculation coefficient was calculated from the blank-corrected average from six replicates. (Bottom right) Correlation between the flocculation coefficient and the copy number of LgFLO1.
DISCUSSION
High-degree aneuploidy in S. pastorianus: a result of domestication?
Changes in chromosome number can result either in euploidy, with multiple complete sets of chromosomes (3n, 4n), or in aneuploidy, with different copy numbers for an individual chromosome(s) (51). Euploidy includes allopolyploidy, which indicates the perfect polyploidy of a hybrid genome that contains subgenomes from different ancestors. In higher eukaryotes, aneuploidy is often related to pathological or developmental anomalies (52, 53). In S. cerevisiae, disomy of individual chromosomes in haploid strains has been implicated in various phenotypes, including defects in cell cycle progression, increased glucose uptake, and increased sensitivity to conditions interfering with protein synthesis and protein folding (54). Different disomic S. cerevisiae mutants also shared a characteristic transcriptional response (55). However, aneuploidy is generally well tolerated in Saccharomyces species (19), and increased ploidy caused by a whole-genome duplication ca. 100 million years ago in an ancestor of Saccharomyces yeasts is seen to be a key factor in shaping their genomes and phenotypes (56). On a shorter time scale, whole-chromosome and segmental aneuploidies are frequently observed in laboratory evolution experiments, in which increased expression of genes carried by duplicated regions confers a selective advantage (57–61).
This study clearly identifies S. pastorianus yeasts to be alloaneuploids in which high-degree aneuploidy affects both subgenomes. In contrast to the other members of the Saccharomyces genera, S. pastorianus has never been found in nature. It represents the ultimate domesticated Saccharomyces species, as it directly arose out of human activity. The conditions auspicious for the hybridization of S. cerevisiae and S. eubayanus were likely fabricated by humans. In wort fermentation, an artificial hybrid of these yeasts already showed superior characteristics inherited from each parent, like the ability to consume maltotriose from S. cerevisiae and fast growth at low temperature from S. eubayanus (62, 63). The diversity of the S. pastorianus genome likely originates from as many different brewing practices (differences in temperature profiles, wort compositions, strain management, etc.) and therefore is a local record of human history.
S. pastorianus: a model for cancer cell biology?
While polyploidy is a common occurrence in nature, high-degree aneuploidy is comparatively rare. In fact, a high-degree aneuploidy like that observed for S. pastorianus has been documented only for tumor cells (64–66). For example, in an MCF7 breast cancer cell line, 17 of 22 autosomes were present at 3 or 4 copies (64). Hitherto, haploid S. cerevisiae strains with defined disomies have been used as models to study the implications of aneuploidy on pathogenesis in humans (54). The present study indicates that S. pastorianus, with its extensive, dynamic aneuploidy, may present a better model with which to unravel the molecular mechanisms that underlie the development and progression of aneuploidy in cancer cell development.
Different chromosome complements in S. pastorianus: phenotypic and taxonomic implications.
The S. pastorianus strains analyzed in this study exhibited a remarkably wide range of genome sizes, with total chromosome numbers ranging from 45 to 79 (Table 4). The significant differences in the chromosomal content of three clonal isolates of a single, commonly used strain (S. pastorianus Weihenstephan 34/70) indicated that genome copy number in brewing yeast strains is highly dynamic.
In several higher eukaryotes, copy number variation has been associated with phenotypic traits and species separation (67–69). This study identified clear correlations between chromosome copy number in S. pastorianus and two brewing-related traits: diacetyl production and flocculence (Fig. 5 and 6). Three nearly isogenic karyotypic variants of S. pastorianus Weihenstephan 34/70 revealed different diacetyl production profiles, although no differences in the DNA sequence of genes involved in the biosynthesis of this important off flavor were found. Instead, the diacetyl production profiles directly correlated with the copy numbers of structural genes involved in the valine/diacetyl biosynthetic pathway (Fig. 5). The biological relevance of this correlation is supported by previous targeted genetic modification studies, in which increased copy numbers of genes involved in this pathway caused altered diacetyl production profiles (44, 45, 47).
The results of the different chromosome complements of the lager yeast strains analyzed in this study were not limited to subtle quantitative changes in chromosome or gene copy number. A particularly pronounced difference between group I and group II strains concerned the complete absence of S. cerevisiae chromosomes III, VI, and XII in group I strains. As a consequence, S. pastorianus group I and II genomes differed by at least 852 genes. One clear impact of this difference is that group I strains exclusively rely on S. eubayanus ribosomal DNA (rDNA), whereas in group II the rRNA is also transcribed from S. cerevisiae rDNA. The spectacular differences in gene content among S. pastorianus strains is in marked contrast to the high degree of genome conservation observed for S. cerevisiae (70, 71). Recently, on the basis of the genomes of the reference lager yeast strains Weihenstephan 34/70 (12) and CBS1513 (13), a change in classification of lager yeasts was proposed. Group I (Saaz) yeasts, originally classified as S. carlsbergensis, were renamed S. pastorianus and thereby assigned to the same species as group II (Frohberg) strains (6). The highly distinct genome content of the Weihenstephan 34/70 and CBS1513 strains might support a reinstatement of different species names for group I and group II S. pastorianus strains, as was recently proposed (72). Such a step would also be consistent with the clear-cut physiological characteristic differences exhibited by Saaz and Frohberg strains (73).
Reconstructing evolution and domestication of S. pastorianus.
The discovery of S. eubayanus, whose genome exhibits over 99.5% identity with the non-S. cerevisiae-type subgenome of lager yeast strains (15–17), represented a scientific breakthrough in understanding the evolutionary history of lager brewing yeasts. Indeed, it recently enabled the recreation, in the laboratory, of the hybridization event between S. cerevisiae and S. eubayanus, which combined the properties of the two parents that are advantageous for brewing (62, 63). Culture collections harbor a vast number of S. pastorianus strains with different phenotypic characteristics (2, 73, 74) and very likely, on the basis of the findings of this study, large genomic variations. This diversity suggests that their genome plasticity enabled these strains to evolve to adapt to a wide range of environmental conditions and/or to be selected for different product properties. The availability of synthetic hybrids of S. cerevisiae and S. eubayanus provides unique opportunities to assess the impact of culture conditions on the genome content and dynamics (22, 75) and thereby reconstruct the evolutionary trajectory of S. pastorianus strains and the role of aneuploidy in their evolution. Similar to a recent study on the laboratory evolution of plant isolates of the lactic acid bacterium Lactococcus lactis in milk (76), such analyses are likely to increase our understanding of how domestication has shaped the genomes of brewing yeasts, which are among the most important industrial microorganisms on Earth.
Conclusion: three-dimensional genomic diversity in S. pastorianus.
The present analysis of the genomes of different S. pastorianus strains provides new insights into the genetic changes that, after the initial hybridization of S. cerevisiae and S. eubayanus, led to current brewing strains. Extensive genome reorganization, involving changes in the copy numbers of genes and chromosomes, has been followed by the generation of allelic diversity and the differential expression of paralogs (77). Together, these processes have led to a remarkable genomic diversity of lager brewing strains, which has important implications for further genetic analysis of brewing-related traits and for strain optimization. In addition to mutations in the original S. cerevisiae and S. eubayanus subgenomes, large variations in chromosome copy number and allelic variation of the thus multiplied genes were observed. These three dimensions, primary sequence, copy number variation, and allelic variation of gene copies within individual strains, should be taken into account in future studies aimed at understanding the domestication of brewing yeasts and their further optimization for beer fermentation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marit Hebly and Pascale Daran-Lapujade for technical assistance and Frank Rosenzweig (University of Montana, Missoula, MT) for kindly providing us the polyploid standard strain FRY153.
D.D.R., J.T.P., J.M.G., and J.-M.D. conceived of and designed the experiments. I.B., J.F.N., E.R., M.A.H.L., and F.K. performed the experiments. M.V.D.B., J.F.N., D.D.R., and J.-M.D. analyzed the data. M.V.D.B., I.B., J.T.P., D.D.R., and J.-M.D. wrote the paper.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01263-15.
REFERENCES
- 1.Gibson BR, Lawrence SJ, Leclaire JP, Powell CD, Smart KA. 2007. Yeast responses to stresses associated with industrial brewery handling. FEMS Microbiol Rev 31:535–569. doi: 10.1111/j.1574-6976.2007.00076.x. [DOI] [PubMed] [Google Scholar]
- 2.Steensels J, Meersman E, Snoek T, Saels V, Verstrepen KJ. 2014. Large-scale selection and breeding to generate industrial yeasts with superior aroma production. Appl Environ Microbiol 80:6965–6975. doi: 10.1128/AEM.02235-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaughan-Martini A, Martini A. 1998. Saccharomyces Myen ex Rees, p 358–371. In Kurtzman CP, Fell JW (ed), The yeasts: a taxonomic study. Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- 4.Rainieri S, Kodama Y, Kaneko Y, Mikata K, Nakao Y, Ashikari T. 2006. Pure and mixed genetic lines of Saccharomyces bayanus and Saccharomyces pastorianus and their contribution to the lager brewing strain genome. Appl Environ Microbiol 72:3968–3974. doi: 10.1128/AEM.02769-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bond U. 2009. Chapter 6: the genomes of lager yeasts. Adv Appl Microbiol 69:159–182. doi: 10.1016/S0065-2164(09)69006-7. [DOI] [PubMed] [Google Scholar]
- 6.Martini AV, Martini A. 1987. Three newly delimited species of Saccharomyces sensu stricto. Antonie van Leeuwenhoek 53:77–84. doi: 10.1007/BF00419503. [DOI] [PubMed] [Google Scholar]
- 7.Vaughan-Martini A, Kurtzman CP. 1985. Deoxyribonucleic acid relatedness among species of the genus Saccharomyces sensu stricto. Int J Syst Evol Microbiol 35:508–511. [Google Scholar]
- 8.Tamai Y, Momma T, Yoshimoto H, Kaneko Y. 1998. Co-existence of two types of chromosome in the bottom fermenting yeast, Saccharomyces pastorianus. Yeast 14:923–933. [DOI] [PubMed] [Google Scholar]
- 9.Yamagishi H, Ogata T. 1999. Chromosomal structures of bottom fermenting yeasts. Syst Appl Microbiol 22:341–353. doi: 10.1016/S0723-2020(99)80041-1. [DOI] [PubMed] [Google Scholar]
- 10.Casaregola S, Nguyen HV, Lapathitis G, Kotyk A, Gaillardin C. 2001. Analysis of the constitution of the beer yeast genome by PCR, sequencing and subtelomeric sequence hybridization. Int J Syst Evol Microbiol 51:1607–1618. [DOI] [PubMed] [Google Scholar]
- 11.Caesar R, Palmfeldt J, Gustafsson JS, Pettersson E, Hashemi SH, Blomberg A. 2007. Comparative proteomics of industrial lager yeast reveals differential expression of the cerevisiae and non-cerevisiae parts of their genomes. Proteomics 7:4135–4147. doi: 10.1002/pmic.200601020. [DOI] [PubMed] [Google Scholar]
- 12.Nakao Y, Kanamori T, Itoh T, Kodama Y, Rainieri S, Nakamura N, Shimonaga T, Hattori M, Ashikari T. 2009. Genome sequence of the lager brewing yeast, an interspecies hybrid. DNA Res 16:115–129. doi: 10.1093/dnares/dsp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walther A, Hesselbart A, Wendland J. 2014. Genome sequence of Saccharomyces carlsbergensis, the world's first pure culture lager yeast. G3 (Bethesda) 27:783–793. doi: 10.1534/g3.113.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hewitt SK, Donaldson IJ, Lovell SC, Delneri D. 2014. Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS One 9:e92203. doi: 10.1371/journal.pone.0092203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libkind D, Hittinger CT, Valerio E, Goncalves C, Dover J, Johnston M, Goncalves P, Sampaio JP. 2011. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc Natl Acad Sci U S A 108:14539–14544. doi: 10.1073/pnas.1105430108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peris D, Sylvester K, Libkind D, Goncalves P, Sampaio JP, Alexander WG, Hittinger CT. 2014. Population structure and reticulate evolution of Saccharomyces eubayanus and its lager-brewing hybrids. Mol Ecol 23:2031–2045. doi: 10.1111/mec.12702. [DOI] [PubMed] [Google Scholar]
- 17.Bing J, Han PJ, Liu WQ, Wang Q M, Bai FY. 2014. Evidence for a Far East Asian origin of lager beer yeast. Curr Biol 24:R380–R381. doi: 10.1016/j.cub.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 18.Dunn B, Sherlock G. 2008. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res 18:1610–1623. doi: 10.1101/gr.076075.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bond U, Neal C, Donnelly D, James TC. 2004. Aneuploidy and copy number breakpoints in the genome of lager yeasts mapped by microarray hybridisation. Curr Genet 45:360–370. doi: 10.1007/s00294-004-0504-x. [DOI] [PubMed] [Google Scholar]
- 20.Fischer G, James SA, Roberts IN, Oliver SG, Louis EJ. 2000. Chromosomal evolution in Saccharomyces. Nature 405:451–454. doi: 10.1038/35013058. [DOI] [PubMed] [Google Scholar]
- 21.Usher J, Bond U. 2009. Recombination between homoeologous chromosomes of lager yeasts leads to loss of function of the hybrid GPH1 gene. Appl Environ Microbiol 75:4573–4579. doi: 10.1128/AEM.00351-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn B, Paulish T, Stanbery A, Piotrowski J, Koniges G, Kroll E, Louis EJ, Liti G, Sherlock G, Rosenzweig F. 2013. Recurrent rearrangement during adaptive evolution in an interspecific yeast hybrid suggests a model for rapid introgression. PLoS Genet 9:e1003366. doi: 10.1371/journal.pgen.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verduyn C, Postma E, Scheffers WA, van Dijken JP. 1992. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- 24.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding preassembled contigs using SSPACE. Bioinformatics 27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol 13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt C, Yandell M. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nijkamp JF, van den Broek MA, Geertman JM, Reinders MJ, Daran JM, de Ridder D. 2012. De novo detection of copy number variation by co-assembly. Bioinformatics 28:3195–3202. doi: 10.1093/bioinformatics/bts601. [DOI] [PubMed] [Google Scholar]
- 29.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramakers C, Ruijter JM, Deprez RH, Moorman AF. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66. doi: 10.1016/S0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 35.Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porro D, Brambilla L, Alberghina L. 2003. Glucose metabolism and cell size in continuous cultures of Saccharomyces cerevisiae. FEMS Microbiol Lett 229:165–171. doi: 10.1016/S0378-1097(03)00815-2. [DOI] [PubMed] [Google Scholar]
- 37.Haase SB, Reed SI. 2002. Improved flow cytometric analysis of the budding yeast cell cycle. Cell Cycle 1:132–136. [PubMed] [Google Scholar]
- 38.Helm E, Nohr B, Thorne R. 2014. The measurement of yeast flocculence and its significance in brewing. Wallerstein Lab Commun 16:315–325. [Google Scholar]
- 39.Bendiak D, Van der Aar P, Barbero F, Benzing P, Berndt R, Carrick K, Dull C, Dunn S, Eto M, Gonzalez M, Hayashi N, Lawrence D, Miller J, Pugh T, Rashel L, Rossmore K, Smart K, Sobczak J, Speers A, Casey G. 1996. Yeast flocculation by absorbance. J Am Soc Brew Chem 54:245–248. [Google Scholar]
- 40.Bolat I, Romagnoli G, Zhu F, Pronk JT, Daran JM. 2013. Functional analysis and transcriptional regulation of two orthologs of ARO10, encoding broad-substrate-specificity 2-oxo-acid decarboxylases, in the brewing yeast Saccharomyces pastorianus CBS1483. FEMS Yeast Res 13:505–517. doi: 10.1111/1567-1364.12051. [DOI] [PubMed] [Google Scholar]
- 41.Solis-Escalante D, Kuijpers NG, Bongaerts N, Bolat I, Bosman L, Pronk JT, Daran JM, Daran-Lapujade P. 2013. amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res 13:126–139. doi: 10.1111/1567-1364.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolat I, Walsh MC, Turtoi M. 2008. Isolation and characterization of two new lager yeast strains from the WS34/70 population. Roum Biotechnol Lett 6:62–73. [Google Scholar]
- 43.Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. 1996. Life with 6000 genes. Science 274:563–567. [DOI] [PubMed] [Google Scholar]
- 44.Duong CT, Strack L, Futschik M, Katou Y, Nakao Y, Fujimura T, Shirahige K, Kodama Y, Nevoigt E. 2011. Identification of Sc-type ILV6 as a target to reduce diacetyl formation in lager brewers' yeast. Metab Eng 13:638–647. doi: 10.1016/j.ymben.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Gjermansen C, Nilsson-Tillgren T, Petersen JG, Kielland-Brandt MC, Sigsgaard P, Holmberg S. 1988. Towards diacetyl-less brewers' yeast. Influence of ilv2 and ilv5 mutations. J Basic Microbiol 28:175–183. [DOI] [PubMed] [Google Scholar]
- 46.Kusunoki K, Ogata T. 2012. Construction of self-cloning bottom-fermenting yeast with low vicinal diketone production by the homo-integration of ILV5. Yeast 29:435–442. doi: 10.1002/yea.2922. [DOI] [PubMed] [Google Scholar]
- 47.Mithieux SM, Weiss AS. 1995. Tandem integration of multiple ILV5 copies and elevated transcription in polyploid yeast. Yeast 11:311–316. doi: 10.1002/yea.320110403. [DOI] [PubMed] [Google Scholar]
- 48.Bauer FF, Govender P, Bester MC. 2010. Yeast flocculation and its biotechnological relevance. Appl Microbiol Biotechnol 88:31–39. doi: 10.1007/s00253-010-2783-0. [DOI] [PubMed] [Google Scholar]
- 49.Verstrepen KJ, Klis FM. 2006. Flocculation, adhesion and biofilm formation in yeasts. Mol Microbiol 60:5–15. doi: 10.1111/j.1365-2958.2006.05072.x. [DOI] [PubMed] [Google Scholar]
- 50.Van Mulders SE, Ghequire M, Daenen L, Verbelen PJ, Verstrepen KJ, Delvaux FR. 2010. Flocculation gene variability in industrial brewer's yeast strains. Appl Microbiol Biotechnol 88:1321–1331. doi: 10.1007/s00253-010-2843-5. [DOI] [PubMed] [Google Scholar]
- 51.Storchova Z. 2014. Ploidy changes and genome stability in yeast. Yeast 31:421–430. doi: 10.1002/yea.3037. [DOI] [PubMed] [Google Scholar]
- 52.Potter H. 1991. Review and hypothesis: Alzheimer disease and Down syndrome—chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet 48:1192–1200. [PMC free article] [PubMed] [Google Scholar]
- 53.Torres EM, Williams BR, Tang YC, Amon A. 2010. Thoughts on aneuploidy. Cold Spring Harbor Symp Quant Biol 75:445–451. doi: 10.1101/sqb.2010.75.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 55.Sheltzer JM, Torres EM, Dunham MJ, Amon A. 2012. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci U S A 109:12644–12649. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolfe KH, Shields DC. 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- 57.Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. 2009. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5:e1000705. doi: 10.1371/journal.pgen.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Kok S, Nijkamp JF, Oud B, Roque FC, de Ridder D, Daran JM, Pronk JT, van Maris AJ. 18 January 2012. Laboratory evolution of new lactate transporter genes in a jen1Δ mutant of Saccharomyces cerevisiae and their identification as ADY2 alleles by whole-genome resequencing and transcriptome analysis. FEMS Yeast Res doi: 10.1111/j.1567-1364.2012.00787.x.. [DOI] [PubMed] [Google Scholar]
- 60.Oud B, van Maris AJ, Daran JM, Pronk JT. 2012. Genome-wide analytical approaches for reverse metabolic engineering of industrially relevant phenotypes in yeast. FEMS Yeast Res 12:183–196. doi: 10.1111/j.1567-1364.2011.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oud B, Guadalupe-Medina V, Nijkamp JF, de Ridder D, Pronk JT, van Maris AJ, Daran JM. 2013. Genome duplication and mutations in ACE2 cause multicellular, fast-sedimenting phenotypes in evolved Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 110:E4223–E4231. doi: 10.1073/pnas.1305949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hebly M, Brickwedde A, Bolat I, Driessen MR, de Hulster EA, van den Broek M, Pronk JT, Geertman JM, Daran JM, Daran-Lapujade P. 2015. S. cerevisiae × S. eubayanus interspecific hybrid, the best of both worlds and beyond. FEMS Yeast Res 15:fov005. doi: 10.1093/femsyr/fov005. [DOI] [PubMed] [Google Scholar]
- 63.Krogerus K, Magalhaes F, Vidgren V, Gibson B. 2015. New lager yeast strains generated by interspecific hybridization. J Ind Microbiol Biotechnol 42:769–778. doi: 10.1007/s10295-015-1597-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davidson JM, Gorringe KL, Chin SF, Orsetti B, Besret C, Courtay-Cahen C, Roberts I, Theillet C, Caldas C, Edwards PA. 2000. Molecular cytogenetic analysis of breast cancer cell lines. Br J Cancer 83:1309–1317. doi: 10.1054/bjoc.2000.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grigorova M, Staines JM, Ozdag H, Caldas C, Edwards PA. 2004. Possible causes of chromosome instability: comparison of chromosomal abnormalities in cancer cell lines with mutations in BRCA1, BRCA2, CHK2 and BUB1. Cytogenet Genome Res 104:333–340. doi: 10.1159/000077512. [DOI] [PubMed] [Google Scholar]
- 66.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 67.Hu G, Wang J, Choi J, Jung WH, Liu I, Litvintseva AP, Bicanic T, Aurora R, Mitchell TG, Perfect JR, Kronstad JW. 2011. Variation in chromosome copy number influences the virulence of Cryptococcus neoformans and occurs in isolates from AIDS patients. BMC Genomics 12:526. doi: 10.1186/1471-2164-12-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bickhart DM, Hou Y, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, Song J, Schnabel RD, Ventura M, Taylor JF, Garcia JF, Van Tassell CP, Sonstegard TS, Eichler EE, Liu GE. 2012. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res 22:778–790. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doan R, Cohen N, Harrington J, Veazey K, Juras R, Cothran G, McCue ME, Skow L, Dindot SV. 2012. Identification of copy number variants in horses. Genome Res 22:899–907. doi: 10.1101/gr.128991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liti G, Barton DB, Louis EJ. 2006. Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174:839–850. doi: 10.1534/genetics.106.062166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, Tsai IJ, Bergman CM, Bensasson D, O'Kelly MJ, A van Oudenaarden A, Barton DB, Bailes E, Nguyen AN, Jones M, Quail MA, Goodhead I, Sims S, Smith F, Blomberg A, Durbin R, Louis EJ. 2009. Population genomics of domestic and wild yeasts. Nature 458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wendland J. 2014. Lager yeast comes of age. Eukaryot Cell 13:1256–1265. doi: 10.1128/EC.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gibson BR, Storgårds E, Krogerus K, Vidgren V. 2013. Comparative physiology and fermentation performance of Saaz and Frohberg lager yeast strains and the parental species Saccharomyces eubayanus. Yeast 30:255–266. doi: 10.1002/yea.2960. [DOI] [PubMed] [Google Scholar]
- 74.Dunn B, Levine RP, Sherlock G. 2005. Microarray karyotyping of commercial wine yeast strains reveals shared, as well as unique, genomic signatures. BMC Genomics 6:53. doi: 10.1186/1471-2164-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Piotrowski JS, Nagarajan S, Kroll E, Stanbery A, Chiotti KE, Kruckeberg AL, Dunn B, Sherlock G, Rosenzweig F. 2012. Different selective pressures lead to different genomic outcomes as newly-formed hybrid yeasts evolve. BMC Evol Biol 12:46. doi: 10.1186/1471-2148-12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bachmann H, Starrenburg MJ, Molenaar D, Kleerebezem M, van Hylckama Vlieg JE. 2012. Microbial domestication signatures of Lactococcus lactis can be reproduced by experimental evolution. Genome Res 22:115–124. doi: 10.1101/gr.121285.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Querol A, Bond U. 2009. The complex and dynamic genomes of industrial yeasts. FEMS Microbiol Lett 293:1–10. doi: 10.1111/j.1574-6968.2008.01480.x. [DOI] [PubMed] [Google Scholar]
- 78.Nijkamp JF, van den Broek MA, Datema E, de Kok S, Bosman L, Luttik MA, Daran-Lapujade P, Vongsangnak W, Nielsen J, Heijne WHM, Klaassen P, Paddon CJ, Platt D, Kotter P, van Ham RC, Reinders MJ, Pronk JT, de Ridder D, Daran JM. 2012. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 11:36. doi: 10.1186/1475-2859-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Entian KD, Kötter P. 2007. Yeast genetic strain and plasmid collections. Method Microbiol 36:629–666. doi: 10.1016/S0580-9517(06)36025-4. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.