

Abstract
The objective of this study was to evaluate the progression of the uterine microbiota from calving until establishment of metritis. Uterine swabs (n = 72) collected at 0, 2, and 6 ± 2 days postpartum (dpp) from 12 metritic and 12 healthy cows were used for metagenomic sequencing of the 16S rRNA gene on the Illumina MiSeq platform. A heat map showed that uterine microbiota was established at calving. The microbiota changed rapidly from 0 to 6 ± 2 dpp, with a decrease in the abundance of Proteobacteria and an increase in the abundance of Bacteroidetes and Fusobacteria, which were dominant in metritic cows. Uterine microbiota composition was shared; however, metritic and healthy cows could be discriminated using relative abundance of bacterial genera at 0, 2, and 6 ± 2 dpp. Bacteroides was the main genus associated with metritis because it was the only genus that showed significantly greater abundance in cows with metritis. As the abundance of Bacteroides organisms increased, the uterine discharge score, a measure of uterine health, worsened. Fusobacterium was also an important genus associated with metritis because Fusobacterium abundance increased as Bacteroides abundance increased and the uterine discharge score worsened as the abundance increased. The correlation with uterine discharge score and the correlation with Bacteroides or Fusobacterium showed that other bacteria, such as Helcoccocus, Filifactor, and Porphyromonas, were also associated with metritis. There were also bacteria associated with uterine health, such as “Candidatus Blochmannia,” Escherichia, Sneathia, and Pedobacter.
INTRODUCTION
Metritis is a huge concern for the dairy industry worldwide because it is highly prevalent (25 to 40%) and negatively affects the productivity, survival, and welfare of dairy cows (1). Diverse bacteria, including anaerobes and facultative anaerobes, were observed in the uteri of dairy cows within the first 2 weeks postpartum, but they were naturally cleared out within 60 days postpartum (dpp) (1). Culture-based studies observed that Escherichia coli, Trueperella pyogenes, Fusobacterium necrophorum, and Bacteroides spp. (e.g., Prevotella melaninogenica, formerly Bacteroides melaninogenicus) were commonly associated with endometritis or pyometra (1–3).
Although culture-based studies have laid out the foundation of our understanding of the uterine microbiota, previous studies might have underestimated the microbial complexity of the intrauterine environment of cows postpartum, given that less than 1% of the microorganisms in many environments are readily cultured under standard laboratory conditions (4). In recent years, culture-independent techniques such as clone library sequencing (5, 6) and pyrosequencing (7, 8) have been used to characterize the uterine microbiota of cows with metritis (5–7) and endometritis (7, 8). Sequencing using the Illumina platform allows for deeper sequencing than has previously been feasible even with pyrosequencing (9). Indeed, evaluating the rarefaction curves from previous 16S rRNA sequencing studies (5–7), we can state that a larger number of sequences would be required to reflect the complete diversity of the microbiota of several samples.
Therefore, although the uterine microbiota of metritic and healthy cows have previously been compared, use of a more powerful sequencing platform may allow us to have more complete coverage and a better understanding of the uterine microbiota. Furthermore, no one has evaluated the uterine microbiota at the time of calving. Our hypothesis was that uterine microbiota would have a different progression in healthy cows than in cows that develop metritis. Our objective was to evaluate the progression of the uterine microbiota composition from calving until establishment of metritis using the Illumina MiSeq platform to sequence the 16S rRNA gene.
MATERIALS AND METHODS
Animals.
All animal procedures were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC protocol no. 201207405). Holstein cows from a commercial dairy in central Florida that milks 5,000 cows were used in this study.
Sampling.
Uterine swab samples were collected from 92 cows at 0 (within 60 min of calving; mean time, 20 min; standard deviation, 14 min), 2, 4, 6, and 8 dpp as previously described (7). Swab samples were collected in March and April 2013. Briefly, cows were restrained and the perineum area was disinfected with 70% ethanol. Then, a sterile swab (McCullough double-guarded uterine culture swab; Jorgensen Labs Inc., Loveland, CO) covered by a sterile pipette (inside a plastic sheath) was introduced into the cranial vagina. To avoid vaginal contamination of the swab, the plastic-sheath-covered pipette was directed into the cervix; inside the cervix, the plastic sheath (first layer of protection) was ruptured and the pipette was then manipulated through the cervix into the uterus. Once inside the uterus, the swab was advanced through the sealed plastic pipette (second layer of protection), exposing the sterile cotton swab to uterine secretion. The swab was pulled inside the pipette and removed while the pipette was maintained inside the uterus to avoid contamination by vaginal fluid. Swabs were transferred to a 15-ml conical sterile polypropylene centrifuge tube, placed on ice, transferred to the laboratory within 4 h, and then frozen at −80°C. Uterine discharge was scored at 4, 6, and 8 (6 ± 2) dpp using a 5-point scale as previously described (10): 1, not fetid normal lochia, viscous, clear, red, or brown; 2, cloudy mucoid discharge with flecks of pus; 3, not fetid mucopurulent discharge with <50% pus; 4, not fetid mucopurulent discharge with ≥50% pus; 5, fetid red-brownish, watery discharge. Cows with a discharge score of ≤4 were classified as healthy, and cows with a score of 5 and a fever (≥39.5°C) were classified as metritic, as previously described (11). For metagenomic sequencing, 72 uterine swab samples collected at 0, 2, and 6 ± 2 dpp from 12 cows that developed metritis and 12 randomly selected healthy cows were used. Samples from healthy cows matched the day a cow was diagnosed with metritis.
Metagenomic DNA extraction.
Frozen swabs were soaked in 1 ml of phosphate-buffered saline (PBS) and incubated on ice for 2 h to release uterine bacteria from swabs. The PBS suspension was used to isolate bacterial genomic DNA (gDNA) using a QIAamp DNA minikit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol, with a minor modification: before AL buffer was added, samples were incubated with 500 μg of lysozyme for 1 h at 37°C to maximize bacterial DNA extraction. The purity and concentration of gDNA were measured by a spectrophotometer (Nanodrop 1000; Thermo Scientific, Waltham, MA, USA). Genomic DNA was isolated in March 2014. All samples had an A260-to-A280 absorbance ratio between 1.7 and 2.0 (average of 1.82).
PCR amplification of the V4 hypervariable region of bacterial 16S rRNA genes.
Sequencing of 16S amplicons was performed as previously described (12). The DNA template was tagged with 12-bp error-correcting Golay barcodes, and the V4 hypervariable region of the bacterial 16S rRNA gene was amplified using 10 μM primer 515F and 806R, 1× GoTaq Green master mix (Promega, Madison, WI, USA), 1 mM MgCl2, and the 5′-barcoded amplicons in triplicate. PCR was carried out as follows: an initial denaturing step at 94°C for 3 min, followed by 35 cycles of 94°C for 45 s, 50°C for 1 min, and 72°C for 90 s, and a final elongation step at 72°C for 10 min. Replicate amplicons were pooled and purified with a QIAquick PCR purification kit (Qiagen). The purified amplicons were quantified and standardized to the same concentration using a Qubit 3.0 fluorometer (Thermo Fisher Scientific), and sequencing was performed on the Illumina MiSeq platform (Illumina, Inc., San Diego, CA, USA) using the MiSeq reagent kit v2 at 300 cycles. Eleven samples (7 from calving [4 healthy and 3 metritic cows], 3 from 2 dpp [all healthy], and 1 from 6 ± 2 dpp [healthy]) failed to amplify and therefore were not sequenced. Additionally, one sequenced sample (metritic at 6 ± 2 dpp) was excluded (12 total) because of a low number of reads (42 reads); therefore, 60 samples (8 healthy and 9 metritic at 0 dpp, 9 healthy and 12 metritic at 2 dpp, and 11 healthy and 11 metritic at 6 ± 2 dpp) were used for data analysis.
Data analysis.
Taxonomy (phylum, class, order, family, genus, and species) and relative abundance were identified using the MiSeq Reporter proprietary metagenomics workflow based on the Greengenes database (http://greengenes.lbl.gov).
Statistical analysis.
Rarefaction curves of 60 samples based on the number of species were built using the Metagenomics RAST server (MG-RAST, http://metagenomics.anl.gov). Chao1 and Shannon indices were calculated using the R software (http://www.r-project.org). All continuous variables, such as relative abundance of bacteria, number of reads and number of species per sample, and species richness and diversity, were analyzed by analysis of variance (ANOVA) for repeated measures using the Mixed procedure of SAS 9.3 (SAS Institute Inc., Cary, NC, USA). Models included the fixed effects of health status, time (0, 2, and 6 ± 2 dpp), and interaction between health status and time. Metritic and healthy cows at each time point were compared when there was a main effect of health status or an interaction between health status and time. Data are presented as means and standard errors of the means (SEM). Heat maps and hierarchical clustering of microbial communities at the phylum and genus level were generated based on Euclidean distance using the R software. To identify shared and unique bacterial genera, Venn diagrams were built using the Bioinformatics & Evolutionary Genomics tool (http://bioinformatics.psb.ugent.be/webtools/Venn/). Discriminant analysis was performed using JMP Pro 11. The relative abundance of the 25 most abundant genera found at 0, 2, or 6 ± 2 dpp was used to discriminate cows that did or did not have metritis at 6 ± 2 dpp. A stepwise selection method was performed, and genera that did not contribute to the discrimination between groups were excluded. Microbial genera with canonical loadings of ≤−0.3 or ≥0.3 were considered important predictors of health status. Correlations were examined using Spearman's rank in SAS. For all statistical comparisons, differences at a P of ≤0.05 were considered significant. P values for comparisons between healthy and metritic cows were adjusted for multiple comparisons using the step-down Bonferroni adjustments available in the Multitest procedure of SAS. Both unadjusted and adjusted (adj) P values are presented.
Metagenome sequence accession numbers.
Metagenome sequences were deposited in the MG-RAST server under the identification numbers listed in Table S1 in the supplemental material (4566112 to 4566156 and 4566158 to 4566172).
RESULTS
Sequencing results, number of reads and species, and Chao1 and Shannon indices.
Sequencing of the V4 region of the bacterial 16S rRNA gene of 60 swab samples resulted in 4,979,168 reads, with an average of 82,986 ± 3,674 (SEM) reads per sample after quality filtering. Rarefaction curves reached plateau in all but one of the samples sequenced, indicating that sampling depth was sufficient to describe uterine microbiota (see Fig. S1 in the supplemental material). Species richness, the mean number of different species, was lower (P = 0.01) for metritic than for healthy cows at 6 ± 2 dpp (Fig. 1A). The Chao1 index, a measure of species richness based on the number of rare species (singletons and doublets), was also significantly lower (P < 0.01) in metritic cows at 0 and 6 ± 2 dpp (Fig. 1B). The mean number of reads, a measure of species abundance, and the Shannon index, a measure of species diversity which combines species abundance and evenness, were not affected (P > 0.15) (see Fig. S2 in the supplemental material). Taken together, these data indicate that uterine microbiota from metritic cows had decreased richness compared with that from healthy cows but the two had similar levels of diversity.
FIG 1.
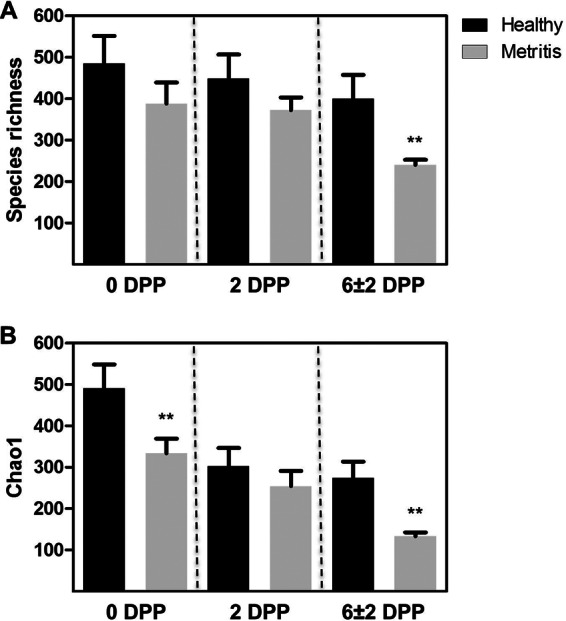
Estimates of alpha diversity. (A) Species richness calculated as the number of different species. (B) Species richness calculated as the Chao1 index. Data are presented as means ± SEM. **, significant difference between the healthy and metritic cows at P < 0.01 using ANOVA.
Comparisons at the phylum level.
We found 28 phyla in all samples; Bacteroidetes (28.9%), Proteobacteria (22.5%), Fusobacteria (20.2%), Firmicutes (15.8%), Tenericutes (10.7%), Actinobacteria (1.0%), Spirochaetes (0.5%), and Verrucomicrobia (0.1%) were the eight most abundant phyla in the uteri of all dairy cows, accounting for 99.7% of bacterial communities. A heat map analysis showed a change in uterine microbiota from calving until establishment of metritis, with an abundance of Proteobacteria at calving and an abundance of Bacteroidetes and Fusobacteria at metritis diagnosis (Fig. 2A). To identify the differences in uterine microbiota between healthy and metritic cows, we compared the relative abundances of the five most abundant phyla between metritic and healthy cows across time (Fig. 2B). Bacteroidetes were more abundant in metritic cows than in healthy cows at 6 ± 2 dpp (53.1 versus 27.3%; P < 0.01; adj P = 0.03) (Fig. 2C). Bacteroidetes increased from 0 to 6 ± 2 dpp in metritic cows (27.5 versus 53.1%; P < 0.01), whereas the abundance was unchanged in healthy cows (26.5 versus 27.3%; P = 0.93). The abundances of Fusobacteria in metritic and healthy cows were similar (P > 0.75) at all time points, but there was an overall increase in abundance at 2 (26.2%) and 6 ± 2 (24.1%) dpp compared with that at 0 dpp (8.4%; P < 0.02) (Fig. 2C).
FIG 2.

Bacterial composition and abundance at phylum level. (A) Heat map analysis using the Euclidean distance. Columns represent 60 dairy cows, and rows represent the eight most abundant bacterial phyla. The color of each cell indicates the relative abundance of bacterial phyla. The heat map shows the shift of uterine microbiota from calving (Calving; 0 dpp) until establishment of metritis (Metritis) at 6 ± 2 dpp. “No metritis” shows samples from healthy and metritic cows at 2 dpp and healthy cows at 6 ± 2 dpp. (B) Distribution of the five most abundant phyla in healthy and metritic cows at 0, 2, and 6 ± 2 dpp. (C) Relative abundances of Bacteroidetes and Fusobacteria in healthy and metritic cows from 0 to 6 ± 2 dpp. **, significant difference between healthy and metritic cows at P < 0.01 using ANOVA.
Comparisons at the genus level.
We found 824 genera in all samples. Table S2 in the supplemental material shows the 30 most abundant genera, accounting for 91.1% of bacterial microbiota. Fusobacterium (15.7%) was the most abundant genus, followed by Bacteroides (13.9%), Coxiella (12.7%), Porphyromonas (9.9%), and Ureaplasma (5.2%). Prevotella (2.9%), Escherichia (2.1%), and Arcanobacterium/Trueperella (0.3%) were the 10th, 12th, and 27th most prevalent genera, respectively. The Illumina MiSeq Reporter metagenomics workflow lumps the genus Trueperella with the genus Arcanobacterium; therefore, we refer to it as Arcanobacterium/Trueperella.
We evaluated whether a unique bacterial microbiota was associated with the development of metritis by examining core genera shared by all of the dairy cows within each health status group (Fig. 3A) and genera that were found in any of the dairy cows within each group (Fig. 3B). Overall, most genera were shared between metritic and healthy cows at 0 and 6 ± 2 dpp. The similarity in bacterial genera between metritic and healthy cows was 48.6% at calving, 62.8% at 2 dpp, and 64.6% at 6 ± 2 dpp when only core genera were evaluated and 77.4% at calving, 78.6% at 2 dpp, and 59.8% at 6 ± 2 dpp when genera found in any of the cows were evaluated (see Fig. S3 in the supplemental material). Among bacterial genera that were shared are those that were prevalent and/or are considered important for the development of metritis, such as Fusobacterium, Bacteroides, Escherichia, and Arcanobacterium/Trueperella. Among genera that were not shared are those that were rare (<0.01% prevalence), such as Pandoraea, Pseudonocardia, Wautersiella, Ethanoligenens, and others. Since most bacterial genera were shared among metritic and healthy cows at all times, it is likely that changes in bacterial abundance and interactions among shared bacteria are more important for the development of metritis than unique differences in bacterial communities.
FIG 3.
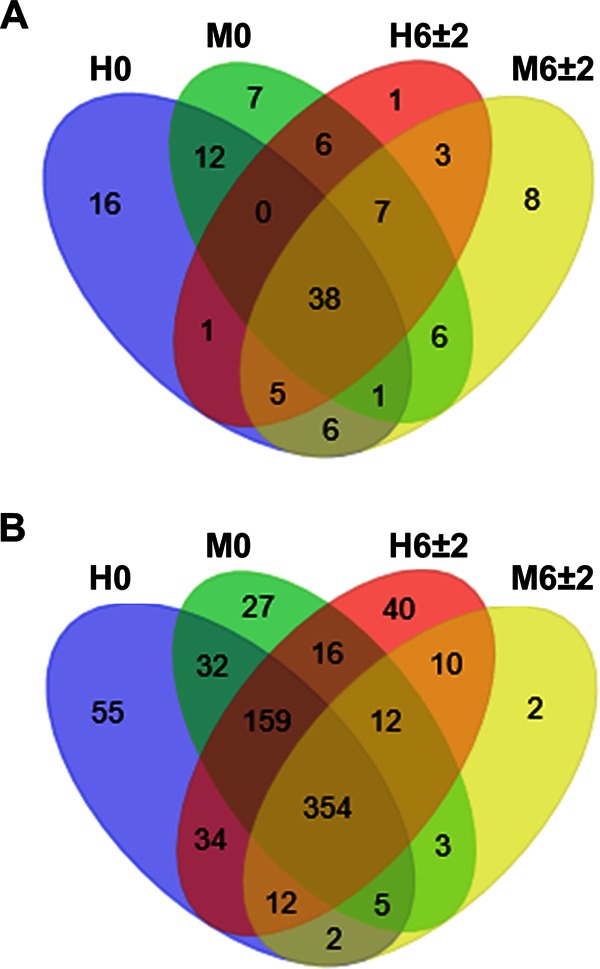
Venn diagrams showing numbers of shared and unique genera among uterine bacterial communities. (A) Number of genera shared by all of the dairy cows within each health status group. (B) Number of genera found in any of the dairy cows within each group. H, healthy; M, metritis; 0, calving; 6 ± 2, 6 ± 2 dpp.
To investigate this hypothesis, we evaluated the progression of the 15 most abundant genera (≥1% abundance; representing 84.3% of uterine microbiota) using a heat map analysis (Fig. 4A) and compared the relative abundances of the five most abundant genera (≥5% abundance) from 0 dpp to 6 ± 2 dpp (Fig. 4B). The overall relative abundances for the 30 most prevalent bacterial genera are presented in Table S2 in the supplemental material. Similarly to the results at the phylum level, the heat map (Fig. 4A) and pie chart (Fig. 4B) showed a change in uterine microbiota from calving until establishment of metritis. There was an overall increase in the relative abundances of Bacteroides, Fusobacterium, and Porphyromonas bacteria from 0 to 6 ± 2 dpp, whereas Ureaplasma increased from 0 to 2 dpp and then decreased from 2 to 6 ± 2 dpp and Coxiella decreased dramatically from 0 to 6 ± 2 dpp. Bacteroides was significantly more abundant in metritic than in healthy cows at 6 ± 2 dpp (28.3 versus 12.5%; P < 0.01; adj P = 0.02). Ureaplasma showed a lower abundance in metritic cows than in healthy cows at 2 dpp (5.8 versus 19.0%; P = 0.04), but that was not significant after adjustment for multiple comparisons (adj P = 0.22). Fusobacterium, Coxiella, and Porphyromonas were similar at all time points (P > 0.05; adj P > 0.25).
FIG 4.

Bacterial composition and abundance at genus level. (A) Heat map analysis using the Euclidean distance. Columns represent 60 dairy cows, and rows represent the 15 most abundant bacterial genera. The color of each cell indicates the relative abundance of bacterial genera. The heat map shows the shift of uterine microbiota from calving (Calving; 0 dpp) until establishment of metritis (Metritis) at 6 ± 2 dpp. “No metritis” shows samples from healthy and metritic cows at 2 dpp and healthy cows at 6 ± 2 dpp. (B) Distribution of the five most abundant genera in healthy and metritic cows at 0, 2, and 6 ± 2 dpp.
Furthermore, we evaluated the correlations between the 30 most prevalent bacterial genera and clinical signs of metritis at 6 ± 2 dpp (Fig. 5; also see Table S3 in the supplemental material). Although cows with a uterine discharge score of ≤4 were classified as healthy, the relative abundance of Bacteroides organisms increased as the uterine discharge score increased (Spearman rs = 0.51; P = 0.02) (Fig. 5A). Likewise, the relative abundance of Fusobacterium bacteria increased as the uterine discharge score increased (Spearman rs = 0.49; P = 0.02) (Fig. 5B). This indicates that Bacteroides and Fusobacterium are highly associated with metritis. On the other hand, Sneathia, Mycoplasma, Escherichia, Pedobacter, Serratia, “Candidatus Blochmannia,” Treponema, Oscillospira, Pasteurella, and Mannheimia were all negatively correlated (P ≤ 0.05) with uterine discharge score (see Table S3). Interestingly, the abundance of Fusobacterium organisms was positively correlated with the abundance of Bacteroides organisms (Spearman rs = 0.51; P = 0.02) (Fig. 5C). Fusobacterium was also positively correlated (P = 0.01) with Helcococcus, which showed a positive correlation (P = 0.05) with uterine discharge score. Meanwhile, Bacteroides was positively correlated (P = 0.03) with Filifactor, which was positively correlated (P ≤ 0.05) with Porphyromonas, Peptoniphilus, Peptostreptococcus, and Campylobacter. Porphyromonas, Peptoniphilus, and Peptostreptococcus were positively correlated (P ≤ 0.05) with Prevotella. Escherichia, Ruminococcus, Blautia, Clostridium, Pedobacter, Serratia, Treponema, Oscillospira, Caloramator, and Pasteurella were negatively correlated (P ≤ 0.05) with Bacteroides and/or Fusobacterium.
FIG 5.

Associations of Bacteroides and Fusobacterium with metritis. (A) Correlation between uterine discharge score and Bacteroides (Spearman rs = 0.51; P = 0.02). Cows were assigned to five groups according to uterine discharge score: 1, not fetid normal lochia, viscous, clear, red, or brown; 2, cloudy mucoid discharge with flecks of pus; 3, not fetid mucopurulent discharge with <50% pus; 4, not fetid mucopurulent discharge with ≥50% pus; 5, fetid red-brownish, watery discharge. Cows with a discharge score of ≤4 were classified as healthy, and cows with a score of 5 were classified as metritic at 6 ± 2 days postpartum. (B) Correlation between uterine discharge score and Fusobacterium (Spearman rs = 0.49; P = 0.02). (C) Correlation between Bacteroides and Fusobacterium (Spearman rs = 0.51; P = 0.02).
Because of the complex interactions among the main bacteria, we used discriminant analysis to identify the most important bacteria involved in the development of metritis. Discriminant analysis was performed using the 25 most abundant genera in the uteri of cows at 0, 2, or 6 ± 2 dpp. Abundance was evaluated at each time point separately. Discriminant analysis is a multivariate procedure used to predict to which group a case belongs. The discriminant function is used to calculate canonical scores, which are plotted to visualize the separation between groups. The goal is to have an equation that has strong discriminatory power between groups. The discriminant analysis showed that the discriminant function was highly significant (P < 0.001), and all cows were correctly discriminated at all time points (Fig. 6A; also see Table S4 in the supplemental material). Canonical structure coefficients (canonical loadings), which represent the correlation between each variable (bacterial genus) and the discriminant function adjusted for intercorrelations among microbial genera, were used to evaluate the importance of each bacterial genus. Canonical structure coefficients for bacterial genera that contributed to the discriminant function are presented in Fig. 6B. A canonical loading cutoff of ±0.3 was used. At 0 dpp, the top three genera that loaded most strongly for the metritis group did not reach the 0.3 cutoff (Helcococcus [0.27], Bacteroides [0.24], and Fusobacterium [0.20]); however, Escherichia (−0.43) loaded significantly for the healthy group. At 2 dpp, Bacteroides (0.30) was the only important variable for the metritis group, whereas “Candidatus Blochmannia” (−0.50) and Ureaplasma (−0.31) were important variables for the healthy group. At 6 ± 2 dpp, Bacteroides (0.54), Filifactor (0.39), Porphyromonas (0.38), and Fusobacterium (0.32) were important variables to predict metritis, whereas Sneathia (−0.41), Acholeplasma (−0.41), “Candidatus Blochmannia” (−0.41), Ruminococcus (−0.31), and Pedobacter (−0.30) were important variables to predict the healthy group.
FIG 6.

Discriminant analysis using the 25 most abundant genera. (A) Canonical discriminant analysis of uterine swab samples from cows at 0, 2, and 6 ± 2 dpp. Metritic cows were significantly discriminated from healthy cows from 0 to 6 ± 2 dpp. An ellipse indicates the 95% confidence region to contain the true mean of the group, and a plus symbol indicates the center (centroid) of each group. (B) Canonical structure coefficients representing the correlation between each bacterial genus and the discriminant function. Bacteria with canonical structure coefficients of ≤−0.3 or ≥0.3 are considered important to predict between healthy and metritic cows.
Taken together, our data indicate that Bacteroides is the main genus associated with metritis because it showed higher abundance in cows with metritis, it was positively correlated with uterine discharge score, and it loaded most strongly in the discriminant analysis at 6 ± 2 dpp. The correlation and discriminant analyses also indicate that other genera, such as Fusobacterium, Helcococcus, Filifactor, Porphyromonas, Peptoniphilus, Peptostreptococcus, Campylobacter, and Prevotella, may be important for the development of metritis. Figure 7 shows the microbial interactions among bacteria that may be important for the development of metritis, highlighting Bacteroides and Fusobacterium, which were positively correlated with uterine discharge score and loaded significantly for the metritis group in the discriminant analysis, but also showing bacteria that were positively correlated with these main bacteria, such as Filifactor and Helcococcus, and other bacteria that were positively correlated with Filifactor and among themselves, such as Peptoniphilus, Porphyromonas, Peptostreptococcus, and Prevotella. There were also several bacteria associated with uterine health, such as Escherichia, “Candidatus Blochmannia,” Sneathia, Pedobacter, Mycoplasma, Ureaplasma, Bifidobacterium, Acholeplasma, Ruminococcus, Serratia, Treponema, Oscillospira, Pasteurella, and Mannheimia. Figure 8 shows the microbial interactions associated with uterine health, highlighting Escherichia, “Candidatus Blochmannia,” Sneathia, and Pedobacter, which were negatively correlated with uterine discharge score and loaded significantly for the healthy group in the discriminant analysis, but also showing bacteria that were positively correlated with these main bacteria, such as Ruminococcus, Treponema, Oscillospira, Pasteurella, Acholeplasma, and Mannheimia, and other bacteria that were positively correlated with them and among themselves, such as Serratia, Bifidobacterium, Ureaplasma, and Mycoplasma.
FIG 7.
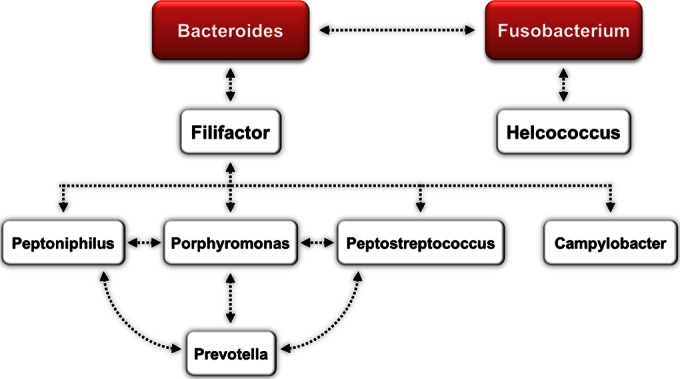
Microbial interactions associated with metritis. Bacteria in red boxes were positively correlated with uterine discharge score (higher score indicates worse uterine health) and loaded significantly for the metritis group in the discriminant analysis. Bacteria in the white boxes were positively correlated with the bacteria in the red boxes. Arrows indicate positive correlations (P ≤ 0.05).
FIG 8.
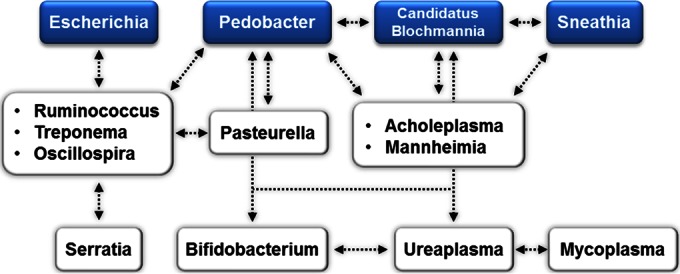
Microbial interactions associated with uterine health. Bacteria in blue boxes were negatively correlated with uterine discharge score (lower score indicates uterine health) and loaded significantly for the healthy group in the discriminant analysis. Bacteria in the white boxes were positively correlated with the bacteria in the blue boxes. Arrows indicate positive correlations (P ≤ 0.05).
DISCUSSION
Our study used high-throughput sequencing on the Illumina MiSeq platform, which is one of the most powerful tools to investigate the microbiome associated with health and disease (13, 14). The rarefaction curves confirmed that the MiSeq platform provided deeper sequencing and greater coverage than what has been previously reported using pyrosequencing (5, 7) or clone library sequencing (6). It is not clear why 11 samples failed to amplify and one sample resulted in a low number of reads, but samples at calving were particularly more likely not to amplify (7/11); therefore, low bacterial abundance at calving may be an important factor contributing to a lack of PCR amplification.
Our data showed that uterine microbiota was already established shortly after calving and that microbiota changed rapidly, favoring anaerobes such as Bacteroidetes and Fusobacteria. This change is probably a consequence of stoppage of blood flow through the placenta, low oxygen tension, and necrosis of caruncular epithelium, which, along with substrates such as remaining blood and amniotic and allantoic fluids, may create the ideal conditions for the growth of anaerobes (1, 15, 16). In this study, Bacteroidetes was the only phylum with significantly higher abundance in metritic than in healthy cows, therefore indicating that Bacteroidetes may be an important phylum for the development of metritis in dairy cows. Two recent studies using clone library sequencing found either an overwhelming predominance of Fusobacteria in metritic cows (5) or an overall predominance of Bacteroidetes in both metritic and healthy cows (6). One study using pyrosequencing observed a predominance of Bacteroidetes and Fusobacteria in both metritic and healthy cows (7). One study using pyrosequencing observed higher abundances of Bacteroidetes, Tenericutes, and Fusobacteria in cows with endometritis than in healthy cows (8). A recent study using MiSeq sequencing found a predominance of the families Fusobacteriaceae, Bacteroidaceae, Porphyromonadaceae, and Pasteurellaceae in the uterine fluid of cows with pyometra (17). Consistent with our findings, Santos et al. described that the microbiota of healthy cows was dominated by Proteobacteria and Tenericutes (5). Since metritis is a complex disease, some conflicting findings are expected; therefore, further studies in different parts of the country and/or the world are needed to further elucidate the role of each phylum in the development of metritis.
Differences in relative abundance, correlation with uterine discharge score, and discriminant analysis showed that Bacteroides, followed by Fusobacterium, was the most important genus associated with metritis. The correlation with uterine discharge score, the correlation with Bacteroides and Fusobacterium, and the discriminant analysis showed that other bacteria, such as Helcococcus, Filifactor, Porphyromonas, Peptoniphilus, Peptostreptococcus, Campylobacter, and Prevotella, may also be important for the development of metritis. Particularly, Helcococcus, Filifactor, and Porphyromonas were directly associated with metritis by either having a positive correlation with the uterine discharge score (Helcococcus) or being associated with the metritis group in the discriminant analysis (Filifactor and Porphyromonas). Therefore, it is possible that Bacteroides acts synergistically with Fusobacterium and other bacteria, such as Helcococcus, Filifactor, and Porphyromonas, to cause metritis in dairy cows. The pathogenicity of Bacteroides in mixed bacterial infections is believed to occur through inhibition of phagocytosis and killing by polymorphonuclear leukocytes (PMN); Bacteroides was found to produce short-chain fatty acids that directly inhibited phagocytosis and killing of bacteria by PMN (18). In models of necrobacillosis, the lethal dose of F. necrophorum could be greatly decreased by coinfection with Bacteroides spp., T. pyogenes, or E. coli (19, 20). Porphyromonas levii has also been shown to produce an immunoglobulin protease that can cleave bovine IgG and reduce PMN phagocytosis (21).
Early culture-based studies found an association between Bacteroides spp. and uterine discharge score; however, P. levii (formerly Bacteroides levii) and Prevotella melaninogenica (formerly Bacteroides melaninogenicus) were all combined into Bacteroides spp., which now can be interpreted as the phylum Bacteroidetes (3). Early culture-based studies also found an association between F. necrophorum and endometritis (i.e., purulent uterine discharge after 21 dpp) or pyometra and a correlation between Bacteroides and F. necrophorum (3, 22, 23). Other studies found that cows with retained placenta, a major risk factor for metritis, usually had mixed bacterial infections containing P. levii, F. necrophorum, and T. pyogenes (24). These studies pointed to the importance of Bacteroides spp. and P. levii for causing endometritis and potentially metritis. Recent studies using clone library sequencing (5, 6) or pyrosequencing (7) focused on characterization of phylum but also tried to identify the main species in healthy and metritic cows. An advantage of the clone library sequencing studies is the ability to sequence large (6) or multiple (5) regions of the 16S rRNA gene; however, a disadvantage is the low sequencing depth and coverage. Pyrosequencing and Illumina sequencing provide greater sequencing depth and coverage, but the shorter reads may provide inaccurate species classification, especially for species with high 16S rRNA gene similarity, such as Escherichia and Shigella species (25). Within the phylum Bacteroidetes, Peng et al. observed that Bacteroides pyogenes, Bacteroides heparinolyticus, and P. levii were the most prevalent species (6). Santos et al. reported that the genus Porphyromonas was the most prevalent in the phylum Bacteroidetes, but species like B. heparinolyticus and Bacteroides denticanum (also known as B. denticanoris) were also identified (5). Within the phylum Fusobacteria, recent studies using clone library sequencing (5, 6) or pyrosequencing (7) observed that F. necrophorum was the most prevalent species in metritic cows (5) or in both healthy and metritic cows (6, 7). Although sequencing of only the V4 region may be less accurate for assigning taxonomy at the species level, our observations at the species level (see Table S5 in the supplemental material) corroborate previous observations (5–7). A recent study using pyrosequencing observed that endometritic cows had higher relative abundances of Ureaplasma, Fusobacterium, Bacteroides, and Porphyromonas bacteria than healthy cows (8); therefore, apart from Ureaplasma, our observations in metritic cows corroborate the findings in endometritic cows.
Members of the genus Helcococcus, found to be associated with metritis in this study, were also associated with endometritis in the study by Machado et al. (8). Herein, Helcococcus was positively correlated with the uterine discharge score, and Helcococcus ovis was identified as the most prevalent species for this genus. H. ovis was recently reported to be an important pathogen associated with bovine valvular endocarditis (26); therefore, it may be an important species associated with uterine disease. In addition, we recognized Filifactor as an important genus because it was associated with the metritis group in the discriminant analysis and it was positively correlated with Bacteroides. Filifactor villosus was the main species from this genus (see Table S5 in the supplemental material). Therefore, further investigation of the role of Helcococcus and Filifactor and the main species from these genera in the development of metritis is warranted. Peptoniphilus was also associated with endometritis in the study by Machado et al. (8). The genus Prevotella was positively correlated with bacteria considered important, such as Porphyromonas, which indicates an indirect association with metritis. The most prevalent species from the genus Prevotella was Prevotella albensis (0.6%) (see Table S5). Previous culture-based studies have identified P. melaninogenica as an important pathogen for the development of endometritis (2, 3); however, none of the metritic cows and only one healthy cow had P. melaninogenica. Similarly, Machado et al. identified Prevotella as an important genus in cows with endometritis; however, the analysis at the species level was not presented (8). Therefore, further investigation into the importance of Prevotella spp. in the development of metritis is warranted.
Intriguingly, the genera Trueperella and Escherichia, which contain species commonly associated with uterine disease, such as Trueperella pyogenes and Escherichia coli (1, 2, 27), were not associated with metritis in this study. In fact, Escherichia was associated with uterine health. Herein, we identified T. pyogenes as the most prevalent species of the genus Trueperella and E. coli as the second most prevalent species of the genus Escherichia (see Table S5 in the supplemental material). It is likely that species-intrinsic factors may be more relevant to the development of uterine disease than abundance. For instance, previous studies have observed that uterine disease is associated with the presence of specific virulence factor genes, such as fimA in T. pyogenes (28) or fimH in E. coli (29).
In conclusion, we observed that a uterine microbiota that was shared among metritic and healthy cows was present in the uterus shortly after calving. The microbiota changed rapidly, favoring anaerobes such as Bacteroidetes and Fusobacteria. Bacteroides was the main genus associated with metritis, followed by Fusobacterium. Other bacteria, such as Helcococcus, Filifactor, Porphyromonas, Peptoniphilus, Peptostreptococcus, Campylobacter, and Prevotella, may also be important for the development of metritis. There were also bacteria associated with uterine health, such as “Candidatus Blochmannia,” Escherichia, Sneathia, and Pedobacter.
Supplementary Material
ACKNOWLEDGMENTS
We thank the owners and staff of Alliance Dairy for allowing the use of their cows in this experiment.
This project was supported by the USDA-NIFA-CRIS program (accession no. 1002880).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01753-15.
REFERENCES
- 1.Sheldon IM, Williams EJ, Miller AN, Nash DM, Herath S. 2008. Uterine diseases in cattle after parturition. Vet J 176:115–121. doi: 10.1016/j.tvjl.2007.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams EJ, Fischer DP, Pfeiffer DU, England GC, Noakes DE, Dobson H, Sheldon IM. 2005. Clinical evaluation of postpartum vaginal mucus reflects uterine bacterial infection and the immune response in cattle. Theriogenology 63:102–117. doi: 10.1016/j.theriogenology.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 3.Dohmen MJW, Lohuis JACM, Huszenicza G, Nagy P, Gacs M. 1995. The relationship between bacteriological and clinical findings in cows with subacute/chronic endometritis. Theriogenology 43:1379–1388. doi: 10.1016/0093-691X(95)00123-P. [DOI] [Google Scholar]
- 4.Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos TM, Gilbert RO, Bicalho RC. 2011. Metagenomic analysis of the uterine bacterial microbiota in healthy and metritic postpartum dairy cows. J Dairy Sci 94:291–302. doi: 10.3168/jds.2010-3668. [DOI] [PubMed] [Google Scholar]
- 6.Peng Y, Wang Y, Hang S, Zhu W. 2013. Microbial diversity in uterus of healthy and metritic postpartum Holstein dairy cows. Folia Microbiol (Praha) 58:593–600. doi: 10.1007/s12223-013-0238-6. [DOI] [PubMed] [Google Scholar]
- 7.Santos TM, Bicalho RC. 2012. Diversity and succession of bacterial communities in the uterine fluid of postpartum metritic, endometritic and healthy dairy cows. PLoS One 7:e53048. doi: 10.1371/journal.pone.0053048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machado VS, Oikonomou G, Bicalho ML, Knauer WA, Gilbert R, Bicalho RC. 2012. Investigation of postpartum dairy cows' uterine microbial diversity using metagenomic pyrosequencing of the 16S rRNA gene. Vet Microbiol 159:460–469. doi: 10.1016/j.vetmic.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 9.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108(Suppl 1):S4516–S4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lima FS, Vieira-Neto A, Vasconcellos GS, Mingoti RD, Karakaya E, Sole E, Bisinotto RS, Martinez N, Risco CA, Galvao KN, Santos JE. 2014. Efficacy of ampicillin trihydrate or ceftiofur hydrochloride for treatment of metritis and subsequent fertility in dairy cows. J Dairy Sci 97:5401–5414. doi: 10.3168/jds.2013-7569. [DOI] [PubMed] [Google Scholar]
- 11.Sheldon IM, Lewis GS, LeBlanc S, Gilbert RO. 2006. Defining postpartum uterine disease in cattle. Theriogenology 65:1516–1530. doi: 10.1016/j.theriogenology.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Lima FS, Oikonomou G, Lima SF, Bicalho MLS, Ganda EK, de Oliveira Filho JC, Lorenzo G, Trojacanec P, Bicalho RC. 2015. Prepartum and postpartum rumen fluid microbiomes: characterization and correlation with production traits in dairy cows. Appl Environ Microbiol 81:1327–1337. doi: 10.1128/AEM.03138-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blainey PC, Milla CE, Cornfield DN, Quake SR. 2012. Quantitative analysis of the human airway microbial ecology reveals a pervasive signature for cystic fibrosis. Sci Transl Med 4:153ra130. doi: 10.1126/scitranslmed.3004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin R, Miquel S, Langella P, Bermudez-Humaran LG. 2014. The role of metagenomics in understanding the human microbiome in health and disease. Virulence 5:413–423. doi: 10.4161/viru.27864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer B, Bavister BD. 1993. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil 99:673–679. doi: 10.1530/jrf.0.0990673. [DOI] [PubMed] [Google Scholar]
- 16.Gier HT, Marion GB. 1968. Uterus of the cow after parturition: involutional changes. Am J Vet Res 29:83–96. [PubMed] [Google Scholar]
- 17.Knudsen LR, Karstrup CC, Pedersen HG, Agerholm JS, Jensen TK, Klitgaard K. 2015. Revisiting bovine pyometra—new insights into the disease using a culture-independent deep sequencing approach. Vet Microbiol 175:319–324. doi: 10.1016/j.vetmic.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Rotstein OD. 1993. Interactions between leukocytes and anaerobic bacteria in polymicrobial surgical infections. Clin Infect Dis 16(Suppl 4):S190–S194. doi: 10.1093/clinids/16.Supplement_4.S190. [DOI] [PubMed] [Google Scholar]
- 19.Smith GR, Till D, Wallace LM, Noakes DE. 1989. Enhancement of the infectivity of Fusobacterium necrophorum by other bacteria. Epidemiol Infect 102:447–458. doi: 10.1017/S0950268800030168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith GR, Barton SA, Wallace LM. 1991. Further observations on enhancement of the infectivity of Fusobacterium necrophorum by other bacteria. Epidemiol Infect 106:305–310. doi: 10.1017/S0950268800048457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lobb DA, Loeman HJ, Sparrow DG, Morck DW. 1999. Bovine polymorphonuclear neutrophil-mediated phagocytosis and an immunoglobulin G2 protease produced by Porphyromonas levii. Can J Vet Res 63:113–118. [PMC free article] [PubMed] [Google Scholar]
- 22.Ruder CA, Sasser RG, Williams RJ, Ely JK, Bull RC, Butler JE. 1981. Uterine infections in the postpartum cow. II. Possible synergistic effect of Fusobacterium necrophorum and Corynebacterium pyogenes. Theriogenology 15:573–580. [Google Scholar]
- 23.Olson JD, Ball L, Mortimer RG, Farin PW, Adney WS, Huffman EM. 1984. Aspects of bacteriology and endocrinology of cows with pyometra and retained fetal membranes. Am J Vet Res 45:2251–2255. [PubMed] [Google Scholar]
- 24.Bekana M, Jonsson P, Ekman T, Kindahl H. 1994. Intrauterine bacterial findings in postpartum cows with retained fetal membranes. Zentralbl Veterinarmed A 41:663–670. doi: 10.1111/j.1439-0442.1994.tb00134.x. [DOI] [PubMed] [Google Scholar]
- 25.Chakravorty S, Helb D, Burday M, Connell N, Alland D. 2007. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods 69:330–339. doi: 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kutzer P, Schulze C, Engelhardt A, Wieler LH, Nordhoff M. 2008. Helcococcus ovis, an emerging pathogen in bovine valvular endocarditis. J Clin Microbiol 46:3291–3295. doi: 10.1128/JCM.00867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bicalho ML, Machado VS, Oikonomou G, Gilbert RO, Bicalho RC. 2012. Association between virulence factors of Escherichia coli, Fusobacterium necrophorum, and Arcanobacterium pyogenes and uterine diseases of dairy cows. Vet Microbiol 157:125–131. doi: 10.1016/j.vetmic.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 28.Santos TM, Caixeta LS, Machado VS, Rauf AK, Gilbert RO, Bicalho RC. 2010. Antimicrobial resistance and presence of virulence factor genes in Arcanobacterium pyogenes isolated from the uterus of postpartum dairy cows. Vet Microbiol 145:84–89. doi: 10.1016/j.vetmic.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Bicalho RC, Machado VS, Bicalho ML, Gilbert RO, Teixeira AG, Caixeta LS, Pereira RV. 2010. Molecular and epidemiological characterization of bovine intrauterine Escherichia coli. J Dairy Sci 93:5818–5830. doi: 10.3168/jds.2010-3550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.